
ABSTRACT
Microbes modify immunometabolism responses linking obesity and type 2 diabetes. Immunity helps maintain a host-microbe symbiosis, but inflammation can promote insulin resistance in tissues that control blood glucose. We were interested in compartmentalization of immune responses during obesity and show here that feeding mice an obesity-causing high-fat diet (HFD) decreased a marker of neutrophil activation and cytokines related to Th17 responses in the gut. A HFD decreased IL-17 and IL-21/22 in the ileum and colon, respectively. A HFD increased IL-17, IL-21/22 and other related Th17 responses in the liver. At the whole tissue level, there is divergence in gut and metabolic tissue Th17 cytokines during diet-induced obesity. Deletion of the bacterial peptidoglycan sensor NOD2 had relatively minor effects on these immune responses. We propose a model where diet-induced obesity promotes a permissive gut immune environment and sets the stage for host genetics to contribute to dysbiosis-driven metabolic tissue inflammation.
Obesity promotes insulin resistance, which often precedes type 2 diabetes. Obesity and eating fat-laden diets are associated with chronic, low-level inflammation in certain tissues. Increased inflammation impairs insulin action in metabolic cells such as those in liver and adipose tissue, which compromises the control of blood glucose.Citation1 The appreciation that inflammation can alter endocrine function has fostered interest in immunometabolism.Citation2 Understanding the connection between the immune and metabolic systems represents a potential way to combat obesity-driven risk of diabetes and cardiovascular disease without necessarily decreasing obesity.Citation3,4 However, the instigating factors of inflammation and potential compartmentalization of immune responses in different tissues are not well understood. Gut bacteria have emerged as a link between immunity and metabolism.Citation5 Obesity-promoting diets alter the intestinal microbiota and gut function, which can contribute to an increase in circulating and tissue resident microbial components.Citation6 This state of metabolic endotoxemia contributes to inflammation-driven insulin resistance during obesity.Citation7,8 Both dietary factors and host bacterial sensing ability can modify gut mucosal immunity and metabolic endotoxemia. Microbes and microbial components are positioned as responders and instigators of compartmentalized immune responses in the gut and metabolic tissues. The contribution of host genetics interacting with dietary cues to promote dysbiosis during obesity should be considered in models trying to understand the immunometabolism of insulin resistance.
Pathogen recognition, innate immunity, inflammation, metabolism
Pattern recognition receptors (PRRs) of the innate immune system mount inflammatory and antimicrobial responses by detecting a range of conserved molecular motifs. Common bacterial compounds such as lipopolysaccharide and peptidoglycan can be activator ligands of toll-like receptors (TLRs) and nucleotide oligomerization domain (NOD)-like receptors (NLRs), respectively. Upon activation, PRRs initiate inflammatory responses in the form of cytokines, chemokines, and antimicrobial peptides to defend the host against perceived microbial threats. PRRs contribute to host protection from pathogenic environmental factors including certain microbes. However, bacterial component, lipid-related, and drug-mediated PRR activation can promote inflammation and insulin resistance in key metabolic tissues.Citation9-13 It is not surprising that deletion of certain PRRs can protect against insulin resistance during obesity.Citation14-16 Paradoxically, PRRs can also protect against excessive inflammation by limiting gut dysbiosis.Citation17,18
Obesity, microbiota, gut and metabolic disease
An obesity-inducing high-fat diet (HFD) compromises gut permeability in mice and permits an increased burden of bacteria, bacterial components, and bacterial metabolites to areas of the body beyond mucosal barriers.Citation8 This metabolic endotoxemia causes inflammation in metabolic tissues that contributes to insulin resistance.Citation7,8 Gut bacteria alone is able to transmit features of gut-derived disease such as experimental colitis and colorectal cancer in mice.Citation19,20 Certain host metabolic phenotypes are also transmissible through the gut microbiota. Adiposity (i.e. fat tissue mass) is the best example to date. Dietary changes in the microbiota can be passed to recipient mice as a stand-alone factor promoting increased fat mass.Citation21 Impressively, microbial transmissibility of adiposity can occur despite similar host genetics.Citation22 However, host genetics is positioned to alter the relationship between microbes, immunity and disease outcomes. Couturier-Maillard and colleagues demonstrated that absence of NOD2 causes dysbiosis that is sufficient to promote colorectal cancer and colitis.Citation19 This adds to strong evidence implicating defective NOD2 signaling in intestinal diseases. Mutations of NOD2 increase susceptibility to Crohn's disease in humans.Citation23 In experimental models, deletion of NOD2 predisposes mice to gut barrier dysfunction and altered intestinal immunity.Citation24
NOD2, gut microbiota, and metabolic disease
Building on these concepts, we recently examined the role of NOD2 in inflammation, gut permeability, and gut bacterial composition in the context of diet-induced insulin resistance.Citation17 We showed that whole-body deletion of NOD2 worsened metabolic inflammation (in the liver and adipose tissue) and lowered insulin sensitivity. A high-fat diet was required to reveal the protective role of NOD2 regarding insulin resistance and metabolic inflammation. We then hypothesized that NOD2 acted as a gut bacterial surveillance protein, limiting diet-induced dysbiosis and the associated insulin resistance. Indeed, NOD2 deletion predisposed mice to increased gut bacterial adherence and dissemination of bacteria within the resident immune cells of the adipose tissue. The added stress of an obesity-causing diet revealed that NOD2-null mice had more bacterial DNA in the liver, implicating NOD2 as a protective sensor limiting invasion of a bacterial component into a key tissue involved in glucose control. Host genetics and diet combined to influence gut microbial composition. Interestingly, the gut bacteria from NOD2-deficient obese mice were able to recapitulate some, but not all, aspects of metabolic disease in germ-free recipient WT mice. This finding reinforced work from other groups showing the potential of a NOD2 knockout-specific microbiota to transmit disease phenotypes.Citation19
Intriguingly, the poor glucose control in obese NOD2-null mice did not occur when NOD2 was deleted from immune cells. This result is important because it suggests that the defect originates in parenchymal cells, which appear sufficient to preserve gut microbial and metabolic balance. The type of cell(s) required for NOD2-mediated protection from exacerbated metabolic disease should be identified. A more important question to address is why an obesity-causing diet was required to elicit worse insulin resistance in NOD2-null mice. How did this nutritional stress set the immunometabolic stage for worse insulin resistance?
Th17 immunity, gut microbiota, metabolic disease
It is not yet clear how NOD2 contributes to immunity that can influence microbial composition, metabolic tissue inflammation and insulin resistance during an obesity-causing diet. NOD2 activation with the bacterial ligand muramyl dipeptide (MDP) stimulates production of IL-17, an important effector cytokine in the Th17 arm of immunity.Citation25 It has been shown that NOD1 and NOD2 are required for early intestinal Th17 responses to enteric infection.Citation26 Th17 responses contribute to host defense against certain bacterial pathogens.Citation27,28 A number of cytokines, including, but not limited to IL-17, IL-21, IL-22, IL-23 are important for the development and maintenance of the Th17 response.Citation27
Increased Th17 responses can restore gut mucosal immunity and barrier function. This is important because both IL-22 and IL-23 can improve host defense to pathogens that cause infectious colitis and these same Th17-related effectors can actually improve insulin sensitivity during obesity.Citation29 Recent evidence showed that HFD-feeding in mice reduced the ability for antigen presenting cells to induce Th17 differentiation in the lamina propria of the small intestine.Citation30 This is important because absence of IL-17 in intestinal T-cells promotes obesity, inflammation in metabolic tissues, and insulin resistance.Citation30 Both intestinal and metabolic tissue dysbiosis were implicated in the association of defective gut Th17 responses to insulin resistance.Citation30 Therefore, certain dietary factors and/or obesity appear to impair gut Th17 immunity linked with dysbiosis, where the resultant metabolic endotoxemia promotes inflammation in metabolic tissues and insulin resistance. These results showing decreased Th17 in the gut during obesity are in direct opposition to increased systemic (and spleen) Th17 bias during obesity.Citation31 In this article, we explored the gut and metabolic tissue Th17 responses during diet-induced obesity. We hypothesized that absence of NOD2 would compromise gut Th17 responses. However, our results show that diet was the major driver of decreased Th17 responses in different gut segments. We propose a model where obesity-causing diets promote a permissive intestinal immune environment allowing dysbiosis and microbial-driven increased inflammation in certain metabolic tissues.
High-fat feeding reduces aspects of gut immunity in mice
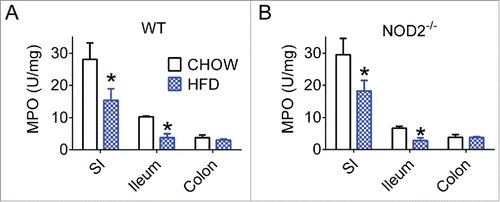
There is evidence that a HFD and/or obesity corresponds with increased markers of certain inflammatory responses and decreases in others throughout the intestine.Citation32,33 We found that all gut segments from HFD-fed WT and NOD2-null mice had decreased myeloperoxidase (MPO), a marker of neutrophil activity during respiratory burst (). Neutrophils can secrete IL-17 and IL-22 and are recruited as part of the Th17 immune response and have potent antibacterial capabilities.Citation34,35 While it is known that other neutrophil products, such as elastase, are capable of worsening diet-induced inflammation and insulin resistance in adipose and liver,Citation36 we postulated that intestinal neutrophil activity is a marker of a permissive intestinal immune environment. Our results suggest that chronic dietary stress and obesity attenuates certain aspects of gut immune responses, setting the stage for decreased gut bacterial surveillance.
Figure 1. High-fat feeding reduces aspects of gut immunity in mice. Quantification of neutrophil myeloperoxidase (MPO) levels in small intestine, ileum, and colon gut segments of WT (A) and NOD2-null (B) 16-week chow- and HFD-fed mice. For all groups, n = 5. * indicates P <0.05.
High-fat diet impairs gut Th17 immune responses
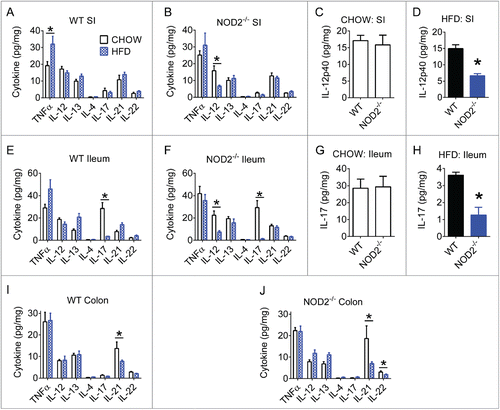
We next investigated if levels of Th1, Th2, or Th17 immunity markers were modified in various gut segments of from chow- and HFD-fed mice. We found that HFD increased TNFα in small intestine of WT mice (). However, the most pronounced effect of a HFD was a decrease in certain Th17-related immune responses in the ileum and colon segments (). Our analysis also highlighted a small role for host genetics and peptidoglycan sensing. Compared to HFD-fed WT mice, HFD-fed NOD2-null mice had significantly lower IL-17 in the ileum (). The biological relevance of these NOD2-driven changes in very low levels of IL-17 is questionable. Diet had a much larger effect on IL-17 levels. In addition to decreased Th17 related immune responses, we also observed that HFD-fed NOD2-null mice also had decreased IL-12p40 in the small intestine and a HFD failed to increase TNFα in NOD2-null mice (). Together, these results corroborate recent findings that HFD-feeding can impair certain antimicrobial intestinal immune responses,Citation30 and also reveal that host genetics can have an additional impact.
Figure 2. High-fat diet impairs gut Th17 immune responses. Quantification of various Th1, Th2, and Th17-related cytokines in small intestine, ileum, and colon gut segments of WT (A, E, I) and NOD2-null (B, F, J) 16-week chow- and HFD-fed mice. Host genotype-specific effects were observed during dietary stress for IL-12p40 in small intestine (C, D) and IL-17A in ileum (G, H). Data is presented as pg cytokine per mg of indicated tissue. For all groups, n = 5. * indicates P <0.05.
High-fat diet has different effects on Th17 immunity in metabolic tissues
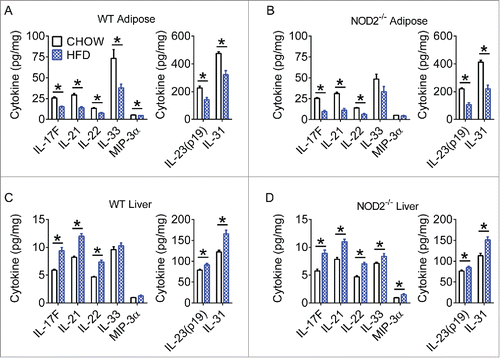
Our data reveals that diet and NOD2-genotype can promote a permissive gut immune environment. This may allow bacteria or bacterial components to evade the gut mucosal barrier. In a situation of decreased Th17 gut microbial surveillance, dysbiosis and subsequent bacterial invasion of the host would be expected to cause additional inflammation in tissues beyond the gut. Indeed, we previously found that HFD and defective NOD2 combine to augment Th1 inflammation in liver and adipose tissue.Citation17 We therefore sought to determine the effect of diet and host genetics on Th17-related immunity in metabolic tissues. We analyzed whole adipose and liver from WT and NOD2-null, chow- and HFD-fed, mice for Th17-related cytokines. Diet was the major driver of changes in tissue Th17-related responses and interestingly, we found that a HFD drove a differential Th17 immune response in metabolic tissues. HFD-feeding was associated with significantly decreased Th17-related cytokines in white adipose tissue (WAT, ), and increased Th17-related cytokines in liver (). Further scrutiny of specific cell populations is warranted, since different cell types can be the major producers of IL-17 in various tissues.Citation37
Figure 3. High-fat diet has different effects on Th17 immunity in metabolic tissues. Quantification of various Th17 cytokines in white adipose (A, B) and liver (C, D) tissue of WT and NOD2-null chow- and HFD-fed mice. Data is presented as pg cytokine per mg of indicated tissue. For chow-fed animals, n = 10. For HFD-fed animals, n = 11 (WT) or n = 8 (NOD2-null). * indicates P <0.05.
Diet, host genotype, and microbes link compartmentalized Th17 immunity
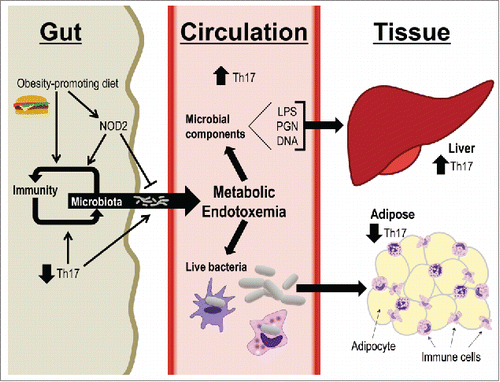
Based on our previous work and the data presented in this report, we propose a model integrating microbes as a link between diet, host genetics, and compartmentalized immunity (). An obesity-promoting diet perturbs the balance between gut immunity and microbiota composition. A permissive gut immune environment promotes bacterial (component) evasion of the gut mucosal barrier. Decreased gut Th17 immune responses are part of this permissive gut immunity and could result from a combination of dietary factors or altered microbiota. Further, defective microbial sensing by loss of NOD2 exacerbates these diet-induced effects. This resultant metabolic endotoxemia can affect distant tissues such as adipose and liver to worsen localized inflammation and whole-body insulin resistance.Citation7,38 The effects of specific immune responses in various metabolic tissues may be related to the type or amount of bacterial components and sensors within each tissue. While we have previously shown (via measurement of bacterial DNA) that HFD and NOD2 deletion combine to increase bacterial load in liver,Citation17 it will be important to identify the specific bacterial species and/or additional bacterial components that invade metabolic tissues during dietary stress. It will be equally important to determine whether the permissive environment created by an interaction between diet and host genetics is favorable to live bacteria or simply allows for an increased abundance of inflammatory bacterial products or components from dead cells.
Figure 4. Diet, host genotype, and microbes link compartmentalized Th17 immunity. A proposed model indicating how dietary factors, host genetics, and microbiota connect gut immunity to metabolic tissue inflammation. An obesity-promoting diet alters the balance between gut immunity and microbiota composition, and defective NOD2 worsens these effects. This environment is characterized by decreased Th17 immune responses that contribute to bacterial cell and component evasion of the gut mucosal barrier and dissemination into host circulation. Destinations for gut bacterial products include tissues such as adipose and liver, which in turn experience increased metabolic inflammation and subsequent impaired insulin sensitivity.
Concluding remarks
We found that increased fat content of the diet causes different immune responses in the gut versus metabolic (liver, adipose) tissues. We observed compartmentalization of Th17 immune responses during diet-induced obesity. At the whole tissue level, this included lower Th17-related cytokines in the gut; lower Th17-related cytokines in the adipose tissue; and higher Th17-related cytokines in the liver of mice. We propose a model where obesity-causing diets promote a permissive immune environment in the gut that allows microbes to relay increased inflammation to metabolic tissues that help control blood glucose, such as the liver. NOD2-genotype did not alter diet-induced changes in Th17 responses. Therefore, we propose that diet is the major driver of gut Th17 responses compared to host genetics. We recently showed that NOD2-deletion promotes dysbiosis-related metabolic disease. Diet-induced obesity was required to reveal the protective role of this bacterial sensor. We have now shown that certain aspects of gut immunity are decreased during feeding of obesity-causing diets, which appears to set the stage for host genetics to modify dysbiosis-driven changes in host metabolism.
Materials and methods
Animals
The McMaster University Animal Research Ethics Board approved all animal procedures. Mice were housed in a 12h light/dark cycle and given free access to food and water. Chow control diet (5% kcal from fat) was from Harlan Laboratories. HFD (45% kcal from fat) was from Research Diets. Male C57BL/6 WT and NOD2-null mice were described previously.Citation17 Animals were fed the indicated diet for 16 weeks, euthanized, and tissues were removed immediately and frozen in liquid nitrogen.
Experimental procedures
MPO assay was performed as previously described.Citation39 Cytokine measurement was performed using the indicated BioPlex kits on a BioPlex-200 instrument (Bio-Rad). Tissues were homogenized in a buffer containing 50 mM Tris-HCl pH 8.0, 150 mM NaCl, and 1% Triton X-100. Protein lysates were analyzed according to BioPlex kit instructions, and cytokine levels were normalized to total protein concentration of tissue lysates.
Statistical analysis
Values are mean + SEM. Statistical significance was determined by unpaired t-test or ANOVA and Tukey's post-hoc test where appropriate (Prism 5–6, GraphPad Software). P < 0.05 was considered significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Supported by an operating grant to JDS from and the Canadian Diabetes Association (CDA; OG-3–12–3745-JS) and by an operating grant to WIK from CIHR. JDS holds CDA Scholar (SC-5–12–3891-JS) and CIHR New Investigator awards (MSH-136665). JFC was supported by a CIHR doctoral scholarship. KPF was supported by an NSERC fellowship.
References
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 2011; 29:415-45; PMID:21219177; http://dx.doi.org/10.1146/annurev-immunol-031210-101322
- Schertzer JD, Steinberg GR. Immunometabolism: the interface of immune and metabolic responses in disease. Immunol Cell Biol 2014; 92:303; PMID:24613974; http://dx.doi.org/10.1038/icb.2014.12
- Steinberg GR, Schertzer JD. AMPK promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: implications for diabetes and cardiovascular disease. Immunol Cell Biol 2014; 92:340-5; PMID:24638063; http://dx.doi.org/10.1038/icb.2014.11
- Fullerton MD, Steinberg GR, Schertzer JD. Immunometabolism of AMPK in insulin resistance and atherosclerosis. Mol Cell Endocrinol 2013; 366:224-34; PMID:22361321; http://dx.doi.org/10.1016/j.mce.2012.02.004
- Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 2012; 3:279-88; PMID:22572877; http://dx.doi.org/10.4161/gmic.19625
- Burcelin R, Serino M, Chabo C, Garidou L, Pomié C, Courtney M, Amar J, Bouloumié A. Metagenome and metabolism: The tissue microbiota hypothesis. Diabetes, Obes. Metab. 2013; 15:61-70
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007; 56:1761-72; PMID:17456850; http://dx.doi.org/10.2337/db06-1491
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008; 57:1470-81; PMID:18305141; http://dx.doi.org/10.2337/db07-1403
- Tamrakar AK, Schertzer JD, Chiu TT, Foley KP, Bilan PJ, Philpott DJ, Klip A. NOD2 activation induces muscle cell-autonomous innate immune responses and insulin resistance. Endocrinology 2010; 151:5624-37; PMID:20926588; http://dx.doi.org/10.1210/en.2010-0437
- Schertzer JD, Tamrakar AK, Magalhães JG, Pereira S, Bilan PJ, Fullerton MD, Liu Z, Steinberg GR, Giacca A, Philpott DJ, et al. NOD1 activators link innate immunity to insulin resistance. Diabetes 2011; 60:2206-15; PMID:21715553; http://dx.doi.org/10.2337/db11-0004
- Chi W, Dao D, Lau TC, Henriksbo BD, Cavallari JF, Foley KP, Schertzer JD. Bacterial Peptidoglycan Stimulates Adipocyte Lipolysis via NOD1. PLoS One 2014; 9:e97675; PMID:24828250; http://dx.doi.org/10.1371/journal.pone.0097675
- Henriksbo BD, Lau TC, Cavallari JF, Denou E, Chi W, Lally JS, Crane JD, Duggan BM, Foley KP, Fullerton MD, et al. Fluvastatin Causes NLRP3 Inflammasome-Mediated Adipose Insulin Resistance. Diabetes 2014; 63:3742-7; PMID:24917577; http://dx.doi.org/10.2337/db13-1398
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 2006; 116:3015-25; PMID:17053832; http://dx.doi.org/10.1172/JCI28898
- Nakamura T, Kunz RC, Zhang C, Kimura T, Yuan CL, Baccaro B, Namiki Y, Gygi SP, Hotamisligil GS. A Critical Role for PKR Complexes with TRBP in Immunometabolic Regulation and eIF2α Phosphorylation in Obesity. Cell Rep 2015; 11(2):295-307; PMID:25843719; http://dx.doi.org/10.1016/j.celrep.2015.03.021
- Vandanmagsar B, Youm Y-H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 2011; 17:179-88; PMID:21217695; http://dx.doi.org/10.1038/nm.2279
- Poggi M, Bastelica D, Gual P, Iglesias MA, Gremeaux T, Knauf C, Peiretti F, Verdier M, Juhan-Vague I, Tanti JF, et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 2007; 50:1267-76; PMID:17426960; http://dx.doi.org/10.1007/s00125-007-0654-8
- Denou E, Lolmede K, Garidou L, Pomie C, Chabo C, Lau TC, Fullerton MD, Nigro G, Zakaroff-Girard A, Luche E, et al. Defective NOD2 peptidoglycan sensing promotes diet-induced inflammation, dysbiosis, and insulin resistance. EMBO Mol Med 2015; 7:259-74; PMID:25666722; http://dx.doi.org/10.15252/emmm.201404169
- Caricilli AM, Picardi PK, de Abreu LL, Ueno M, Prada PO, Ropelle ER, Hirabara SM, Castoldi Â, Vieira P, Camara NOS, et al. Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol 2011; 9; e1001212; PMID:22162948
- Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest 2013; 123(2):700-11; PMID:23281400; http://dx.doi.org/10.1172/JCI62236
- Schulz MD, Atay C, Heringer J, Romrig FK, Schwitalla S, Aydin B, Ziegler PK, Varga J, Reindl W, Pommerenke C, et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 2014; 514(7523):508-12; PMID: 25174708
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 2009; 1:6ra14; PMID:20368178
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013; 341:1241214; PMID:24009397; http://dx.doi.org/10.1126/science.1241214
- Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 2001; 411:603-6; PMID:11385577; http://dx.doi.org/10.1038/35079114
- Barreau F, Madre C, Meinzer U, Berrebi D, Dussaillant M, Merlin F, Eckmann L, Karin M, Sterkers G, Bonacorsi S, et al. Nod2 regulates the host response towards microflora by modulating T cell function and epithelial permeability in mouse Peyer's patches. Gut 2010; 59:207-17; PMID:19837677; http://dx.doi.org/10.1136/gut.2008.171546
- van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, Muller FJ, Hommes DW, Zaat S a J, Kapsenberg ML, de Jong EC. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity 2007; 27:660-9; PMID:17919942; http://dx.doi.org/10.1016/j.immuni.2007.08.013
- Geddes K, Rubino SJ, Magalhaes JG, Streutker C, Le Bourhis L, Cho JH, Robertson SJ, Kim CJ, Kaul R, Philpott DJ, et al. Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat Med 2011; 17:837-44; PMID:21666695; http://dx.doi.org/10.1038/nm.2391
- Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature 2008; 453:1051-7; PMID:18563156; http://dx.doi.org/10.1038/nature07036
- Ouyang W, Kolls JK, Zheng Y. The Biological Functions of T Helper 17 Cell Effector Cytokines in Inflammation. Immunity 2008; 28:454-67; PMID:18400188; http://dx.doi.org/10.1016/j.immuni.2008.03.004
- Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, Zhang J, Lesch J, Lee WP, Ross J, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 2014; 514:237-41; PMID:25119041
- Garidou L, Pomié C, Klopp P, Waget A, Charpentier J, Aloulou M, Giry A, Serino M, Stenman L, Lahtinen S, et al. The Gut Microbiota Regulates Intestinal CD4 T Cells Expressing RORγt and Controls Metabolic Disease. Cell Metab 2015; 22:100-12; PMID:26154056; http://dx.doi.org/10.1016/j.cmet.2015.06.001
- Winer S, Paltser G, Chan Y, Tsui H, Engleman E, Winer D, Dosch HM. Obesity predisposes to Th17 bias. Eur J Immunol 2009; 39:2629-35; PMID:19662632; http://dx.doi.org/10.1002/eji.200838893
- Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NMJ, Magness S, Jobin C, Lund PK. High-fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One 2010; 5: e12191; PMID: 20808947
- Johnson AMF, Costanzo A, Gareau MG, Armando AM, Quehenberger O, Jameson JM, Olefsky JM. High Fat Diet Causes Depletion of Intestinal Eosinophils Associated with Intestinal Permeability. PLoS One 2015; 10:e0122195; PMID:25837594; http://dx.doi.org/10.1371/journal.pone.0122195
- Zindl CL, Lai J-F, Lee YK, Maynard CL, Harbour SN, Ouyang W, Chaplin DD, Weaver CT. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci U S A 2013; 110:12768-73; PMID:23781104; http://dx.doi.org/10.1073/pnas.1300318110
- Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol 2012; 5:354-66; PMID:22491176; http://dx.doi.org/10.1038/mi.2012.24
- Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 2012; 18:1407-12; PMID:22863787; http://dx.doi.org/10.1038/nm.2885
- Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, Bruce AT. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol 2011; 187:490-500; PMID:21606249; http://dx.doi.org/10.4049/jimmunol.1100123
- McPhee JB, Schertzer JD. Immunometabolism of obesity and diabetes: microbiota link compartmentalized immunity in the gut to metabolic tissue inflammation. Clin Sci 2015; 129:1083-96; PMID:26464517; http://dx.doi.org/10.1042/CS20150431
- Kim JJ, Shajib MS, Manocha MM, Khan WI. Investigating Intestinal Inflammation in DSS-induced Model of IBD. J Vis Exp 2012; 60; PMID: 22331082