
Abstract
Clostridium difficile TcdB harbors a glucosyltransferase that targets host Rho GTPases. However, the role of the enzyme activity in the induction of host intestinal disease has not been demonstrated. In this study, we established a mouse acute intestinal disease model by cecum injection of wild type and glucosyltransferase-deficient TcdB and a chronic model by delivering toxin intraluminally via engineered surrogate host Bacillus megaterium. We demonstrated, for the first time, that the glucosyltransferase activity of TcdB is essential for inducing disease symptoms and intestinal pathological responses that resemble human disease, highlighting the importance of targeting toxin glucosyltransferase activity for future therapy.
Introduction
Clostridium difficile is an anaerobic gram-positive bacterial species that can induce serious and potentially fatal inflammatory disease of the colon, and is the most prevalent cause of antibiotic-associated diarrhea and pseudomembranous colitis in nosocomial settings. Disease in patients with C. difficile infection is strongly associated with the two toxins, TcdA and TcdB.Citation1 Both toxins are glucosyltransferases targeting host Rho GTPase family proteinsCitation2,3 that induce cell rounding after disruption of the actin cytoskeleton and tight junctions in human colonocytes.Citation4 TcdB appears to be more clinically relevant for C. difficile virulence, as it is invariably associated with clinically isolated pathogenic strains.Citation5 The high potency of TcdB may in part be attributed to the efficient enzymatic activities of its glucosyltransferase domains.Citation6 Recently, several studies have indicated that glucosyltransferase activity of TcdB may be not required for inducing cell death process.Citation7,8 These in vitro findings raised debate over the role of the glucosyltransferases of the toxins in virulence, thus clinical relevant animal models are critical to evaluate the key effector activity in disease pathogenesis. In this study, utilizing two novel murine intestinal disease models, we demonstrated that TcdB glucosyltransferase activity is critical for the induction of intestinal histopathology and disease symptoms associated with CDI.
Results
Induction of chronic murine CDI in a Bacillus megaterium toxin delivery model
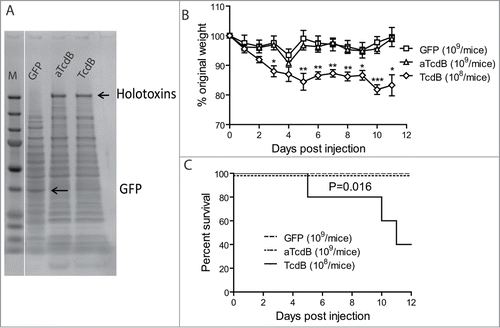
To study capacity of wild type and mutant toxins on inducing diseases after intraluminal toxin delivery, we developed a toxigenic CDI model by orally administrating mice with Bacillus megaterium cells expressing either green fluorescent protein (GFP) alone, wild-type TcdB, or the glucosyltransferase-deficient TcdB mutant (TcdB-W102A/D288N). SDS-PAGE analysis of total bacterial lysates demonstrated that similar levels of toxin were produced by these bacteria (). Repeated gavage of B. megaterium expressing wild type TcdB (108 bacteria/mouse, twice per day) induced persistent weight loss (), whereas gavage of 109 control GFP-expressing B. megaterium or glucosyltransferase-deficient aTcdB did not induce overall weight loss () or any other signs of clinical disease, although there was a transient weight loss on day 4 post gavage. 60% of mice succumbed in TcdB group whereas none of the mice treated with aTcdB died (, p < 0.05).
Figure 1. Disease induction of mice with oral injecting Bacillus megaterium expressing toxins. Mice were inoculated with live Bacillus megaterium bacterial cells expressing wild-type TcdB (108 bacteria/injection), aTcdB (109), or GFP (109) twice a day for 10 d. Mouse weights (A) and overall survival (B) were monitored over time. The statistical analysis of TcdB in comparison with GFP group. * represents p < 0.05 whereas ** shows p < 0.01 and *** shows p < 0.001. aTcdB group showed no statistical significance in either weight loss or survival curves when compared with GFP group.
Induction of acute murine CDI in a cecum toxin injection model
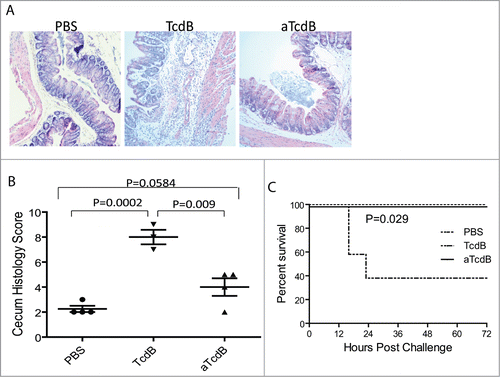
We further used acute murine CDI modelCitation9 to study the role of glucosyltransferase in disease pathogenesis. In this model, purified wild type or mutant toxins were directly injected into mouse ceca. Wild type TcdB (15 µg/mouse) induced acute intestinal tissue damages and inflammations. Compared with PBS injection, TcdB induced significant epithelial damages, submucosal edema, and immune cell infiltrations (). More than 60% of mice succumbed within 24 hr of TcdB injection (). On the contrary, cecum injection of glucosyltransferase-deficient TcdB (25 µg/mouse) did not induce any discernable clinical disease symptoms and all mice survived from challenge of a significantly higher dose than wild type TcdB. On intestinal histopathology level (), aTcdB at 25 μg per mouse induced a histopathology score higher than PBS group (p = 0.0586) but substantially lower when compared with ceca from mice injected with 15 μg of wild type TcdB (p = 0.009). Unlike wild type TcdB, aTcdB injection failed to induce neutrophil influx (), a hallmark of C. difficile induced intestinal inflammation.
Figure 2. Disease induction of mice with cecum injection of wild type and mutant toxins. (A) Histology of cecum tissues from mice injected with PBS, TcdB, or aTcdB. (B) Histology scores of cecum tissue sections. P values between the groups were shown. (C) Survival curves of mice after toxin cecum injection. P < 0.05 between TcdB and PBS groups.
Discussion
TcdB, one of the major virulence factors of the C. difficile, is a glucosyltransferase that glucosylates host Rho GTPases for intoxication. However, the role of this critical enzymatic activity in the induction of host intestinal diseases has not been demonstrated in animal disease models. Since recent in vitro studies have found glucosyltransferase-independent cytotoxic activities of TcdB,Citation7,8 it is now critically important to validate the key toxin virulence mechanisms in vivo. In this study, we utilized two murine intestinal disease models to study the virulence determinant of TcdB in mice. Our studies unequivocally demonstrate a major virulence role for TcdB glucosyltransferase domain in gastrointestinal disease pathogenesis.
Although a spore-challenge CDI model testing enzymatic deficient holotoxins has not yet been described, wild type and mutant C. difficile holotoxins have been successfully expressed in non-pathogenic, environmental B. megaterium,Citation4,10 which may also be used as a potential surrogate host to deliver recombinant toxin into the gastrointestinal tract. Multiple oral gavage of mice with TcdB-expressing B. megaterium over days were required to induce disease. Thus, although in this model it is difficult to know exactly how much toxin actually reaches target intestinal epithelial cells, the animals were likely exposed to low doses of the toxin that caused an accumulative toxin effect over time. B. megaterium expressing wild TcdB caused persistent weight loss and over 50% of mice eventually succumbed. Although the glucosyltransferase-deficient mutant aTcdB was expressed at similar levels to wild type TcdB in B. megaterium, no discernable clinical disease was evident in these animals even when administered at a 10-fold higher bacterial load. Weight loss and survival curves from mice treated with aTcdB are similar with those from mice treated with B. megaterium expressing GFP control. These data demonstrate that induction of CDI by delivering TcdB into intestine via B. megaterium is dependent on TcdB glucosyltransferase activity.
Comparing to B. megaterium toxin delivery model, the recent reported cecum toxin injection modelCitation9 allows injection of a precisely controlled amount of the toxins into the ceca of mice. Injection of mice with 15 μg of wild type TcdB induced around 50% mortality whereas 25 μg of wild type TcdB led to rapid death of all mice (data not shown). Thus we chose 15 μg of TcdB in this study. Although in this acute disease model after TcdB injection, mice did not develop intestinal pathology in early time points but significant intestinal tissue damages and inflammation were evident after 24 hr of toxin treatment. These results are consistent with previous study that typical intestinal epithelial damages and inflammation were observed 16 hr post toxin exposure.Citation9 Consistent with mouse spore-challenge induced CDI, mice developed intestinal tissue damage, colitis, and death. Thus, these data demonstrate that the purified TcdB alone causes disease symptoms and pathology that resemble CDI in animals and humans.
Consistent with the results from B. megaterium toxin delivery model, cecum injection of glucosyltransferase-deficient TcdB did not induce any discernable clinical disease symptoms and all mice survived from challenge of a significantly higher dose than wild type TcdB. Although aTcdB at 25 μg induced detectable histopathologic changes, the histopathological score is not significantly higher than that of PBS group but is substantially lower when compared with mice injected with 15 μg of wild type TcdB. Moreover, unlike wild type TcdB, aTcdB injection failed to induce neutrophil influx, a hallmark of C. difficile induced intestinal inflammation. At the lethal dose for wild type TcdB, aTcdB caused mild histological changes in mouse cecum, which may be due to possible residual glucosyltransferase activitiesCitation10 or non-glucosyltransferase activities that otherwise induce a rapid cell death in in vitro culture settings.Citation7,8 It is possible that the toxin in vitro exposure represents a significantly different environment as what actually occurs in vivo, thus observations from in vitro experimentations warrant verification by using clinically relevant disease models. Given that CDI represents a significant public health threat and novel therapeutic strategies that precisely target the bacterial virulence mechanisms are urgently needed, it is significant to study C. difficile toxin virulence determinants in disease pathogenesis using clinical relevant models.
In summary, by using two independent animal intestinal disease models, we consistently demonstrated that TcdB in vivo potency is critically dependent on the glucosyltransferase activity. These results are consistent with our previous studies using systemic toxemia model.Citation10,11 Thus our studies confirm the critical role of the glucosyltransferase in clinical disease pathogenesis, highlighting the importance of targeting this enzyme activity for future therapy.
Materials and Methods
Mice and toxins
6 to 8 week-old CD1 mice were purchased from Harlan Laboratory. All mice were housed in dedicated pathogen-free facilities in groups of 5 mice per cage under the same conditions. Food, water, bedding, and cages were autoclaved. All procedures involving mice were conducted under protocols approved by the Institutional Animal Care and Use Committee.
The full-length wild-type recombinant TcdB and glucosyltransferase-deficient aTcdB (TcdB-W102A/D288N) were purified from total crude extract of Bacillus megaterium as described previously.Citation4,10 Bacterial pellets from Bacillus megaterium expressing wild-type TcdB and aTcdB were harvested after xylose inductionCitation4 and aliquoted and stored at −80°C until used. These bacteria expressed an equal amount of toxin as determined by SDS-PAGE. B. megaterium expressing GFP under the same shuttle vector pHis-1522Citation4 was used as a control.
Cecum toxin injection surgery
Mice were anesthetized with ketamine : xylazine (20mg/ml : 2mg/ml, 0.1ml/mouse, i.m.). A midline laparotomy was performed and the cecum, ileum and colon were exposed. An insulin syringe (29G1/2) was inserted into the connection part between ileum and cecum and 100 µl of PBS or toxin was injected into the cecum. The gut was then returned to the abdomen, and the incision was closed with suture silk. Mice were put under heat lamp to recovery and returned to its home cage when they woke up from the anesthesia. Mice were closely monitored for 72 hr post-surgery and the survival was monitored.
Intestinal inflammation and histopathology
Four mice from each treatment group were sacrificed 24 hr post-surgery. Two sections of cecum were collected from each mouse. Sections were flushed with PBS and fixed inside 10% Phosphate Buffer Formalin. The tissues were sectioned and stained with hematoxylin and eosin for histopathological analysis. Overall damage was analyzed by a histologist blinded to the identity of each sample. Damage scores were graded based on 5 criteria each graded on a scale from 0 to 3 (normal, mild, moderate to severe) and added together to generate a score with a maximum value of 15. The criteria were: epithelial cell and architectural disruption, hemorrhagic congestion, mucosal edema, mucosal depletion, inflammatory cell infiltration and inflammation.
Oral challenge of mice with toxin-expressing B. megaterium
CD1 mice were orally administered with B. megaterium via gavage (0.1 ml/mouse) twice daily for 10 d. Mice were monitored closely for sign of disease and moribund animals were sacrificed. Weight loss was expressed as a percentage of body weight before challenge. Mice lost 20% of body weight were sacrificed.
Statistical analysis
Data were analyzed by Kaplan–Meier survival analysis with Logrank test of significance, analysis of variance, or by one-way ANOVA followed by Bonferroni post-tests using the Prism statistic software program. Results are expressed as mean ± standard error of mean. P < 0.05 was regarded as significantly different between groups.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by awards R01AI088748, R01DK084509, R56AI99458, and U19 AI109776 funded from the National Institute of Allergy and Infectious Diseases and National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health (NIH).
References
- Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 2005; 18:247-63; PMID:15831824; http://dx.doi.org/10.1128/CMR.18.2.247-263.2005.
- Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995; 375:500-3; PMID:7777059; http://dx.doi.org/10.1038/375500a0.
- Just I, Wilm M, Selzer J, Rex G, von Eichel-Streiber C, Mann M, Aktories K. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem 1995; 270:13932-6; PMID:7775453; http://dx.doi.org/10.1074/jbc.270.23.13932.
- Yang G, Zhou B, Wang J, He X, Sun X, Nie W, Tzipori S, Feng H. Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol 2008; 8:192; PMID:18990232; http://dx.doi.org/10.1186/1471-2180-8-192.
- King AM, Mackin KE, Lyras D. Emergence of toxin A-negative, toxin B-positive Clostridium difficile strains: epidemiological and clinical considerations. Future Microbiol 2015; 10:1-4; PMID:25598331; http://dx.doi.org/10.2217/fmb.14.115.
- Chaves-Olarte E, Weidmann M, Eichel-Streiber C, Thelestam M. Toxins A and B from Clostridium difficile differ with respect to enzymatic potencies, cellular substrate specificities, and surface binding to cultured cells. J Clin Invest 1997; 100:1734-41; PMID:9312171; http://dx.doi.org/10.1172/JCI119698.
- Chumbler NM, Farrow MA, Lapierre LA, Franklin JL, Haslam DB, Goldenring JR, Lacy DB. Clostridium difficile Toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog 2012; 8:e1003072; PMID:23236283; http://dx.doi.org/10.1371/journal.ppat.1003072.
- Wohlan K, Goy S, Olling A, Srivaratharajan S, Tatge H, Genth H, Gerhard R. Pyknotic cell death induced by Clostridium difficile TcdB: Chromatin condensation and nuclear blister are induced independently of the glucosyltransferase activity. Cell Microbiol 2014; 16(11):1678-92; PMID:24898616.
- D'Auria KM, Kolling GL, Donato GM, Warren CA, Gray MC, Hewlett EL, Papin JA. In vivo physiological and transcriptional profiling reveals host responses to Clostridium difficile toxin A and toxin B. Infect Immun 2013; 81:3814-24; PMID:23897615; http://dx.doi.org/10.1128/IAI.00869-13.
- Wang H, Sun X, Zhang Y, Li S, Chen K, Shi L, Nie W, Kumar R, Tzipori S, Wang J, et al. A chimeric toxin vaccine protects against primary and recurrent Clostridium difficile infection. Infect Immun 2012; 80:2678-88; PMID:22615245; http://dx.doi.org/10.1128/IAI.00215-12.
- Li S, Shi L, Yang Z, Zhang Y, Perez-Cordon G, Huang T, Ramsey J, Oezguen N, Savidge TC, Feng H. Critical Roles of Clostridium difficile Toxin B Enzymatic Activities in Pathogenesis. Infect Immun 2015; 83:502-13; PMID:25404023; http://dx.doi.org/10.1128/IAI.02316-14.