
Abstract
Helicobacter pylori is the dominant member of the gastric microbiota in over half of the human population of which 5–15% develop gastritis or gastric malignancies. Immune responses to H. pylori are characterized by mixed T helper cell, cytotoxic T cell and NK cell responses. The presence of Tregs is essential for the control of gastritis and together with regulatory CX3CR1+ mononuclear phagocytes and immune-evasion strategies they enable life-long persistence of H. pylori. This H. pylori-induced regulatory environment might contribute to its cross-protective effect in inflammatory bowel disease and obesity. Here we review host-microbe interactions, the development of pro- and anti-inflammatory immune responses and how the latter contribute to H. pylori's role as beneficial member of the gut microbiota. Furthermore, we present the integration of existing and new data into a computational/mathematical model and its use for the investigation of immunological mechanisms underlying initiation, progression and outcomes of H. pylori infection.
Introduction
H. pylori is a Gram-negative, microaerophilic bacterium within the class of Epsilonproteobacteria. It chronically colonizes the human gastric microenvironment and is one of the most genetically diverse bacterial species that has colonized the stomach since early in human evolution.Citation1,2 The study of H. pylori populations such as hpAfrica, hpEurope, hspEAsia, and hspAmerindCitation3 has delineated competition between strains and suggests co-adaptation and co-evolution with its host that parallels human migration throughout the globe.
Since its initial discovery by Marshall and Warren,Citation4 H. pylori has been associated with the development of chronic gastritis, peptic ulcer disease, gastric adenocarcinoma, and gastric mucosa-associated lymphoid tissue (MALT) lymphoma.Citation5,6 Although it is present in the stomach of over 50% of the human population worldwide, only 15% develop serious gastric and duodenal pathologies. Mounting clinical and epidemiological evidence suggests that the presence of H. pylori can protect from the development of various diseases including esophageal and cardial pathologies,Citation7-10 childhood asthma and allergies.Citation11-14 Originally viewed solely as a pathogen, recent studies are unveiling commensal and symbiotic roles for H. pylori as the predominant member of the human gastric microbiota.Citation15 Therefore, investigating the complex tolerance mechanisms that facilitate the co-existence between H. pylori and its human host may yield a deeper understanding of mechanisms of immunoregulation at the mucosal sites. This is a first step toward predicting pathogenic versus beneficial health outcomes of host-H. pylori interactions.
The life-long persistence of H. pylori in the human stomach suggests that the host response fails to clear the infection and induces an underlying regulatory response. Indeed, H. pylori induces a mixed immune response characterized by T helper (Th) 1, Th17 and regulatory T cell (Treg) responses. Whether H. pylori exerts a protective effect in the context of a dysregulated immune response or whether it contributes to cell damage and malignant transformation is dependent on host-microbial interactions.
This review sheds new light on the complex interactions of host- and microbial-factors in immune responses to H. pylori specifically emphasizing the role of inflammasome and TLR signaling in shaping the innate and adaptive T cell responses. We will further discuss novel mechanistic insights leading to H. pylori-induced gastritis and mechanisms of immune evasion by H. pylori and how this might help explain the role of H. pylori as an amphibiont at the interface between commensalism/symbiosis and pathogenicity. Furthermore, we will discuss the use of novel computational modeling approaches to systematically integrate existing knowledge and varied datasets into information processing representations of the mucosal immune system. These computational approaches have facilitated characterizing emerging behaviors and improving our systems-wide understanding of the mechanisms of host-H. pylori interactions implicated in the initiation, progression and health outcomes of infection.
H. pylori and its human host: An intimate relationship
H. pylori can persist in the human stomach for a lifetime.Citation16 Its ability to survive in the gastric niche is tightly connected to the expression of various pathogenicity factors that enable adherence and penetration of the epithelial cell layer and manipulation of innate and adaptive immune responses at the gastric mucosa. Dozens of bacterial factors are involved in H. pylori-mediated colonization and molecular pathogenesis including adhesins, outer membrane proteins,Citation17 urease, catalase, neutrophil-activating protein A (NapA), peptidoglycan (PG), vacuolating cytotoxin A (VacA) and the cag pathogenicity island (cagPAI) including cytotoxin-associated gene A (CagA).Citation18 provides an overview of H. pylori's colonization process and highlights bacterial factors that are important for persistence, survival and the initiation of immune responses. For a detailed description of relevant pathogenicity factors we would like to refer the reader to recent review articles on this topic.Citation19,20
Figure 1. Colonization of the gastric niche and initiation of immune responses by H. pylori. H. pylori possesses a number of virluence factors that aid in its ability to colonize and exploit the gastric niche for survival and replication. It elevates the gastric pH by secretion of urease and traverses the mucus layer through flagellar movement and urease-induced gel-sol transition. Once in contact with the epithelial cell layer it binds to the apical cell surface using various adhesins (SabA, BabA/B, AlpA/B, HopZ, OMP) causing cell damage and facilitating the delivery of toxins. H. pylori disrupts the cell barrier by breaking up tight- and adherent junctions (TJ, AJ) through HtrA and enters the lamina propria to replicate at the basolateral cell surface. H. pylori further exploits gastric glands and the apical cell surface as replicative niche for which the proteins Chepep and the virulence factor CagA respectively are essential. At the basolateral cell surface H. pylori forms the type 4 secretion system (T4SS) encoded by the cag pathogenicity island and causes integrin clustering at lipid rafts to inject CagA into epithelial cells. Peptidoglycan (PG) leaks through the T4SS and is recognized by the pathogen recognition receptor (PRR) NOD1. These two virulence factors induce the transcription of host-cell genes including IL-8, chemokines and type-I interferons (type-I IFN) through NFκB/AP1 and IRF7, respectively. H. pylori further causes host cell remodeling and damage through VacA. This toxin either heterodimerizes and forms pores in the cell membrane or enters by receptor binding to cause cell-vacuolization, pro-inflammatory cytokine signaling and cytochrome C (CytC)- induced apoptosis. Epithelial cells at the mucosal barrier are involved in initiating the immune response to H. pylori by antigen-induced TLR-2 and -4 signaling through NFκB. This is further amplified by the induction of TLR-4 transcription via NF-Y-mediated TLR-2 signaling. The subsequent secretion of chemokines attracks peripheral mononuclear cells (PMN) including neutrophils and dendritic cells to the site of infection.
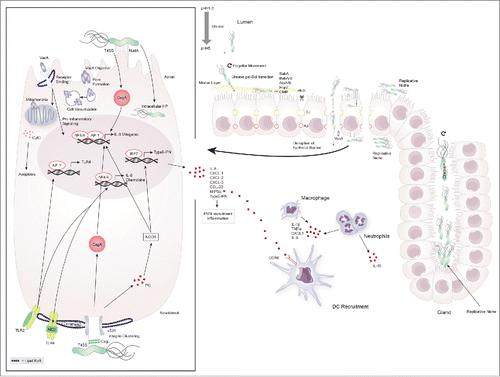
Physical contact and the localization of H. pylori within in the gastric mucosa are critical determinants for immunopathology. Approximately 20% of H. pylori organisms in the stomach adhere to the surface of gastric epithelial cells.Citation6 This physical contact causes cellular damage to the epithelium, induces inflammation and facilitates the delivery of toxins,Citation21,22 which in turn promotes bacterial invasion and persistence. Upon CagA injection H. pylori exploits the apical epithelial cell surface as a replicative niche.Citation23 Furthermore, H. pylori can establish colonies deep in the gastric glands (). The protein ChePep is necessary for this ability by regulating flagellar rotation through the chemotaxis system,Citation24 and mutant H. pylori strains lacking this chemotaxis ability are excellent tools for forming a comprehensive picture of how microscopic biogeography shapes the type and intensity of mucosal immune responses. Despite its initial classification as extracellular pathogen, recent studies revealed the facultative intracellular nature of H. pylori. It invades and multiplies in gastric epithelial cellsCitation25,26 and adult epithelial progenitor cellsCitation27 thereby serving as a repository for H. pylori and protecting it from antibiotic eradication. Although the mechanisms of invasion and persistance are not well understood, the importance of H. pylori invasin NudA for host cell uptake was reportedCitation28 in vitro and dependence on c-Met and the type IV secretion system (T4SS) were determined.Citation29
Whether H. pylori exerts a protective effect in the context of a dysregulated immune response or whether it contributes to immunopathology, cell damage and malignant transformation is dependent on host-pathogen interactions. The genetic background of both H. pylori and the host contribute to disease etiology. On the microbial side the presence of an intact cagPAICitation30 and the s1m1 variant of VacA among other factors have been associated with more severe pathology and an increased risk for gastroduodenal disease.Citation31 The importance of the host's immune response in delineating the outcome of chronic H. pylori infection is reflected in the association of genetic polymorphisms with a higher risk for developing gastric cancer. These include IL-1β,Citation32 genes involved in Toll like receptor (TLR) and NOD-like receptor (NLR) signaling pathwaysCitation33,34 and the immunoregulatory transciption factor peroxisome proliferator activated receptor γ (PPAR-γ).Citation35 The relevance of these host factors will become apparent in the following sections discussing the current knowledge on innate and adaptive immune responses to H. pylori and how its virulence factors influence these processes.
Initiating the immune response to H. pylori
Sensing of pathogen associated molecular patterns (PAMPs) including PG, lipopolysaccharide (LPS), flagellin and unmethylated CpG DNA by pathogen recognition receptors (PRRs) present on epithelial cells, antigen presenting cells and neutrophils is required for the initiation of immune responses to H. pylori. Recognition of PAMPs is mediated by membrane-bound TLRs, retinoic acid-inducible gene 1 (RIG-I)-like receptors, cytosolic NLRs and C-type lectins.Citation36
The T4SS plays a crucial role in initiating immune responses to H. pylori. Upon contact with the host cell it forms a needle-like structure which protrudes from the bacterial surface and delivers virulence factors (e.g. CagA and PG) into the host cell.Citation37,38 Intracellular PG is specifically recognized by NOD1 and causes the phosphorylation of MAPK, the activation of NF-κB as well as AP-1 and subsequent release of IL-8 by epithelial cells.Citation39,40 NOD1-mediated NF-κB activation is dependent on the delivery of PG into the host cell via the T4SS and requires the interaction with host α5β1 integrin localized to lipid rafts of AGS cells.Citation41 Cholesterol-rich microdomains, also known as lipid rafts, are important sites for receptor complex formation upon H. pylori LPS recognition by TLRs. In AGS cells the induction of the TLR4/MD2 complex by H. pylori is dependent on the integrity of lipid rafts, where H. pylori, TLR4 and ceramide co-localize.Citation42 Furthermore, H. pylori LPS can induce the recruitment of TLR2 to lipid rafts and trigger the formation of receptor clusters involving TLR1, CD11b/CD18 and CD36 in human vascular endothelial cells.Citation43
Several studies have demonstrated that H. pylori LPS only signals via TLR2Citation44-47 while others demonstrated signaling via TLR4.Citation32,48 Despite these controversial results it has been established that both TLR2 and 4 play a role in H. pylori LPS-mediated immune responses. Yokota et al.Citation49 were able to provide an experimental link between TLR2 and TLR4 signaling in a gastric epithelial cell line. They show that H. pylori LPS binding to TLR2 activates the MEK1/2-ERK1/2 pathway which leads to NF-Y-mediated transcription of TLR4 and results in IL-8 secretion.Citation49
Overall, recognition of H. pylori antigens by epithelial cells culminates in chemokine secretion (IL-8, CXCL1, CXCL2, CXCL3 and CCL20) and subsequently leads to the recruitment of neutrophils, eosinophils, monocytes, dendritic cells and macrophages.Citation50,51 Macrophages located at sites of neutrophil infiltration in the lamina propria, gastric epithelium infiltrated by neutrophils as well as neutropils themselves have been reported to express high levels of Gro-α and IL-8, two neutrophil-attractant chemokines.Citation52 Furthermore, the upregulation of MIP-3α expression in gastric epithelial cells induces an influx of myeloid dendritic cells (DC) into the lamina propria as demonstrated in neonatally thymectomized BALB/c mice.Citation53
In the following section we will discuss the current knowledge on sensing of H. pylori by innate immune cells and how this paves the way for the mixed pro- and anti-inflammatory adaptive immune responses typically observed in H. pylori infection.
Innate immune responses to H. pylori
In cultured human neutrophils H. pylori initiates an early innate immune response, which is partially mediated by TLR2 and TLR4, and characterized by rapid upregulation of IL-8, IL-1β and TNF-α followed by an increase in IL-10 production.Citation54 Furthermore, H. pylori NapA, an agonist of TLR2, induces IL-12 and IL-23 secretion by neutrophils as well as monocytes and elicits an antigen-specific Th1 response in the gastric mucosa of H. pylori infected patients.Citation55 Recognition of H. pylori PAMPs by antigen presenting cells, triggers the production of pro- and anti-inflammatory cytokines including IL-1, IL-6, IL-8, IL-10, IL-12 and type 1 IFN.Citation56 Rad et al.Citation57 identified 3 gene clusters that were upregulated in H. pylori-stimulated DC, i. MyD88 independent and MyD88-dependent mediated by ii. surface TLRs and iii. endosomal TLRs (). MyD88-independent expression of type-I IFNs and interferon-stimulated genes in DC is elicited by binding of H. pylori RNA to RIG-I ().Citation57 The main surface PRRs involved in H. pylori sensing by DC are TLR2 and to a lesser extent TLR4.Citation57 Both surface TLRs play a dual role in H. pylori pathogenesis. Although TLR2 signaling participates in the induction of pro-inflammatory responses, transcriptome analysis of H. pylori-treated DC also revealed a TLR2-dependent anti-inflammatory signature in mice.Citation57 Indeed, the lack of TLR2 in H. pylori infected mice induced a higher IFN-γ-mediated Th1 response and lower Treg/Th17 cell responses resulting in increased bacterial clearance at the expense of lesion development.Citation58 In human DC this dual pro-and anti-inflammatory role seems to be conferred by TLR4 which has been shown to be the main contributor to H. pylori-induced IL-10 and IL-12p70 mediated Th1 induction. Neutralization of TLR-4 led to reduced IFN-γ, IL-17A and IL-10 secretion as well as FOXP3 expression in H. pylori-primed DC-T cell co-cultures.Citation59 The endosomal TLR9 recognizing H. pylori DNA contributes significantly to the cytokine response (IL-6, IL-12) elicited by the bacterium. Interestingly, it also plays a role in the suppression of H. pylori-induced gastritis in the early phase by downregulating Th1 cytokines mediated by IFN-α (Type-I IFN).Citation60
Figure 2. Innate immune sensing of H. pylori. The innate immune response to H. pylori is initiated by epithelial cells and tissue resident macrophages. Upon chemokine secretion dendritic cells are recruited to the gastric mucosa, where they encounter, recognize and process various known and unknown H. pylori antigens. Myd88-dependent TLR2 (LPS, NapA) and TLR4 (LPS) antigen binding results in both pro- and anti-inflammatory cytokine production which reflects the complexity of the immune response initiated by H. pylori. Following phagocytosis bacterial DNA is bound by TLR9 on endosomes and induces transcription of pro-inflammatory cytokines and type-I interferons (IFN) through NFkB and IRF7/IRF8, respectively. Bacterial RNA is recognized by RIG-I and elicits Myd88-independent signaling to induce transcription of type-I IFN. Furthermore, inflammasome priming is mediated by binding of peptidoglycan (PG) to NOD2 and subsequent activation of NFκB-mediated transcription of pro-IL-1β. Subsequently, the inflammasome complex NLRP3/ASC/pro-Casp1 is activated by potassium efflux and phagocytosis-induced ROS production, which results in Casp1-mediated cleavage of pro-IL-1β to IL-1β.
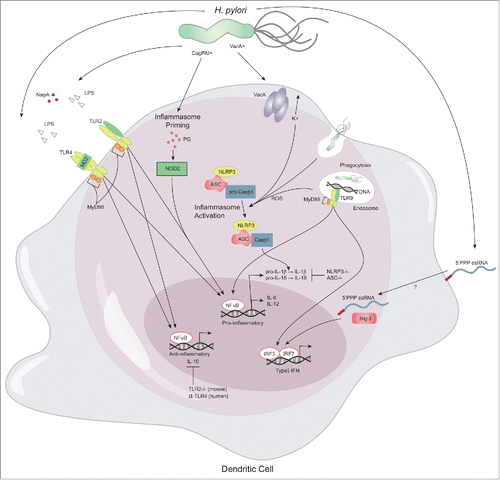
Another immune sensing mechanism playing an important role in immune responses toward H. pylori is the inflammasome. As a multi-protein complex it mediates the activation of CASP1 which promotes the secretion of the pro-inflammatory cytokines IL-1β and IL-18. Members of the NOD-like receptor family including NLRP1, NLRP3 and NLRC4 and the adaptor ASC are critical components of the inflammasome which link microbial and endogenous danger signals to caspase 1 (CASP1) activation.Citation61 In the context of H. pylori infection, the concomitant activation of TLR2 and NOD2 has been shown to induce pro-IL-1β expression and subsequent IL-1β secretion by H. pylori-infected DC. Dependency on CASP1 activation through priming of the NLRP3 inflammasome was demonstrated in vitro ().Citation62 A recent study by Semper et al.Citation63 further corroborates these findings and identifies potassium influx, phagocytosis and ROS production as mechanisms involved in H. pylori-mediated NLRP3 activation. Adhesion of H. pylori to the host cell and the cagPAI, but not CagA, were important for inflammasome activation in murine DC and human monocytes/macrophages (). Moreover, H. pylori infected NLRP3-deficient mice showed increased bacterial colonization and decreased inflammation associated with decreased levels of IL-1β, IL-18, IL-17 and IFN-γ in the gastric mucosa.Citation63 Similarly ASC-deficient mice showed higher colonization and less inflammation upon H. pylori infection.Citation64
Furthermore, the expression of NLRP12 and NLRX1, two negative regulators of NF-κB, were downregulated in THP-1-derived macrophages upon infection with the highly virulent H. pylori strain GC026 isolated from a gastric cancer patient. In concordance, NF-κB and several of its target genes including IFNB1, IL12B, IL6, TNF, CXCL1, CXCL2, CCL5, PTGS2 and BIRC3 were upregulated.Citation65 In line with these findings, we recently validated that intracellular H. pylori modulates the dynamics of NLRX1 and NLRC3 expression, another regulatory NLR, and simultaneously induces NF-κB responsive genes in bone marrow derived macrophages. Interestingly, NLRX1 was necessary for maintaining high levels of intracellular H. pylori at the cellular level and in a murine model of infection.Citation34 The contribution of NLRX1 in modulating host immunity to H. pylori may be associated with the combined effect of disruption of RIG-I signaling and inhibition of NF-κB activation by TRAF6 as observed in other infectious disease models, or a yet undiscovered pathway.Citation66,67 Suppression of regulatory NLRs is associated with worsened immunopathology in both immune mediated and infectious diseases but mechanisms of negative regulation have not been described.Citation68-70
The following section reviews the molecular mechanisms and complex cellular interactions that underlie the pro-and anti-inflammatory adaptive immune responses to H. pylori and how this contributes to chronic gastritis.
Th1/Th17 responses to H. pylori infection
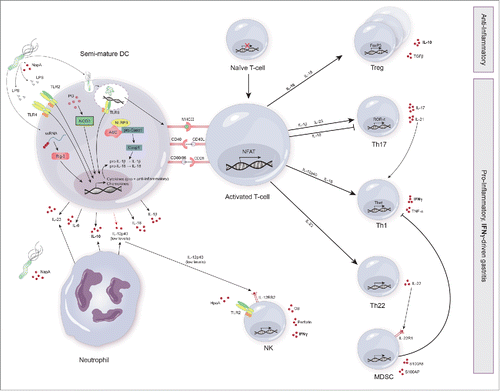
Consistent with the induction of Th1 and Th17 responses, H. pylori-treated DC stimulate the secretion of IFN-γ and high levels of IL-17 by CD4+ T cells. Furthermore, murine splenic macrophages stimulated with H. pylori signficantly upregulated gene expression of Th1- and Th17-inducing cytokines including IL-6, TGF-β, IL-23 p19, IL-12/IL-23 p40 and IL-12 p35 ().Citation71 Even though H. pylori-pulsed DC secrete only low levels of IL-12 (p35/p40), robust IL-23 (p19/p40) expression is induced.Citation72 Importantly, IL-23 was also detected in gastric myeloid DC of H. pylori-infected patientsCitation72 and expression of IL-23 p19 was increased in gastric epithelial cells of patients with H. pylori-associated gastritis, suggesting a potential role for epithelial cells in shaping mucosal immunity.Citation73 IL-23 belongs to the IL-12 family of cytokines and is known to promote the expansion and survival of Th17 cells.Citation74 Indeed, blocking of endogenous IL-23 in cultures of lamina propria mononuclear cells from H. pylori-infected patients inhibited STAT3 phosphorylation and consequently diminished IL-17 secretion.Citation75 The virulence factors CagA and CagE have been implicated in the induction of IL-17 and IL-23 secretion further corroborating the significance of the cagPAI in H. pylori immunopathology. The cytokine BAFF, released by macrophages upon H. pylori infection, has recently been implicated in the induction of H. pylori-mediated Th17 responses in humans. BAFF has been shown to induce Th17 differentiation directly or indirectly through the creation of a pro-Th17 cytokine milieu by innate immune cells.Citation76
Figure 3. Shaping adaptive immune responses to H. pylori. Dendritic cells (DC) encountering H. pylori have been shown to possess a semi-mature phenotype. The adaptive immune response to H. pylori is characterized by a mixed anti- and pro-inflammatory response. Upon pathogen receptor recognition, DC and neutrophils secrete a number of cytokines including IL-1β, IL-6, IL-10, IL-18, IL-23 and low levels of IL-12p40. These cytokines and concomitant presentation of H. pylori antigens by semi-mature DC to naive T cells via MHC-II results in the expansion of H. pylori-specific Treg. Furthermore, T cells differentiate into Th17, Th1 and Th22. The secretion of IL-22 by Th22 induces the secretion of anti-bacterial proteins by MDSC (monocytic-myeloid derived suppressor cells). IL-21 secretion by Th17 supports Th1 differentiation and thus maintains the Th17/Th1 axis during gastritis. In contrast, MDSC exert an inhibitory effect on Th1 cells. Upon binding of HpaA to TLR2 and IL-12RB2 signaling, NK cells secrete granzyme B (GB), perforin and IFN-γ. Importantly, IFN-γ secreted by NK cells, Th1 cells and possibly other cell types (CD8+ T cells) has been identified as the main driver of gastritis.
Regulatory T cell responses to H. pylori
Despite the induction of mixed pro-inflammatory T cell responses, the immune system is unable to clear H. pylori resulting in life-long colonization. An underlying regulatory immune response which is induced by known and unknown bacterial factors and executed by regulatory T cells has been attributed to H. pylori's ability to persist in its host. Although H. pylori-pulsed DC increase the production of IL-12 and induce secretion of Th1 cytokines (TNF-α, IFN-γ) as well as proliferation of murine splenocytes (), this response is much lower compared to DC pulsed with the Gram-negative bacillus Acinetobacter iwoffii. A heat stable soluble factor released by H. pylori has been found to suppress IL-12 secretion by A. iwoffii-stimulated DC.Citation77 Another study recently demonstrated that H. pylori VacA suppressed IFN-β and IL-12 expression in murine bone marrow derived macrophages stimulated with Lactobacillus acidophilus.Citation78 Furthermore, gastric and intestinal stroma-conditioned medium has been shown to down-regulate DC responsiveness to H. pylori in vitro. A yet unknown stromal factor inhibited monocyte-derived DC activation and impaired IL-12 release upon H. pylori infection consequently resulting in the reduced ability of H. pylori-pulsed DC to induce an IFN-γ mediated Th1 response.Citation79 Together, these studies suggest that both H. pylori- and host-derived factors might hinder the induction of a robust Th1 response through the suppression of IL-12 release by DC.
In line with the observation that H. pylori interferes with DC maturation in vitro, DC infiltrating the gastric mucosa of human H. pylori carriers exhibit a semimature DC-SIGN+HLA-DRhiCD80loCD86lo phenotype.Citation80 Specifically, H. pylori can trigger a reprogramming of DC by downregulating MHC-II, inducing IL-10 and inhibiting IL-12 secretion thereby inducing H. pylori specific Treg cells.Citation81-83 CagA has been identified as the main virulence factor responsible for induction of semi-mature DC, IL-10-mediated STAT3 activation and subsequent Treg induction.Citation84 Increased numbers of Treg cells are typically found in the stomachs of infected patients, suggesting that they play an important role in regulating inflammatory responses to H. pylori.Citation85 In children, H. pylori infection favors the induction of mucosal Treg responses over Th17 and is thus associated with reduced gastric inflammatory lesions compared to adults.Citation86,87 Overall, patients with fewer or less functional Treg cells are more likely to develop peptic ulcers and are afflicted by more intense gastritis.Citation88 B cells also play a regulatory role by promoting IL-10 production in co-cultured CD4+ cells and subsequent conversion into a T regulatory 1 (Tr1)-like phenotype.Citation89 The importance of Treg cells for the control of H. pylori-induced gastritis has been highlighted in several studies. Adoptive transfer of C57BL/6 derived CD4+ splenocytes into H. pylori-infected RAG-KO mice lacking Treg induced more severe gastritis compared to H. pylori-infected C57BL/6 mice.Citation90 Moreover, depletion of Treg cells resulted in decreased bacterial colonization at the expense of increased gastric inflammation.Citation91
One host cell factor on the intersection between pro- and anti-inflammatory responses toward H. pylori is the enzyme CASP1. Upon activation by the inflammasome complex it dynamically regulates immune responses through IL-1β and IL-18, respectively.Citation92 IL-1 signaling is required for the induction of Th17 responsesCitation72 and IL-18 together with IL-12 is known to induce Th1 responses (reviewed in Citation93). Albeit its pro-inflammatory role, the presence of IL-18 prevented excessive immunopathologyCitation92 and induced H. pylori-specific tolerance through the induction of Treg differentiation (), while its absence promoted unrestricted Th17 responses to H. pylori in mice.Citation80 Thus, H. pylori-induced IL-18 secretion exerts an immune-regulatory function by counteracting the activities of IL-1β.
Drivers of H. pylori-induced gastritis
To better understand, prevent and treat H. pylori-mediated disease one should identify the main factors involved in the development and sustainance of gastritis. A recent study by Gray et al.Citation90 has shed further light on the complex interactions leading to H. pylori-induced gastritis. They determined that H. pylori-specific CD4+ T cells, innate immune cells expressing the common cytokine gamma chain and IFN-γ were essential for the development of gastritis in mice, whereas Th1 cells were not. This study suggests that IFN-γ secreted by other cell subsets (possibly functional innate immune cells) is the main driver of gastritis ().Citation90 Thus far, NK cells and CD8+ cytotoxic T cells have been identified as additional source for IFN-γ in H. pylori infection. Yun et al.Citation94 demonstrated that stimulation of human NK cells with H. pylori antigens induced IFN-γ production and increased the expression of granzyme B and perforin. Interestingly, H. pylori-induced IFN-γ secretion was dramatically increased in the presence of small doses of IL-12 p40. This synergistic effect was dependent on antigen exposure prior to addition of IL-12 and was associated with increased expression of IL-12 receptor β 2 ().Citation94 H. pylori specific membrane bound lipoprotein A (HpaA) has been implicated in the induction of IFN-γ secretion by NK cells, which was dependent on the recognition by TLR-2 and MyD88 as well as p38 MAPK signaling ().Citation95 CD8+ T cells have also been implicated as a source for IFN-γ. A study assessing proliferation and cytokine secretion of circulating T cells from H. pylori-infected patients demonstrated induction of IFN-γ secretion from both CD4+ T cells and CD8+ T cells upon H. pylori antigen exposure. Interestingly, CD8+ T cells secreted even more IFN-γ than CD4+ T cells on a per cell basis.Citation96 Studies in H. pylori infected children with grade I-III gastritis showed an increased number of CD8+ T cells in the gastric epithelium and lamina propria.Citation97,98 Another study demonstrated an increase of CD8+HLA-DR+ chronically activated memory T cells in peripheral blood of H. pylori-colonized children with duodenal ulcers.Citation99 We have recently demonstrated the expansion of circulating NK cells, CD8+ T cells and concomitant increases in cytotoxic markers including CD16, granzyme B and perforin in a pig model of H. pylori infection.Citation100 Thus, a cytotoxic cell response might play a major role in H. pylori-mediated pathology at least in part due to the cytokine response induced by the bacterium.
While it has been established that IL-17A secretion and Th17 cells play a role in H. pylori-mediated gastritisCitation71 their contribution is less dramatic than that of IFN-γ.Citation90 Interestingly, recent studies have established an association between Th17 and Th1 responses. IL-17 deficiency in mice signficantly reduced IFN-γ, T-bet and IL-12 p40 mRNA expression in gastric tissue of H. pylori infected animals suggesting that the Th17/IL-17 pathway plays a role in driving Th1 responses.Citation71 Further to the association between Th17 and Th1 cells, Caruso et al.Citation75 demonstrated that IL-23 does not only enhance IL-17 but also IFN-γ secretion by ex vivo stimulated normal human gastric lamina propria mononuclear cells. Blocking IL-23 in these cells significantly reduced IL-17 and IFN-γ secretion.Citation75 Another molecule that has been implicated in H. pylori-induced gastritis is IL-21. IL-21 is a functional hallmark of T follicular helper (Tfh) cells, can also be produced by NKT cells, Th17 cells, and has been linked to Th1 and Th17 differentiation at the expense of Treg cell formation.Citation101,102 Of note, we have recently identified the accumulation of cells with a Tfh phenotype (CD4+ T cells expressing CXCR5, ICOS, PD-1, BCL6, and IL-21) in the mesenteric lymph nodes and gastric lamina propria upon H. pylori infection in C57BL/6 mice.Citation103
Deficiency of IL-21 in H. pylori-infected mice resulted in increased bacterial burden and decreased inflammation characterized by highly reduced infiltration of neutrophils, B cells, CD4+ and CD8+ T cells into the stomach. The study also presented data showing that the mRNA expression of chemokines and cytokines including TNF-α, IL-1β, IL-17A and IFN-γ was inhibited in IL-21-deficient mice compared to their wild-type littermates upon H. pylori infection. This was associated with reduced expression of TBX1 and RORγt as well as reduced levels of activated STAT1 and STAT3. Thus, IL-21 contributes to the maintenance of both Th1 and Th17 responses.Citation104
Another recently described T cell subset that contributes to H. pylori induced gastritis are Th22 cells. Zhuang et al.Citation105 show that Th22 cells are enriched in the gastric mucosa of H. pylori-infected patients and mice. They demonstrated that IL-23-secreting DC encountering H. pylori CagA+ strains highly induce secretion of IL-22 by CD4+ T cells. This leads to the recruitment of CXCR2+ monocytic-myeloid derived suppressor cells (MDSC) into the gastric mucosa via IL-22R1 signaling and subsequent secretion of CXCL2 by gastric epithelial cells. Furthermore, MDSC secrete the two pro-inflammatory proteins S100A8/S100A9 upon IL-22 stimulation and suppress Th1 cell development. ().Citation105
Mechanisms of immune evasion
Consistent with its ability to chronically colonize the gastric microenvironment, H. pylori is equipped with efficient ways to evade recognition and clearance by the immune system. This is in part attributed to the expression of modified, less immunogenic PAMPs and to the secretion of certain pathogenicity factors, which alter the function of innate immune cells. Hence, H. pylori has the ability to evade, attenuate and modify innate and adaptive immune responses.
Modification of LPS, e.g., by adding positively charged substituents or removing phosphate groups from the backbone, renders H. pylori much less immunogenic than LPS from other gram-negative enteric bacteria such as Escherichia coli.Citation106 The LPS component lipid A has been shown to bind to and activate the hTLR4-MD2 complex. Lipid A dephosphorylation by the enzymes LpxE and LpxF results in attenuated hTLR4-MD2 activation and confers resistance to the actions of cationic antimicrobial peptidesCitation48 thereby limiting the host's immune response and favoring H. pylori's persistence.
Another mechanism of immune evasion is H. pylori's ablity to manipulate the function of innate immune cells. After successful phagocytosis by neutrophils, H. pylori disrupts the NADPH oxidative system thus enabling unopsonized H. pylori to escape phagocytic killing.Citation107 Furthermore, H. pylori impairs neutrophil migration by decreasing the expression of CXCR1 and CXCR2.Citation108 H. pylori induces the expression of arginase 2 (Arg2) in lamina propria macrophages thereby restricting iNOS upregulation, NO production and enhancing macrophage apoptosis. Accordingly, the expression of IFN-γ, IL17A and IL-12 p40 was increased and levels of IL-10 were decreased in the gastric antrum of Arg2-/- mice.Citation109 Cyclooxygenase (COX)-2 is a key enzyme in arachidonic acid metabolism and induced by H. pylori via MEK/ERK signaling independent of the T4SS.Citation110 Inhibition of COX-2 and its metabolic product prostaglandine E2 (PGE2) resulted in the increased production of IL-12 and IFN-γ and a decrease in IL-10 levels upon H. pylori infectionCitation111 suggesting that it contributes to the anti-inflammatory response against the bacterium.
Survival of H. pylori inside lamina propria macrophagesCitation112 also contributes to its ability to evade elimination by manipulating phagosome and autophagosome maturation.Citation113,114 It impairs the antimicrobial activity of macrophages through the production of catalaseCitation115 and inhibits NO production by decreasing the availability of L-arginine through the bacterial arginase rocFCitation116,117 as well as inhibiting L-arginine uptake by inducing spermine oxidase.Citation118 Once engulfed, H. pylori is present in phagosomesCitation119 and escape from phagocytic killing is dependent on cagPAI.Citation120,121 VacA can arrest phagosome maturation by preventing its fusion with lysosomes, thus contributing to the pathogen's persistence inside the cell.Citation121 H. pylori uses host cholesterol to synthesize cholesterol-α-D-glucopyranoside (CG). This process is mediated through the enzyme cholesterol-α-glucosyltransferase (CGT), encoded by capJ. The presence of capJ has been shown to confer resistance to phagocytic endocytosis, phagolysosome-fusion and -maturation. Furthermore, the synthesis of CG by CGT is responsible for retarded entry through the lipid-raft endocytic route and prolonged intracellular survival in a PI3K-dependent manner.Citation122 H. pylori induces the formation of autophagosomes which are adapted for its replication and thus contribute to increased antimicrobial resistance.Citation113 In primary human macrophages, virulent H. pylori strains can survive for an extended period due to the formation of megasomes, which are large structures arising from homotypic fusion of phagosomes.Citation123
Additionally, H. pylori can directly limit adaptive immune responses by limiting nutrients essential for T-lymphocytes. The depletion of L-arginine by H. pylori arginase decreases the expression of CD3 zeta chains associated with the T cell receptor, thus inhibiting T cell proliferation.Citation124 H. pylori γ-glutamyltranspeptidase depletes extracellular glutamine and consequently inhibits T lymphocyte proliferation, impairs activation and inhibits cytokine secretion involving IRF4.Citation125 Endocytosis of VacA into activated human primary T cellsCitation126 inhibits cell proliferation and the clonal expansion of H. pylori antigen specific T-cells.Citation127 It has also been shown that VacA can block T cell activation by preventing influx of extracellular calcium and subsequent nuclear translocation of NF-AT and by stimulating disordered actin polymerization through Rac activation.Citation128
The beneficial role of H. pylori in extra-gastric disease
Since the identification of H. pylori as a causative agent of peptic ulcers and gastritis in 1983Citation4, the bacterium has been considered primarily a pathogen. However, infection with H. pylori is asymptomatic in 85% of individuals, only 15% develop symptomatic peptic ulcer, and less than 1% develop gastric cancer.Citation129 A potential role of H. pylori as a gastric commensal or symbiotic bacterium is beginning to emerge in recent years. It has been demonstrated that H. pylori colonization even exerts beneficial effects in allergic diseases, such as asthmaCitation130 and eczema,Citation131 as well as gastroesophageal reflux disease,Citation132 obesity and diabetes.Citation133 H. pylori's ability to evade the immune system and to persist in the gastric mucosa through induction of Treg cells, regulatory CX3CR1+ mononuclear phagocytes, and anti-inflammatory molecules as discussed above lends further support to its potential role as a commensal organism.
It has been demonstrated that CD4+CD25+ T cells from H. pylori-infected neonatal WT mice are able to prevent the development of allergen-induced asthma in mice.Citation80 Furthermore, H. pylori NapA has been shown to skew the immune response from a Th2 to a Th1 response in a mouse model of OVA-induced allergic asthma.Citation134 In the context of immune-mediated diseases H. pylori infection protects against Salmonella typhi-induced colitis by suppressing Th17 responses in the lower intestinal tract and upregulating IL-10 expression in lymph nodes.Citation135 Subsequent studies, demonstrated how H. pylori DNA decreases pro-inflammatory cytokine production by DC and attenuates sodium sulfate (DSS)-induced colitis in mice.Citation136 Using a mouse model of DSS-induced and T-cell-transfer-induced colitis Engler et al.Citation137 recently demonstrated a beneficial effect of H. pylori infection on clinical and histopathological features of colitis. They showed that H. pylori-mediated protection against DSS-induced colitis was dependent on the NLRP3 inflammasome and IL-18 signaling.Citation137
Results from a study on the protective role of H. pylori in obesity, provide in vivo evidence that gastric colonization with a cagPAI negative H. pylori strain ameliorates glucose tolerance possibly by activating the transcription factor PPAR-γ. The presence of cagPAI negative H. pylori modulated appetite-controlling hormones (i.e., ghrelin and leptin), suppressed adipose tissue inflammation, and ameliorated glucose homeostasis. Interestingly, these effects were abrogated upon conditional knockout of PPAR-γ in immune- and epithelial cells.Citation133 The inverse relationship between obesity and H. pylori colonization was recently studied in a North American cohort of inner city children revealing a 50% reduction in the odds of being obese when colonized with H. pylori.Citation138
Recent epidemiological and clinical data indicate that the incidence of obesityCitation139 and inflammatory bowel disease (IBD)Citation140 is greater in areas with low prevalence of H. pylori infection. Whether this is due to a „spontaneous eradication“ of H. pylori caused by immunomodulatory drugs and antibiotics taken by IBD patients or due to an actual protective effect of H. pylori is still controversial.Citation141 A study supporting the causal association of H. pylori and IBD, reports that the rising incidence of IBD in H. pylori-endemic regions corresponds to the use of anti-H. pylori therapy for the treatment of peptic ulcer disease.Citation142 Therefore, the evidence for H. pylori's beneficial effects in allergic, immune-mediated and metabolic diseases seriously questions the validity of widespread H. pylori eradication strategies. Thus far, little is known about the relevance of virulence factors in H. pylori-mediated protection and this is likely to differ between diseases. In a study using data from 7663 participants of a large US National Health and Nutrition Examination Survey (NHANES III) Chen and BlaserCitation12 show that the infection with a more virulent CagA+ H. pylori strain was inversely associated with ever having had asthma in younger study participants. The beneficial effect of CagA+ strains in allergic disease and asthma can be at least partly explained by the increased IL-10 responses inducing semi-mature DC and higher Treg responses.Citation84,143 To date, no conclusive association between CagA status and Crohn's disease has been made. Although a high frequency of Crohn's disease patients was infected with CagA+ strains, this was similar to controls.Citation144 Thus, further pre-clinical and clinical studies as well as modeling studies are needed to strengthen and mechanistically characterize H. pylori's role as beneficial member of the gut microbiota.
H. pylori infection provides an excellent avenue to probe mucosal immune responses and study mechanisms of immunoregulation and host tolerance that reduce the negative impact of infection on host fitness. Unlike resistance mechanisms, tolerance does not directly affect microbial burden. Rather, tolerance decreases the host susceptibility to tissue damage, or other fitness costs, caused by the pathogens or by the immune response against them. Given the co-evolution of H. pylori and its human host, studying mechanisms of H. pylori-host interaction may yield novel insights on how the human host can protect itself from infectious diseases by reducing the negative impact of infection on host fitness, independently of modulating the microbial burden. New integrative analyses can help synthesize and transform data, procedural knowledge and theory into information processing representations of immune responses to H. pylori with the potential to yield transformative systems-wide immunological knowledge.
Computational modeling to advance our understanding of the complex interactions between H. pylori and the gastric immune system
The interaction between H. pylori and the stomach mucosa is a complex and dynamic process. Computational modeling can be used to support pre-clinical and clinical research in H. pylori and to identify emerging behaviors of the immune response. We developed a tissue-model of the immune response to infection that represents the gastric lumen, epithelium, lamina propria and lymph nodes.Citation145 The model was calibrated with data from an in vivo time-course study of H. pylori-challenged mice and reproduced the effector and regulatory pathways in the gastric lamina propria, including the induction of a Th17 response, a dominant Th1 response and high levels of mucosal Treg.Citation145 Computational models can be used to study new therapeutic targets in the context of disease processes. To shed light on the potential role of the transcription fator PPAR-γ in modulating host reponses to H. pylori we used a loss-of-function approach. In silico results predicted a predominance of Th17 and Th1 responses in cell-specific PPAR-γ knockout compared to WT mice.Citation145 Sensitivity analysis was used to rank immunological parameters by their relevance in the system's dynamics and outcomes. Global sensitivity analysis has revealed that epithelial cells and macrophages have the highest influence in the mucosal immune response to H. pylori. The prediction of the impact of epithelial cells support the model itself, since most of H. pylori is found in contact with epithelium,Citation24,146,147 and the epithelial cell has been implicated in the initiation of innate responses (as discussed above).
With regards to macrophages, the analysis predicts the initial presence of macropage precursors, followed by the accumulation of a subset of M2 regulatory cells, which peaks between days 21 and 35. Interestingly, through the guidance of our computer modeling work, we have recently identified a CX3CR1+CD64+CD11b+F4/80+ subset of macrophagesCitation148 (unpublished data) that starts accumulating in the stomach of mice at day 15, peaks around day 24 and it is maintained at a steady state for at least 16 weeks. These macrophages produce IL-10 and by depletion studies we have demonstrated that they facilitate colonization by H. pylori.
In addition to the tissue-level computational model that can scale up to 10 billion cells and molecules at a time, we have engineered a model representing signaling events leading to CD4+ T cell differentiation and plasticity.Citation104,149,150 Our cell-based model is generic in the sense that it represents CD4+ T cell differentiation out of any particular context,Citation149,150 although it can be recalibrated with data obtained during infection. To investigate the role of IL-21 during H. pylori infection, we re-calibrated the model with CD4 data obtained from mice infected with H. pylori and used to construct an IL-21 knockout system.Citation104 The main prediction of the model was the reciprocal interplay between IL-21 and IL-10 whereby, in the context of an H. pylori infection, IL-21 promotes differentiation of CD4+ T cells into Th1 and Th17 and opposes IL-10, and vice versa: lack of IL-21 turns the system in favor of IL-10. The predictions of the model were the validated in vivo using IL-21 KO mice. Others have also used mathematical modeling to evaluate the expression of pro and anti-inflammatory cytokines, and how their levels are affected by sonic hedgehog (SHH),Citation151 a gene product that is upregulated in gastric parietal cells early post-H. pylori infection and has been proposed to be a macrophage chemoattractant.Citation152
In summary, integrating computational and mathematical approaches to guide pre-clinical and clinical experimentation into the analysis of the host response to H. pylori can help accelerate the discovery of the mechanisms of action that explain the role of this microbe as pathogen or commensal microbe. Modeling and informatics tools that create information processing representations of immunological processes allow connecting immunology and microbiology to the world above the skin, testing interventions in virtual laboratories to guide human studies.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported in part by NIAID Contract No. HHSN272201000056C to J.B.-R. and funds from the Nutritional Immunology and Molecular Medicine Laboratory.
References
- Ndip RN, MacKay WG, Farthing MJG, Weaver LT. Culturing Helicobacter pylori from clinical specimens: review of microbiologic methods. J Pediatr Gastroenterol Nutr 2003; 36:616-22; PMID:12717085; http://dx.doi.org/10.1097/00005176-200305000-00005
- McNulty SL, Mole BM, Dailidiene D, Segal I, Ally R, Mistry R, Secka O, Adegbola RA, Thomas JE, Lenarcic EM, et al. Novel 180- and 480-base-pair insertions in African and African-American strains of Helicobacter pylori. J Clin Microbiol 2004; 42:5658-63; PMID:15583296; http://dx.doi.org/10.1128/JCM.42.12.5658-5663.2004
- Mane SP, Dominguez-Bello MG, Blaser MJ, Sobral BW, Hontecillas R, Skoneczka J, Mohapatra SK, Crasta OR, Evans C, Modise T, et al. Host-interactive genes in Amerindian Helicobacter pylori diverge from their Old World homologs and mediate inflammatory responses. J Bacteriol 2010; 192:3078-92; PMID:20400544; http://dx.doi.org/10.1128/JB.00063-10
- Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984; 1:1311-5; PMID:6145023; http://dx.doi.org/10.1016/S0140-6736(84)91816-6
- Danesh J. Helicobacter pylori infection and gastric cancer: systematic review of the epidemiological studies. Aliment Pharmacol Ther 1999; 13:851-6; PMID:10383517; http://dx.doi.org/10.1046/j.1365-2036.1999.00546.x
- Permin H, Andersen LP. Inflammation, immunity, and vaccines for Helicobacter infection. Helicobacter 2005; 10 Suppl 1:21-5; PMID:16178967; http://dx.doi.org/10.1111/j.1523-5378.2005.00337.x
- Blaser MJ. Disappearing microbiota: Helicobacter pylori protection against esophageal adenocarcinoma. Cancer Prev Res (Phila) 2008; 1:308-11; PMID:19138974; http://dx.doi.org/10.1158/1940-6207.CAPR-08-0170
- Vieth M, Masoud B, Meining A, Stolte M. Helicobacter pylori infection: protection against Barrett's mucosa and neoplasia? Digestion 2000; 62:225-31; PMID:11070405; http://dx.doi.org/10.1159/000007820
- Vaezi MF, Falk GW, Peek RM, Vicari JJ, Goldblum JR, Perez-Perez GI, Rice TW, Blaser MJ, Richter JE. CagA-positive strains of Helicobacter pylori may protect against Barrett's esophagus. Am J Gastroenterol 2000; 95:2206-11; PMID:11007219; http://dx.doi.org/10.1111/j.1572-0241.2000.02305.x
- Chow WH, Blaser MJ, Blot WJ, Gammon MD, Vaughan TL, Risch HA, Perez-Perez GI, Schoenberg JB, Stanford JL, Rotterdam H, et al. An inverse relation between cagA+ strains of Helicobacter pylori infection and risk of esophageal and gastric cardia adenocarcinoma. Cancer Res 1998; 58:588-90; PMID:9485003
- Lang L. Childhood acquisition of Helicobacter pylori linked to reduced asthma and allergy risk. Gastroenterology 2007; 133:6; PMID:17631119; http://dx.doi.org/10.1053/j.gastro.2007.05.011
- Chen Y, Blaser MJ. Inverse associations of Helicobacter pylori with asthma and allergy. Arch Intern Med 2007; 167:821-7; PMID:17452546; http://dx.doi.org/10.1001/archinte.167.8.821
- Blaser MJ, Chen Y, Reibman J. Does Helicobacter pylori protect against asthma and allergy? Gut 2008; 57:561-7; PMID:18194986; http://dx.doi.org/10.1136/gut.2007.133462
- McCune A, Lane A, Murray L, Harvey I, Nair P, Donovan J, Harvey R. Reduced risk of atopic disorders in adults with Helicobacter pylori infection. Eur J Gastroenterol Hepatol 2003; 15:637-40; PMID:12840675; http://dx.doi.org/10.1097/00042737-200306000-00010
- Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology 2009; 136:1863-73; PMID:19457415; http://dx.doi.org/10.1053/j.gastro.2009.01.073
- Kuipers EJ, Israel DA, Kusters JG, Gerrits MM, Weel J, van Der Ende A, van Der Hulst RW, Wirth HP, Höök-Nikanne J, Thompson SA, et al. Quasispecies development of Helicobacter pylori observed in paired isolates obtained years apart from the same host. J Infect Dis 2000; 181:273-82; PMID:10608776; http://dx.doi.org/10.1086/315173
- Hazell SL, Lee A, Brady L, Hennessy W. Campylobacter pyloridis and gastritis: association with intercellular spaces and adaptation to an environment of mucus as important factors in colonization of the gastric epithelium. J Infect Dis 1986; 153:658-63; PMID:3950447; http://dx.doi.org/10.1093/infdis/153.4.658
- Zanotti G, Cendron L. Structural and functional aspects of the Helicobacter pylori secretome. World J Gastroenterol 2014; 20:1402-23; PMID:24587618; http://dx.doi.org/10.3748/wjg.v20.i6.1402
- Sgouras DN, Trang TTH, Yamaoka Y. Pathogenesis of Helicobacter pylori Infection. Helicobacter 2015; 20 Suppl 1:8-16; PMID:26372819; http://dx.doi.org/10.1111/hel.12251
- Roesler BM, Rabelo-Gonçalves EMA, Zeitune JMR. Virulence Factors of Helicobacter pylori: A Review. Clin Med Insights Gastroenterol 2014; 7:9-17; PMID:24833944; http://dx.doi.org/10.4137/CGast.S13760
- Petersson C, Forsberg M, Aspholm M, Olfat FO, Forslund T, Borén T, Magnusson KE. Helicobacter pylori SabA adhesin evokes a strong inflammatory response in human neutrophils which is down-regulated by the neutrophil-activating protein. Med Microbiol Immunol 2006; 195:195-206; PMID:16758245; http://dx.doi.org/10.1007/s00430-006-0018-x
- Guruge JL, Falk PG, Lorenz RG, Dans M, Wirth HP, Blaser MJ, Berg DE, Gordon JI. Epithelial attachment alters the outcome of Helicobacter pylori infection. Proc Natl Acad Sci U S A 1998; 95:3925-30; PMID:9520469; http://dx.doi.org/10.1073/pnas.95.7.3925
- Tan S, Tompkins LS, Amieva MR. Helicobacter pylori usurps cell polarity to turn the cell surface into a replicative niche. PLoS Pathog 2009; 5:e1000407; PMID:19412339; http://dx.doi.org/10.1371/journal.ppat.1000407
- Howitt MR, Lee JY, Lertsethtakarn P, Vogelmann R, Joubert L-M, Ottemann KM, Amieva MR. ChePep controls Helicobacter pylori Infection of the gastric glands and chemotaxis in the Epsilonproteobacteria. MBio 2011; 2:e00098-11; PMID:21791582; http://dx.doi.org/10.1128/mBio.00098-11
- Necchi V, Candusso ME, Tava F, Luinetti O, Ventura U, Fiocca R, Ricci V, Solcia E. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology 2007; 132:1009-23; PMID:17383424; http://dx.doi.org/10.1053/j.gastro.2007.01.049
- Chu YT, Wang YH, Wu JJ, Lei HY. Invasion and multiplication of Helicobacter pylori in gastric epithelial cells: implications for antibiotic resistance. Infect Immun 2010; 78:4157-65; PMID:20696835; http://dx.doi.org/10.1128/IAI.00524-10
- Oh JD, Karam SM, Gordon JI. Intracellular Helicobacter pylori in gastric epithelial progenitors. Proc Natl Acad Sci U S A 2005; 102:5186-91; PMID:15795379; http://dx.doi.org/10.1073/pnas.0407657102
- Liu H, Semino-Mora C, Dubois A. Mechanism of H. pylori intracellular entry: an in vitro study. Front Cell Infect Microbiol 2012; 2:13; PMID:22919605
- Oliveira MJ, Costa AC, Costa AM, Henriques L, Suriano G, Atherton JC, Machado JC, Carneiro F, Seruca R, Mareel M, et al. Helicobacter pylori induces gastric epithelial cell invasion in a c-Met and type IV secretion system-dependent manner. J Biol Chem 2006; 281:34888-96; PMID:16990273; http://dx.doi.org/10.1074/jbc.M607067200
- Nilsson C, Sillén A, Eriksson L, Strand M-L, Enroth H, Normark S, Falk P, Engstrand L. Correlation between cag pathogenicity island composition and Helicobacter pylori-associated gastroduodenal disease. Infect Immun 2003; 71:6573-81; PMID:14573679; http://dx.doi.org/10.1128/IAI.71.11.6573-6581.2003
- Atherton JC, Cao P, Peek RM, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem 1995; 270:17771-7; PMID:7629077; http://dx.doi.org/10.1074/jbc.270.30.17771
- Basak C, Pathak SK, Bhattacharyya A, Mandal D, Pathak S, Kundu M. NF-kappaB- and C/EBPbeta-driven interleukin-1beta gene expression and PAK1-mediated caspase-1 activation play essential roles in interleukin-1beta release from Helicobacter pylori lipopolysaccharide-stimulated macrophages. J Biol Chem 2005; 280:4279-88; PMID:15561713; http://dx.doi.org/10.1074/jbc.M412820200
- Castaño-Rodríguez N, Kaakoush NO, Mitchell HM. Pattern-recognition receptors and gastric cancer. Front Immunol 2014; 5:336
- Philipson CW, Bassaganya-Riera J, Viladomiu M, Kronsteiner B, Abedi V, Hoops S, Michalak P, Kang L, Girardin SE, Hontecillas R. Modeling the Regulatory Mechanisms by Which NLRX1 Modulates Innate Immune Responses to Helicobacter pylori Infection. PLoS One 2015; 10:e0137839; PMID:26367386; http://dx.doi.org/10.1371/journal.pone.0137839
- Lee JM, Kim SS, Cho Y-S. The Role of PPARγ in Helicobacter pylori Infection and Gastric Carcinogenesis. PPAR Res 2012; 2012:687570; PMID:22936949; http://dx.doi.org/10.1155/2012/687570
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140:805-20; PMID:20303872; http://dx.doi.org/10.1016/j.cell.2010.01.022
- Rohde M, Püls J, Buhrdorf R, Fischer W, Haas R. A novel sheathed surface organelle of the Helicobacter pylori cag type IV secretion system. Mol Microbiol 2003; 49:219-34; PMID:12823823; http://dx.doi.org/10.1046/j.1365-2958.2003.03549.x
- Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, Misselwitz R, Berger J, Sewald N, König W, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 2007; 449:862-6; PMID:17943123; http://dx.doi.org/10.1038/nature06187
- Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Mémet S, Huerre MR, Coyle AJ, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 2004; 5:1166-74; PMID:15489856; http://dx.doi.org/10.1038/ni1131
- Allison CC, Kufer TA, Kremmer E, Kaparakis M, Ferrero RL. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol 2009; 183:8099-109; PMID:20007577; http://dx.doi.org/10.4049/jimmunol.0900664
- Hutton ML, Kaparakis-Liaskos M, Turner L, Cardona A, Kwok T, Ferrero RL. Helicobacter pylori exploits cholesterol-rich microdomains for induction of NF-kappaB-dependent responses and peptidoglycan delivery in epithelial cells. Infect Immun 2010; 78:4523-31; PMID:20713621; http://dx.doi.org/10.1128/IAI.00439-10
- Lu DY, Chen HC, Yang MS, Hsu YM, Lin HJ, Tang CH, Lee CH, Lai CK, Lin CJ, Shyu WC, et al. Ceramide and Toll-like receptor 4 are mobilized into membrane rafts in response to Helicobacter pylori infection in gastric epithelial cells. Infect Immun 2012; 80:1823-33; PMID:22354030; http://dx.doi.org/10.1128/IAI.05856-11
- Triantafilou M, Gamper FGJ, Lepper PM, Mouratis MA, Schumann C, Harokopakis E, Schifferle RE, Hajishengallis G, Triantafilou K. Lipopolysaccharides from atherosclerosis-associated bacteria antagonize TLR4, induce formation of TLR2/1/CD36 complexes in lipid rafts and trigger TLR2-induced inflammatory responses in human vascular endothelial cells. Cell Microbiol 2007; 9:2030-9; PMID:17419716; http://dx.doi.org/10.1111/j.1462-5822.2007.00935.x
- Smith MF, Mitchell A, Li G, Ding S, Fitzmaurice AM, Ryan K, Crowe S, Goldberg JB. Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori-induced NF-kappa B activation and chemokine expression by epithelial cells. J Biol Chem 2003; 278:32552-60; PMID:12807870; http://dx.doi.org/10.1074/jbc.M305536200
- Mandell L, Moran AP, Cocchiarella A, Houghton J, Taylor N, Fox JG, Wang TC, Kurt-Jones EA. Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect Immun 2004; 72:6446-54; PMID:15501775; http://dx.doi.org/10.1128/IAI.72.11.6446-6454.2004
- Yokota S, Ohnishi T, Muroi M, Tanamoto K, Fujii N, Amano K. Highly-purified Helicobacter pylori LPS preparations induce weak inflammatory reactions and utilize Toll-like receptor 2 complex but not Toll-like receptor 4 complex. FEMS Immunol Med Microbiol 2007; 51:140-8; PMID:17645528; http://dx.doi.org/10.1111/j.1574-695X.2007.00288.x
- Smith SM, Moran AP, Duggan SP, Ahmed SE, Mohamed AS, Windle HJ, O'Neill LA, Kelleher DP. Tribbles 3: a novel regulator of TLR2-mediated signaling in response to Helicobacter pylori lipopolysaccharide. J Immunol 2011; 186:2462-71; PMID:21220698; http://dx.doi.org/10.4049/jimmunol.1000864
- Cullen TW, Giles DK, Wolf LN, Ecobichon C, Boneca IG, Trent MS. Helicobacter pylori vs. the host: remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog 2011; 7:e1002454; PMID:22216004; http://dx.doi.org/10.1371/journal.ppat.1002454
- Yokota SI, Okabayashi T, Rehli M, Fujii N, Amano KI. Helicobacter pylori lipopolysaccharides upregulate toll-like receptor 4 expression and proliferation of gastric epithelial cells via the MEK1/2-ERK1/2 mitogen-activated protein kinase pathway. Infect Immun 2010; 78:468-76; PMID:19858308; http://dx.doi.org/10.1128/IAI.00903-09
- Backert S, Naumann M. What a disorder: proinflammatory signaling pathways induced by Helicobacter pylori. Trends Microbiol 2010; 18:479-86; PMID:20863705; http://dx.doi.org/10.1016/j.tim.2010.08.003
- Nagy TA, Allen SS, Wroblewski LE, Flaherty DK, Slaughter JC, Perez-Perez G, Israel DA, Peek RM. Helicobacter pylori induction of eosinophil migration is mediated by the cag pathogenicity island via microbial-epithelial interactions. Am J Pathol 2011; 178:1448-52; PMID:21406172
- Eck M, Schmausser B, Scheller K, Toksoy A, Kraus M, Menzel T, Müller-Hermelink HK, Gillitzer R. CXC chemokines Gro(α)/IL-8 and IP-10/MIG in Helicobacter pylori gastritis. Clin Exp Immunol 2000; 122:192-9; PMID:11091274; http://dx.doi.org/10.1046/j.1365-2249.2000.01374.x
- Nishi T, Okazaki K, Kawasaki K, Fukui T, Tamaki H, Matsuura M, Asada M, Watanabe T, Uchida K, Watanabe N, et al. Involvement of myeloid dendritic cells in the development of gastric secondary lymphoid follicles in Helicobacter pylori-infected neonatally thymectomized BALB/c mice. Infect Immun 2003; 71:2153-62; PMID:12654837; http://dx.doi.org/10.1128/IAI.71.4.2153-2162.2003
- Alvarez-Arellano L, Camorlinga-Ponce M, Maldonado-Bernal C, Torres J. Activation of human neutrophils with Helicobacter pylori and the role of Toll-like receptors 2 and 4 in the response. FEMS Immunol Med Microbiol 2007; 51:473-9; PMID:17892476; http://dx.doi.org/10.1111/j.1574-695X.2007.00327.x
- Amedei A, Cappon A, Codolo G, Cabrelle A, Polenghi A, Benagiano M, Tasca E, Azzurri A, D'Elios MM, Del Prete G, et al. The neutrophil-activating protein of Helicobacter pylori promotes Th1 immune responses. J Clin Invest 2006; 116:1092-101; PMID:16543949; http://dx.doi.org/10.1172/JCI27177
- Uno K, Kato K, Shimosegawa T. Novel role of toll-like receptors in Helicobacter pylori - induced gastric malignancy. World J Gastroenterol 2014; 20:5244-51; PMID:24833854; http://dx.doi.org/10.3748/wjg.v20.i18.5244
- Rad R, Ballhorn W, Voland P, Eisenächer K, Mages J, Rad L, Ferstl R, Lang R, Wagner H, Schmid RM, et al. Extracellular and intracellular pattern recognition receptors cooperate in the recognition of Helicobacter pylori. Gastroenterology 2009; 136:2247-57; PMID:19272387; http://dx.doi.org/10.1053/j.gastro.2009.02.066
- Sun X, Zhang M, El-Zataari M, Owyang SY, Eaton KA, Liu M, Chang YM, Zou W, Kao JY. TLR2 mediates Helicobacter pylori-induced tolerogenic immune response in mice. PLoS One 2013; 8:e74595; PMID:24058595; http://dx.doi.org/10.1371/journal.pone.0074595
- Käbisch R, Mejías-Luque R, Gerhard M, Prinz C. Involvement of Toll-Like Receptors on Helicobacter pylori-Induced Immunity. PLoS One 2014; 9:e104804; http://dx.doi.org/10.1371/journal.pone.0104804
- Otani K, Tanigawa T, Watanabe T, Nadatani Y, Sogawa M, Yamagami H, Shiba M, Watanabe K, Tominaga K, Fujiwara Y, et al. Toll-like receptor 9 signaling has anti-inflammatory effects on the early phase of Helicobacter pylori-induced gastritis. Biochem Biophys Res Commun 2012; 426:342-9; PMID:22940550; http://dx.doi.org/10.1016/j.bbrc.2012.08.080
- Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 2009; 10:241-7; PMID:19221555; http://dx.doi.org/10.1038/ni.1703
- Kim DJ, Park JH, Franchi L, Backert S, Núñez G. The Cag pathogenicity island and interaction between TLR2/NOD2 and NLRP3 regulate IL-1β production in Helicobacter pylori infected dendritic cells. Eur J Immunol 2013; 43:2650-8; PMID:23818043; http://dx.doi.org/10.1002/eji.201243281
- Semper RP, Mejías-Luque R, Groß C, Anderl F, Müller A, Vieth M, Busch DH, Prazeres da Costa C, Ruland J, Groß O, et al. Helicobacter pylori-Induced IL-1β Secretion in Innate Immune Cells Is Regulated by the NLRP3 Inflammasome and Requires the Cag Pathogenicity Island. J Immunol 2014; 193:3566-76; PMID:25172489; http://dx.doi.org/10.4049/jimmunol.1400362
- Benoit BN, Kobayashi M, Kawakubo M, Takeoka M, Sano K, Zou J, Itano N, Tsutsui H, Noda T, Fukuda M, et al. Role of ASC in the mouse model of Helicobacter pylori infection. J Histochem Cytochem 2009; 57:327-38; PMID:19064716; http://dx.doi.org/10.1369/jhc.2008.952366
- Castaño-Rodríguez N, Kaakoush NO, Goh K-L, Fock KM, Mitchell HM. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: a case-control study and gene expression analyses. PLoS One 2014; 9:e98899; http://dx.doi.org/10.1371/journal.pone.0098899
- Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, Gris D, Roney KE, Zimmermann AG, Bowzard JB, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-κB signaling pathways. Immunity 2011; 34:854-65; PMID:21703540; http://dx.doi.org/10.1016/j.immuni.2011.03.026
- Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, Yang X, Hong J, Songyang Z, Chen ZJ, et al. NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. Immunity 2011; 34:843-53; PMID:21703539; http://dx.doi.org/10.1016/j.immuni.2011.02.022
- Kang MJ, Yoon CM, Kim BH, Lee CM, Zhou Y, Sauler M, Homer R, Dhamija A, Boffa D, West AP, et al. Suppression of NLRX1 in chronic obstructive pulmonary disease. J Clin Invest 2015; 125:2458-62; PMID:25938787; http://dx.doi.org/10.1172/JCI71747
- Lord CA, Savitsky D, Sitcheran R, Calame K, Wright JR, Ting JPY, Williams KL. Blimp-1/PRDM1 mediates transcriptional suppression of the NLR gene NLRP12/Monarch-1. J Immunol 2009; 182:2948-58; PMID:19234190; http://dx.doi.org/10.4049/jimmunol.0801692
- Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson K V, Wen H, Davis BK, Allen IC, Holl EK, Ye Z, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-κB. Nat Immunol 2012; 13:823-31; PMID:22863753; http://dx.doi.org/10.1038/ni.2378
- Shi Y, Liu XF, Zhuang Y, Zhang JY, Liu T, Yin Z, Wu C, Mao XH, Jia KR, Wang FJ, et al. Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J Immunol 2010; 184:5121-9; PMID:20351183; http://dx.doi.org/10.4049/jimmunol.0901115
- Khamri W, Walker MM, Clark P, Atherton JC, Thursz MR, Bamford KB, Lechler RI, Lombardi G. Helicobacter pylori stimulates dendritic cells to induce interleukin-17 expression from CD4+ T lymphocytes. Infect Immun 2010; 78:845-53; PMID:19917709; http://dx.doi.org/10.1128/IAI.00524-09
- Al-Sammak F, Kalinski T, Weinert S, Link A, Wex T, Malfertheiner P. Gastric epithelial expression of IL-12 cytokine family in Helicobacter pylori infection in human: is it head or tail of the coin? PLoS One 2013; 8:e75192; PMID:24069393; http://dx.doi.org/10.1371/journal.pone.0075192
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005; 201:233-40; PMID:15657292; http://dx.doi.org/10.1084/jem.20041257
- Caruso R, Fina D, Paoluzi OA, Del Vecchio Blanco G, Stolfi C, Rizzo A, Caprioli F, Sarra M, Andrei F, Fantini MC, et al. IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. Eur J Immunol 2008; 38:470-8; PMID:18200634; http://dx.doi.org/10.1002/eji.200737635
- Munari F, Fassan M, Capitani N, Codolo G, Vila-Caballer M, Pizzi M, Rugge M, Della Bella C, Troilo A, D'Elios S, et al. Cytokine BAFF released by Helicobacter pylori-infected macrophages triggers the Th17 response in human chronic gastritis. J Immunol 2014; 193:5584-94; PMID:25339679; http://dx.doi.org/10.4049/jimmunol.1302865
- Kao JY, Rathinavelu S, Eaton K a, Bai L, Zavros Y, Takami M, Pierzchala A, Merchant JL. Helicobacter pylori-secreted factors inhibit dendritic cell IL-12 secretion: a mechanism of ineffective host defense. Am J Physiol Gastrointest Liver Physiol 2006; 291:G73-81; PMID:16469828; http://dx.doi.org/10.1152/ajpgi.00139.2005
- Weiss G, Forster S, Irving A, Tate M, Ferrero RL, Hertzog P, Frøkiær H, Kaparakis-Liaskos M. Helicobacter pylori VacA suppresses Lactobacillus acidophilus-induced interferon β signaling in macrophages via alterations in the endocytic pathway. MBio 2013; 4:e00609-12; PMID:23760466; http://dx.doi.org/10.1128/mBio.00609-12
- Bimczok D, Grams JM, Stahl RD, Waites KB, Smythies LE, Smith PD. Stromal regulation of human gastric dendritic cells restricts the Th1 response to Helicobacter pylori. Gastroenterology 2011; 141:929-38; PMID:21699795; http://dx.doi.org/10.1053/j.gastro.2011.06.006
- Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, Maxeiner J, Hansson M, Taube C, Quiding-Järbrink M, et al. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest 2012; 122:1082-96; PMID:22307326; http://dx.doi.org/10.1172/JCI61029
- Mitchell P, Germain C, Fiori PL, Khamri W, Foster GR, Ghosh S, Lechler RI, Bamford KB, Lombardi G. Chronic exposure to Helicobacter pylori impairs dendritic cell function and inhibits Th1 development. Infect Immun 2007; 75:810-9; PMID:17101659; http://dx.doi.org/10.1128/IAI.00228-06
- Wang YH, Gorvel JP, Chu YT, Wu JJ, Lei HY. Helicobacter pylori lmpairs murine dendritic cell responses to infection. PLoS One 2010; 5:e10844; PMID:20523725; http://dx.doi.org/10.1371/journal.pone.0010844
- Zhang M, Liu M, Luther J, Kao JY. Helicobacter pylori directs tolerogenic programming of dendritic cells. Gut Microbes 2010; 1:325-9; PMID:21327041; http://dx.doi.org/10.4161/gmic.1.5.13052
- Kaebisch R, Mejías-Luque R, Prinz C, Gerhard M. Helicobacter pylori cytotoxin-associated gene A impairs human dendritic cell maturation and function through IL-10-mediated activation of STAT3. J Immunol 2014; 192:316-23; PMID:24293633; http://dx.doi.org/10.4049/jimmunol.1302476
- Lundgren A, Trollmo C, Edebo A, Svennerholm AM, Lundin BS. Helicobacter pylori-specific CD4+ T cells home to and accumulate in the human Helicobacter pylori-infected gastric mucosa. Infect Immun 2005; 73:5612-9; PMID:16113278; http://dx.doi.org/10.1128/IAI.73.9.5612-5619.2005
- Serrano C, Diaz MI, Valdivia A, Godoy A, Peña A, Rollan A, Kirberg A, Hebel E, Fierro J, Klapp G, et al. Relationship between Helicobacter pylori virulence factors and regulatory cytokines as predictors of clinical outcome. Microbes Infect 2007; 9:428-34; PMID:17336120; http://dx.doi.org/10.1016/j.micinf.2006.12.012
- Harris PR, Wright SW, Serrano C, Riera F, Duarte I, Torres J, Peña A, Rollán A, Viviani P, Guiraldes E, et al. Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology 2008; 134:491-9; PMID:18242215; http://dx.doi.org/10.1053/j.gastro.2007.11.006
- Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest 2009; 119:2475-87; PMID:19729845; http://dx.doi.org/10.1172/JCI38605
- Müller A, Oertli M, Arnold IC. H. pylori exploits and manipulates innate and adaptive immune cell signaling pathways to establish persistent infection. Cell Commun Signal 2011; 9:25; PMID:22044597; http://dx.doi.org/10.1186/1478-811X-9-25
- Gray BM, Fontaine C A, Poe S A, Eaton K A. Complex T cell interactions contribute to Helicobacter pylori gastritis in mice. Infect Immun 2013; 81:740-52; PMID:23264048; http://dx.doi.org/10.1128/IAI.01269-12
- Luther J, Dave M, Higgins PDR, Kao JY. Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm Bowel Dis 2010; 16:1077-84; PMID:19760778; http://dx.doi.org/10.1002/ibd.21116
- Hitzler I, Sayi A, Kohler E, Engler DB, Koch KN, Hardt W-D, Müller A. Caspase-1 has both proinflammatory and regulatory properties in Helicobacter infections, which are differentially mediated by its substrates IL-1β and IL-18. J Immunol 2012; 188:3594-602; PMID:22403439; http://dx.doi.org/10.4049/jimmunol.1103212
- Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol 2001; 19:423-74; PMID:11244043; http://dx.doi.org/10.1146/annurev.immunol.19.1.423
- Yun CH, Lundgren A, Azem J, Sjöling A, Holmgren J, Svennerholm AM, Lundin BS. Natural killer cells and Helicobacter pylori infection: bacterial antigens and interleukin-12 act synergistically to induce gamma interferon production. Infect Immun 2005; 73:1482-90; PMID:15731046; http://dx.doi.org/10.1128/IAI.73.3.1482-1490.2005
- Lindgren A, Pavlovic V, Flach CF, Sjöling A, Lundin S. Interferon-gamma secretion is induced in IL-12 stimulated human NK cells by recognition of Helicobacter pylori or TLR2 ligands. Innate Immun 2011; 17:191-203; PMID:20130107; http://dx.doi.org/10.1177/1753425909357970
- Quiding-Järbrink M, Lundin BS, Lönroth H, Svennerholm AM. CD4+ and CD8+ T cell responses in Helicobacter pylori-infected individuals. Clin Exp Immunol 2001; 123:81-7; http://dx.doi.org/10.1046/j.1365-2249.2001.01427.x
- Krauss-Etschmann S, Gruber R, Plikat K, Antoni I, Demmelmair H, Reinhardt D, Koletzko S. Increase of antigen-presenting cells in the gastric mucosa of Helicobacter pylori-infected children. Helicobacter 2005; 10:214-22; PMID:15904479; http://dx.doi.org/10.1111/j.1523-5378.2005.00313.x
- Lopes AI, Victorino RMM, Palha AM, Ruivo J, Fernandes A. Mucosal lymphocyte subsets and HLA-DR antigen expression in paediatric Helicobacter pylori-associated gastritis. Clin Exp Immunol 2006; 145:13-20; PMID:16792668; http://dx.doi.org/10.1111/j.1365-2249.2006.03100.x
- Figueiredo Soares T, Aguiar Rocha G, Camargos Rocha AM, Corrêa-Oliveira R, Martins-Filho OA, Teles Carvalho AS, Souto Bittencourt PF, Afonso Oliveira C, Ferreira Nogueira AMM, Alvares Cabral MMD, et al. Differences in peripheral blood lymphocyte phenotypes between Helicobacter pylori-positive children and adults with duodenal ulcer. Clin Microbiol Infect 2007; 13:1083-8; PMID:17727687; http://dx.doi.org/10.1111/j.1469-0691.2007.01814.x
- Kronsteiner B, Bassaganya-Riera J, Philipson C, Viladomiu M, Carbo A, Pedragosa M, Vento S, Hontecillas R. Helicobacter pylori infection in a pig model is dominated by Th1 and cytotoxic CD8+ T cell responses. Infect Immun 2013; 81:3803-13; PMID:23897614; http://dx.doi.org/10.1128/IAI.00660-13
- Stolfi C, Rizzo A, Franzè E, Rotondi A, Fantini MC, Sarra M, Caruso R, Monteleone I, Sileri P, Franceschilli L, et al. Involvement of interleukin-21 in the regulation of colitis-associated colon cancer. J Exp Med 2011; 208:2279-90; PMID:21987656; http://dx.doi.org/10.1084/jem.20111106
- Pallone F, Fina D, Caruso R, Monteleone G. Role of IL-21 in inflammatory bowel disease. Expert Rev Clin Immunol 2010; 6:537-41; PMID:20594126; http://dx.doi.org/10.1586/eci.10.44
- Leber A, Abedi V, Hontecillas R, Viladomiu M, Hoops S, Ciupe S, Caughman J, Andrew T, Bassaganya-Riera J. Bistability analysis of CD4+ T follicular helper and regulatory cells during Helicobacter pylori infection. J Theor Biol; under review
- Carbo A, Olivares-Villagómez D, Hontecillas R, Bassaganya-Riera J, Chaturvedi R, Piazuelo MB, Delgado A, Washington MK, Wilson KT, Algood HMS. Systems modeling of the role of interleukin-21 in the maintenance of effector CD4+ T cell responses during chronic Helicobacter pylori infection. MBio 2014; 5:e01243-14; PMID:25053783; http://dx.doi.org/10.1128/mBio.01243-14
- Zhuang Y, Cheng P, Liu XF, Peng LS, Li BS, Wang TT, Chen N, Li WH, Shi Y, Chen W, et al. A pro-inflammatory role for Th22 cells in Helicobacter pylori-associated gastritis. Gut 2015; 64:1368-78; PMID:25134787; http://dx.doi.org/10.1136/gutjnl-2014-307020
- Muotiala A, Helander IM, Pyhälä L, Kosunen TU, Moran AP. Low biological activity of Helicobacter pylori lipopolysaccharide. Infect Immun 1992; 60:1714-6; PMID:1548097
- Allen LAH, Beecher BR, Lynch JT, Rohner OV, Wittine LM. Helicobacter pylori disrupts NADPH oxidase targeting in human neutrophils to induce extracellular superoxide release. J Immunol 2005; 174:3658-67; PMID:15749904; http://dx.doi.org/10.4049/jimmunol.174.6.3658
- Schmausser B, Josenhans C, Endrich S, Suerbaum S, Sitaru C, Andrulis M, Brändlein S, Rieckmann P, Müller-Hermelink HK, Eck M. Downregulation of CXCR1 and CXCR2 expression on human neutrophils by Helicobacter pylori: a new pathomechanism in H. pylori infection? Infect Immun 2004; 72:6773-9; PMID:15557597; http://dx.doi.org/10.1128/IAI.72.12.6773-6779.2004
- Lewis ND, Asim M, Barry DP, de Sablet T, Singh K, Piazuelo MB, Gobert AP, Chaturvedi R, Wilson KT. Immune evasion by Helicobacter pylori is mediated by induction of macrophage arginase II. J Immunol 2011; 186:3632-41; PMID:21296975; http://dx.doi.org/10.4049/jimmunol.1003431
- Jüttner S, Cramer T, Wessler S, Walduck A, Gao F, Schmitz F, Wunder C, Weber M, Fischer SM, Schmidt WE, et al. Helicobacter pylori stimulates host cyclooxygenase-2 gene transcription: critical importance of MEK/ERK-dependent activation of USF1/-2 and CREB transcription factors. Cell Microbiol 2003; 5:821-34; PMID:14531897; http://dx.doi.org/10.1046/j.1462-5822.2003.00324.x
- Meyer F, Ramanujam KS, Gobert AP, James SP, Wilson KT. Cutting edge: cyclooxygenase-2 activation suppresses Th1 polarization in response to Helicobacter pylori. J Immunol 2003; 171:3913-7; PMID:14530307; http://dx.doi.org/10.4049/jimmunol.171.8.3913
- Ozbek A, Ozbek E, Dursun H, Kalkan Y, Demirci T. Can Helicobacter pylori invade human gastric mucosa?: an in vivo study using electron microscopy, immunohistochemical methods, and real-time polymerase chain reaction. J Clin Gastroenterol 2010; 44:416-22; PMID:19904218
- Wang Y, Wu J, Lei H. When Helicobacter pylori invades and replicates in the cells. Autophagy 2009; 5:540-2; PMID:19270492; http://dx.doi.org/10.4161/auto.5.4.8167
- Greenfield LK, Jones NL. Modulation of autophagy by Helicobacter pylori and its role in gastric carcinogenesis. Trends Microbiol 2013; 21:602-12; PMID:24156875; http://dx.doi.org/10.1016/j.tim.2013.09.004
- Basu M, Czinn SJ, Blanchard TG. Absence of catalase reduces long-term survival of Helicobacter pylori in macrophage phagosomes. Helicobacter 2004; 9:211-6; PMID:15165256; http://dx.doi.org/10.1111/j.1083-4389.2004.00226.x
- Gobert AP, McGee DJ, Akhtar M, Mendz GL, Newton JC, Cheng Y, Mobley HL, Wilson KT. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc Natl Acad Sci U S A 2001; 98:13844-9; PMID:11717441; http://dx.doi.org/10.1073/pnas.241443798
- Chaturvedi R, Asim M, Lewis ND, Algood HMS, Cover TL, Kim PY, Wilson KT. L-arginine availability regulates inducible nitric oxide synthase-dependent host defense against Helicobacter pylori. Infect Immun 2007; 75:4305-15; PMID:17562760; http://dx.doi.org/10.1128/IAI.00578-07
- Chaturvedi R, Asim M, Barry DP, Frye JW, Casero RA, Wilson KT. Spermine oxidase is a regulator of macrophage host response to Helicobacter pylori: enhancement of antimicrobial nitric oxide generation by depletion of spermine. Amino Acids 2014; 46:531-42; PMID:23820617; http://dx.doi.org/10.1007/s00726-013-1531-z
- Rittig MG, Shaw B, Letley DP, Thomas RJ, Argent RH, Atherton JC. Helicobacter pylori-induced homotypic phagosome fusion in human monocytes is independent of the bacterial vacA and cag status. Cell Microbiol 2003; 5:887-99; PMID:14641174; http://dx.doi.org/10.1046/j.1462-5822.2003.00328.x
- Ramarao N, Meyer TF. Helicobacter pylori resists phagocytosis by macrophages: quantitative assessment by confocal microscopy and fluorescence-activated cell sorting. Infect Immun 2001; 69:2604-11; PMID:11254625; http://dx.doi.org/10.1128/IAI.69.4.2604-2611.2001
- Zheng PY, Jones NL. Helicobacter pylori strains expressing the vacuolating cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining TACO (coronin 1) protein. Cell Microbiol 2003; 5:25-40; PMID:12542468; http://dx.doi.org/10.1046/j.1462-5822.2003.00250.x
- Du SY, Wang HJ, Cheng HH, Chen SD, Wang LHC, Wang WC. Cholesterol glucosylation by Helicobacter pylori delays internalization and arrests phagosome maturation in macrophages. J Microbiol Immunol Infect 2014
- Borlace GN, Jones HF, Keep SJ, Butler RN, Brooks DA. Helicobacter pylori phagosome maturation in primary human macrophages. Gut Pathog 2011; 3:3; PMID:21426584; http://dx.doi.org/10.1186/1757-4749-3-3
- Zabaleta J, McGee DJ, Zea AH, Hernández CP, Rodriguez PC, Sierra RA, Correa P, Ochoa AC. Helicobacter pylori arginase inhibits T cell proliferation and reduces the expression of the TCR zeta-chain (CD3zeta). J Immunol 2004; 173:586-93; PMID:15210820; http://dx.doi.org/10.4049/jimmunol.173.1.586
- Wüstner S, Mejías-Luque R, Koch MF, Rath E, Vieth M, Sieber SA, Haller D, Gerhard M. H. pylori γ-glutamyltranspeptidase impairs T-lymphocyte function by compromising metabolic adaption through inhibition of cMyc and IRF4 expression. Cell Microbiol 2014; 17(1):51-61; PMID:23910799
- Sewald X, Jiménez-Soto L, Haas R. PKC-dependent endocytosis of the Helicobacter pylori vacuolating cytotoxin in primary T lymphocytes. Cell Microbiol 2011; 13:482-96; PMID:21083636; http://dx.doi.org/10.1111/j.1462-5822.2010.01551.x
- Sundrud MS, Torres VJ, Unutmaz D, Cover TL. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc Natl Acad Sci U S A 2004; 101:7727-32; PMID:15128946; http://dx.doi.org/10.1073/pnas.0401528101
- Boncristiano M, Paccani SR, Barone S, Ulivieri C, Patrussi L, Ilver D, Amedei A, D'Elios MM, Telford JL, Baldari CT. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J Exp Med 2003; 198:1887-97; PMID:14676300; http://dx.doi.org/10.1084/jem.20030621
- Owyang SY, Luther J, Kao JY. Helicobacter pylori: beneficial for most? Expert Rev Gastroenterol Hepatol 2011; 5:649-51; PMID:22017690; http://dx.doi.org/10.1586/egh.11.69
- Chen Y, Blaser MJ. Helicobacter pylori colonization is inversely associated with childhood asthma. J Infect Dis 2008; 198:553-60; PMID:18598192; http://dx.doi.org/10.1086/590158
- Herbarth O, Bauer M, Fritz GJ, Herbarth P, Rolle-Kampczyk U, Krumbiegel P, Richter M, Richter T. Helicobacter pylori colonisation and eczema. J Epidemiol Community Health 2007; 61:638-40; PMID:17568058; http://dx.doi.org/10.1136/jech.2006.046706
- Raghunath A, Hungin APS, Wooff D, Childs S. Prevalence of Helicobacter pylori in patients with gastro-oesophageal reflux disease: systematic review. BMJ 2003; 326:737; PMID:12676842; http://dx.doi.org/10.1136/bmj.326.7392.737
- Bassaganya-Riera J, Dominguez-Bello MG, Kronsteiner B, Carbo A, Lu P, Viladomiu M, Pedragosa M, Zhang X, Sobral BW, Mane SP, et al. Helicobacter pylori Colonization Ameliorates Glucose Homeostasis in Mice through a PPAR γ-Dependent Mechanism. PLoS One 2012; 7:e50069
- Codolo G, Mazzi P, Amedei A, Del Prete G, Berton G, D'Elios MM, de Bernard M. The neutrophil-activating protein of Helicobacter pylori down-modulates Th2 inflammation in ovalbumin-induced allergic asthma. Cell Microbiol 2008; 10:2355-63; PMID:18671823; http://dx.doi.org/10.1111/j.1462-5822.2008.01217.x
- Higgins PDR, Johnson LA, Luther J, Zhang M, Sauder KL, Blanco LP, Kao JY. Prior Helicobacter pylori infection ameliorates Salmonella typhimurium-induced colitis: mucosal crosstalk between stomach and distal intestine. Inflamm Bowel Dis 2011; 17:1398-408; PMID:21560200; http://dx.doi.org/10.1002/ibd.21489
- Luther J, Owyang SY, Takeuchi T, Cole TS, Zhang M, Liu M, Erb-Downward J, Rubenstein JH, Chen CC, Pierzchala AV, et al. Helicobacter pylori DNA decreases pro-inflammatory cytokine production by dendritic cells and attenuates dextran sodium sulphate-induced colitis. Gut 2011; 60:1479-86; PMID:21471567; http://dx.doi.org/10.1136/gut.2010.220087
- Engler DB, Leonardi I, Hartung ML, Kyburz A, Spath S, Becher B, Rogler G, Müller A. Helicobacter pylori-specific protection against inflammatory bowel disease requires the NLRP3 inflammasome and IL-18. Inflamm Bowel Dis 2015; 21:854-61; PMID:25742401; http://dx.doi.org/10.1097/MIB.0000000000000318
- Vo HD, Goli S, Gill R, Anderson V, Stefanov DG, Xu J, Kulsum-Mecci N, Schwarz SM, Rabinowitz SS. Inverse correlation between Helicobacter pylori colonization and obesity in a cohort of inner city children. Helicobacter 2015; 20:64-8; PMID:25308209; http://dx.doi.org/10.1111/hel.12154
- Lender N, Talley NJ, Enck P, Haag S, Zipfel S, Morrison M, Holtmann GJ. Review article: Associations between Helicobacter pylori and obesity-an ecological study. Aliment Pharmacol Ther 2014; 40:24-31; PMID:24832176; http://dx.doi.org/10.1111/apt.12790
- Cho I, Blaser MJ, François F, Mathew JP, Ye XY, Goldberg JD, Bini EJ. Helicobacter pylori and overweight status in the United States: data from the Third National Health and Nutrition Examination Survey. Am J Epidemiol 2005; 162:579-84; PMID:16093294; http://dx.doi.org/10.1093/aje/kwi237
- Papamichael K, Konstantopoulos P, Mantzaris GJ. Helicobacter pylori infection and inflammatory bowel disease: is there a link? World J Gastroenterol 2014; 20:6374-85; PMID:24914359; http://dx.doi.org/10.3748/wjg.v20.i21.6374
- Thia KT, Loftus EV, Sandborn WJ, Yang SK. An update on the epidemiology of inflammatory bowel disease in Asia. Am J Gastroenterol 2008; 103:3167-82; PMID:19086963; http://dx.doi.org/10.1111/j.1572-0241.2008.02158.x
- Robinson K, Kenefeck R, Pidgeon EL, Shakib S, Patel S, Polson RJ, Zaitoun AM, Atherton JC. Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut 2008; 57:1375-85; PMID:18467372; http://dx.doi.org/10.1136/gut.2007.137539
- Wagtmans MJ, Witte AM, Taylor DR, Biemond I, Veenendaal RA, Verspaget HW, Lamers CB, van Hogezand RA. Low seroprevalence of Helicobacter pylori antibodies in historical sera of patients with Crohn's disease. Scand J Gastroenterol 1997; 32:712-8; PMID:9246713; http://dx.doi.org/10.3109/00365529708996523
- Carbo A, Bassaganya-Riera J, Pedragosa M, Viladomiu M, Marathe M, Eubank S, Wendelsdorf K, Bisset K, Hoops S, Deng X, et al. Predictive Computational Modeling of the Mucosal Immune Responses during Helicobacter pylori Infection. PLoS One 2013; 8:e73365; PMID:24039925; http://dx.doi.org/10.1371/journal.pone.0073365
- Kronsteiner B, Bassaganya-Riera J, Philipson N, Hontecillas R. Novel insights on the role of CD8+ T cells and cytotoxic responses during Helicobacter pylori infection. Gut Microbes 2014; 5:357-62; PMID:24755940; http://dx.doi.org/10.4161/gmic.28899
- Lertsethtakarn P, Howitt MR, Castellon J, Amieva MR, Ottemann KM. Helicobacter pylori CheZHP and ChePep form a novel chemotaxis-regulatory complex distinct from the core chemotaxis signaling proteins and the flagellar motor. Mol Microbiol 2015; 97:1063-78; PMID:26061894; http://dx.doi.org/10.1111/mmi.13086
- Viladomiu M, Bassaganya-Riera J, Kronsteiner-Dobramysl B, Washington Philipson C, Michalak P, Eden K, Hontecillas R. Macrophages modulate bacterial persistence and gastric pathology during Helicobacter pylori infection (INC4P.346). J Immunol 2015; 194:125.25-125.25; PMID:25404365; http://dx.doi.org/10.4049/jimmunol.1401644
- Carbo A, Hontecillas R, Kronsteiner B, Viladomiu M, Pedragosa M, Lu P, Philipson CW, Hoops S, Marathe M, Eubank S, et al. Systems modeling of molecular mechanisms controlling cytokine-driven CD4+ T cell differentiation and phenotype plasticity. PLoS Comput Biol 2013; 9:e1003027; PMID:23592971; http://dx.doi.org/10.1371/journal.pcbi.1003027
- Carbo A, Hontecillas R, Andrew T, Eden K, Mei Y, Hoops S, Bassaganya-Riera J. Computational modeling of heterogeneity and function of CD4+ T cells. Front cell Dev Biol 2014; 2:31; PMID:25364738; http://dx.doi.org/10.3389/fcell.2014.00031
- Marwaha S, Schumacher MA, Zavros Y, Eghbalnia HR. Crosstalks between cytokines and Sonic Hedgehog in Helicobacter pylori infection: a mathematical model. PLoS One 2014; 9:e111338; PMID:25364910; http://dx.doi.org/10.1371/journal.pone.0111338
- Schumacher MA, Donnelly JM, Engevik AC, Xiao C, Yang L, Kenny S, Varro A, Hollande F, Samuelson LC, Zavros Y. Gastric sonic hedgehog acts as a macrophage chemoattractant during the immune response to Helicobacter pylori. Gastroenterology 2012; 142:1150-9.e6; PMID:22285806; http://dx.doi.org/10.1053/j.gastro.2012.01.029