
ABSTRACT
Enterococci are members of the gastrointestinal tract of humans and most animals that, over the past 3 decades, have emerged as leading causes of multidrug resistant hospital acquired infection (HAI). In addition to their general hardiness, many traits have entered enterococcal lineages through horizontal gene transfer, which has led to the evolution of pathogenic hospital-associated lineages uniquely adapted for survival and proliferation in the antibiotic perturbed ecology of the gastrointestinal tract. We recently observed that the accretion of mobile genetic elements in the prototype vancomycin resistant E. faecalis, clinical isolate V583, renders it unable to co-exist with native enterococci in healthy human fecal flora. In this addendum, we discuss how these findings inform our understanding of how multidrug resistant enterococci evolve, and the implications for the development of treatments that limit colonization and spread of highly antibiotic refractory microbes of this type.
Introduction
Enterococci are non-spore forming, low G+C Gram-positive bacteria that we propose evolved early as constituents of the animal gastrointestinal tract consortium. As a result, they are found in an unusually wide array of hosts, ranging from insects to humans.Citation1 Although typically constituting less than 1% of the overall gut microbial population of humans, enterococci are highly ubiquitous. They occur throughout the gastrointestinal tract, being represented in high proportion in the ileum and jejunum,Citation2 and are found in the feces at densities of approximately 10Citation6 CFU/gram.Citation3 Although they can be found and survive well in the environment, enterococci are highly adapted to living in the complex nutritional environment of the gut and possess a large number of auxotrophies for amino acids and vitamins. Streamlining of the genome was likely selected to maximize growth efficiency in the gut, despite the dependence on external sources for essential nutrients.Citation1 Instead, they have invested a large proportion of their genetic resources to encode mechanisms for uptake and metabolism of carbohydrates.Citation4,5
Despite a long history of evolution as a streamlined gastrointestinal commensal, enterococci also can cause life threatening hospital acquired infections of surgical sites, the urinary tract, and bloodstream.Citation6 The appearance of vancomycin-resistant enterococci (VRE) in the late 1980sCitation7,8 removed an important last line of therapy and led to a dramatic increase in VRE prevalence over the following decades.Citation9 Despite the introduction of linezolid, daptomycin, tigecycline, and ceftaroline, expanding the repertoire of last line antimicrobial treatments for drug resistant enterococcal infections, along with the implementation of antibiotic stewardship and infection control programs, multidrug resistant enterococcal infection accounts for almost 10% of HAI in the US.Citation6
Broad spectrum antibiotics cause dramatic changes to the gut microbiota by reducing diversity and changing the functional capacity of the community,Citation10,11 which can have long-lasting impacts on the bacterial community structure.Citation12 In addition to being linked to both chronic and acute diseases, the antibiotic-perturbed ecology provides unique opportunities for the evolution of hospital pathogens.Citation13
Enterococcal infection is a multistep process beginning with the acquisition of VRE following hospital admission and antibiotic use.Citation14 The acquisition of a few cells via the oral route from residual contamination of hospital surfaces or healthcare workers, can lead to tremendous amplification in the antibiotic treated colon, reaching numbers between 10Citation7 CFU/gramCitation15 to a near monoculture (10Citation14 CFU/g of feces).Citation16,17 This amplification proportionally increases the likelihood of these organisms reaching the bloodstream, urinary tract and surgical sites.Citation14 Gut colonization with VRE also persists following cessation of antibiotic treatment,Citation15 which can perpetuate the spread of VRE in hospital wards.
Multidrug resistant hospital isolates of enterococci are characterized by the accumulation of mobile elements
The mechanisms utilized by VRE to exploit the antibiotic-destabilized GI tract habitat are not well understood. Compared to commensal strains, the genomes of VRE show a paucity of CRISPR defenses and the resulting accumulation of mobile genetic elements (MGE), including plasmids, pathogenicity islands, resistance transposons, other fitness islands, and phages.Citation4,18,19 MGE found in multidrug resistant pathogenic enterococci encode proteins with diverse functions, including antibiotic resistances, adhesins, bacteriocins, and alternative carbon metabolism pathways that appear to have created new lineages well positioned to colonize and persist in the dysbiotic GI tract resulting from antibiotic treatment (). For the prototype vancomycin resistant E. faecalis isolate strain V583, which was the first VRE reported in the US,Citation8 these MGE constitute over 25% of the genome including: 3 freely replicating plasmids, 3 elements annotated as plasmid-like remnants integrated in to the chromosome, a 136 Kb pathogenicity island (PAI), 7 phage, and over 30 insertion (IS) elements.Citation4
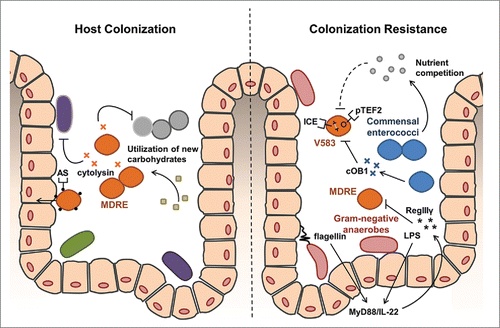
Figure 1. The convergence of acquired traits that can facilitate host colonization by VRE and mechanisms limiting it. Left panel. Several traits have evolved in drug-resistant enterococcal lineages that may work cooperatively to enhance the gastrointestinal colonization and persistence. Cytolysin, a bipartite bacteriocin, has broad spectrum activity against Gram-positive bacteria that likely eliminates commensals in the local environment to reduce competition.Citation35 The surface protein aggregation substance (AS) can promote adherence and invasion of intestinal epithelial cells which promote intestinal persistence.Citation1 Additional carbohydrate uptake and metabolism functions associated with the enterococcal pathogenicity island may allow for more efficient exploitation of available carbohydrate resulting from depletion of normal gut flora.Citation36 Right Panel. Gram-negative anaerobic commensals provide generalized resistance against VRE through the surface factors lipopolysaccharide (LPS) and flagellin activate MyD88 and IL-22 signaling pathways to stimulate host production of the defensin RegIIIγ, which has broad activity against Gram-positives.Citation37 Commensal enterococci may also exert more specific resistance against VRE colonization by competing for shared nutrients.Citation21 In some cases, the accumulation of mobile genetic elements, like the conjugative plasmid pTEF2 and an integrated chromosomal element (ICE), can make VRE incompatible with commensal strains that produce otherwise harmless peptide pheromones.Citation21
The downside to the rapid accumulation of MGE
The acquisition of multiple drug resistances and colonization enhancing traits undeniably contributes to fitness for invading and infecting the hospitalized patient. However, VRE colonization of healthy individuals in the community in the US is rare,Citation20 suggesting that the genome expansion selected for in hospitals may exert a fitness cost in the native GI ecology.
In the research article featured in this commentary,Citation21 we designed an in vitro assay to mimic some of the environmental features encountered in the intestinal tract, to determine if all of the MGE acquisition imposed a burden on VRE in the context of healthy human flora. We also wanted to know whether all of the mobile elements conferring fitness traits, including vitamin biosynthetic pathways, carbohydrate utilization pathways, and other traits enabled VRE E. faecalis V583 to occupy a position in the cross-feeding food web in the GI tract distinct from that of commensal enterococci. To test this prospect, we developed a medium consisting of crude hog gastric mucin dissolved in minimal salts. We found that despite their many auxotrophies, V583 and a strain chosen to represent commensals, OG1RF or its sibling OG1X, could grow reasonably well in this medium, although not to the same extent as in more traditional rich media. We then added competing flora from pooled human feces. With no further additions, both VRE and commensal strains grew in this medium, but to a lesser extent, presumably because of competition for nutrients from the competing flora.
To determine whether VRE strain V583 and commensal strain OG1X occupied the same position in the food web, we then warped the composition of the fecal communities spiked with either V583 or OG1X, by adding in separate experiments various carbohydrates – some like glucose utilizable by many in the community, and others like panose, utilizable by only a few. We reasoned that if VRE and OG1 occupied the same ecological space in the community, and were dependent on the same members of the community for cross-feeding relationships, their numbers would ebb and flow similarly in parallel experiments. On the other hand, if they had distinct interdependencies because of the new pathways gained by V583, this VRE strain might benefit in one case (i.e., in the presence of one type of carbohydrate), where the commensal OG1X strain would not, and vice versa.
What we found was the latter – the commensal strain generally benefitted from the addition of any carbohydrate to the mixed community, whether they could be utilized directly, or whether enhanced growth stemmed from indirect benefits derived from the community. In contrast, not only was the VRE limited in its ability to proliferate in some cases, it was actively killed when some carbohydrates were added to the mixed flora. Specifically, the addition of glucose, N-acetylgalactosamine, galactose, or panose resulted in the selective killing of V583, but enhanced proliferation of commensal E. faecalis in the presence of the fecal consortium. This suggested that these carbohydrates either favored the outgrowth among normal fecal flora of a strain or species that was specifically antagonistic to the VRE strain V583, or that these carbohydrates altered the physiology of V583 in a way that somehow rendered it vulnerable to something in the consortium. In pure culture, when the hog gastric medium was supplemented with any of these carbohydrates, growth of V583 was stimulated, so there was no intrinsic toxicity of their addition – killing only occurred in the presence of the fecal consortium.
To try to deplete the killing element from the fecal consortium, we precultured it in the presence of graded levels of tetracycline or erythromycin. Antibiotic depleted fecal flora was then added to the VRE in coculture, and the ability of the fecal consortium to kill VRE was re-examined. Low level erythromycin treatment was effective in depleting the killing component of the fecal consortium, whereas low level tetracycline treatment enhanced its V583-killing ability. We found that killing correlated with the enrichment or absence of native enterococci in the fecal consortium, specifically the presence of a native commensal E. faecalis strain we termed, Pan7. This intra-species killing was suggestive of a possible bacteriocin mediated mechanism, as enterococci have long been known to be prolific producers of bacteriocins.Citation22 We showed that we could recapitulate all of the behaviors observed in mixtures of VRE V583 with fecal flora, using only mixtures of V583 and Pan7. However, in the process of recapitulating killing of V583 by Pan7, we found that V583 was also killed by control coculture partners OG1X or another lab strain, FA2-2. In other words, V583 seemed to be killed by every plasmid-free strain tested, even those known not to contain anything annotated as a bacteriocin in its genome.
Not knowing how V583 was being killed, since its genome sequence and mobile element content were known,Citation4 we began to get at the mechanism by asking instead ‘what makes V583 vulnerable to this killing?’. As noted above, in addition to carrying about 2.7 Mb of core E. faecalis genes, V583 also possesses about 600 Kb of mobile elements.Citation4 Since commensal strains with only the core genes of the species were not vulnerable to this killing, we immediately suspected it was something about the mobile element content. We started by winnowing down the possibilities in as large of chunks as possible. First we asked whether it might be the presence of the 136 Kb pathogenicity island. In another recent paper,Citation23 we had recently mobilized the pathogenicity island into the commensal OG1RF background. It did not move precisely, but rather brought with it, various large stretches of the V583 chromosome flanking either end,Citation23 in one case, transferring a total of 857 Kb of the V583 genome into OG1RF. We therefore asked whether these hybrid strains possessing the pathogenicity island and varying amounts of flanking V583 chromosomal sequences in the OG1RF background conferred susceptibility to killing by commensal E. faecalis Pan7. They did not.
CollaboratorsCitation24 had recently cured V583 of the 3 resident plasmids, pTEF1, pTEF2, and pTEF3, collectively contributing a total of approximately 130 Kb to the genome size of V583. We then asked whether the plasmid-free cured derivative of V583 remained susceptible to killing. It was now not killed by Pan7. We systematically reintroduced the plasmids back into V583 one at a time, and retested. One plasmid, pTEF2, was found to reconstitute the killing-susceptible phenotype in the V583 chromosomal background.
Having shown that pTEF2 was necessary for the killing-susceptible phenotype, we then asked whether it was sufficient by transferring it into OG1RF and testing OG1RF(pTEF2) for killing susceptibility. These transconjugants were found not to be susceptible to killing, demonstrating that pTEF2 was not sufficient, and there was something else in the V583 chromosomal background that was necessary. As a side experiment, we tested whether OG1RF(pTEF2) could still kill V583 in coculture. Unexpectedly, it could not. The presence of pTEF2, which appeared from the sequence to be a relatively typical E. faecalis pheromone responsive conjugative plasmid,Citation4 somehow suppressed the ability of OG1RF to kill V583. This observation reminded us of the effect that a conjugative plasmid has on its host: In the absence of the plasmid, the host secretes a variety of pheromonesCitation25 which are specific proteolytically processed octapeptide fragments of signal peptides cleaved from the amino termini of cell wall anchored lipoproteins encoded by the core genome of E. faecalis. However, in the presence of the plasmid, a plasmid-encoded inhibitor specifically inhibits the activity of the cognate pheromone to which the plasmid conjugative machinery responds. Presumably, this prevents the futile exchange of plasmids among plasmid-containing siblings.Citation26
If a pheromone were somehow responsible for killing V583, presumably by inducing expression of some factor that, in the V583 chromosomal background, initiates a cascade of events that leads to killing, then we reasoned that mutants of the commensal strain defective in processing the lipoprotein signal peptides, or mutants in the putative pheromone receptor encoded within pTEF2 should not lead to killing. Both possibilities turned out to be correct. A mutation in the eep membrane metalloprotease that, in part, processes the lipoprotein signal peptides in the commensal pheromone secreting strain were partially defective in killing. In addition, a mutation in the gene annotated as traC2, encoding the putative pheromone receptor on pTEF2, reduced the killing-susceptible phenotype of V583.
Having obtained 2 clear lines of evidence that killing of V583 resulted, at least in part, from the response of pTEF2 to a pheromone produced by a commensal strain, we began to identify the octapeptide putatively responsible. By comparing the sequence of several known pheromones to the OG1RF genome sequence and identifying all possible lipoprotein-encoding genes, we inferred rules that allowed us to deduce 81 possible octapeptide pheromone candidates, which were synthesized chemically. Of the 81 octapeptides, picomolar concentrations of only 1 was found to kill VRE strain V583 – a known pheromone termed cOB1.Citation27 Moreover, this killing was specific to V583 variants possessing pTEF2, and the peptide had no effect on control plasmid-free strains. Having this strongly inhibitory, highly specific peptide in hand, we then selected mutants of V583 resistant to killing. Resistant mutants arose at an unexpectedly high rate of 1 × 10−6 cells in the V583 population, which was more in line with the rates of movement of IS elements than with known rates of spontaneous mutation. We resequenced one resistant mutant and found that indeed, it had spontaneously lost a 33 Kb chromosomal element originally annotated as a defective plasmid. In fact, rather than a defective plasmid, this appears to be a fully functional IS element capable of frequent spontaneous excision. We selected 10 additional independently derived V583 mutants resistant to the cOB1 pheromone, and all 10 possessed the same 33 Kb deletion. We also scanned a large collection of E. faecalis strains for which we had collaboratively determined the genome sequence (https://olive.broadinstitute.org/projects/enterogenome), and identified several possessing homologs of this element in their chromosome. We introduced pTEF2 into these strains and showed that they were now rendered susceptible to cOB1 killing.
Exactly how the otherwise harmless pheromone cOB1 kills E. faecalis strains possessing the combination of pTEF2 and the 33 Kb chromosomally-integrated mobile element is the subject of ongoing study. It is known in B. subtilis, that imprecise excision of an integrated conjugative element, ICEBs1, can lead to cell death, presumably by the induction of irreparable breaks in the DNA.Citation28 We speculateCitation21 that somehow, induction of conjugative and perhaps co-transcribed functions on pTEF2, induced by cOB1, results in a lethal crosstalk with the 33 Kb IS element, leading to its imprecise excision and a double stranded break in the chromosome that leads to cell death.
Implications and strategies for managing VRE colonization
Although details of the killing mechanism remain to be determined, there are clear implications of these findings related to patient management and the evolution of VRE in the hospital environment. That lethal crosstalk occurs between mobile elements that have accumulated in V583 shows that following the acquisition of both pTEF2 and the necessary 33 Kb IS element, this strain does not occur in the presence of commensal E. faecalis in the gut. This means that antibiotic elimination of commensal E. faecalis is a precondition for colonization and proliferation of this VRE strain. It has been long known that antibiotic use is an important predisposing factor for VRE infection, and this is a clear example of why that is the case. The second implication is that IS elements have accumulated in at least some VRE in a seemingly haphazard way in the absence of the competition by commensals that would impose a fitness cost. These observations highlight the importance of maintaining to the extent possible, native competing commensal flora of the patient GI tract as a means of excluding colonization by highly hospital adapted pathogenic and often antibiotic resistant strains. Other recent studies also have provided insight into the contribution of native Gram-negative anaerobic commensals in excluding VRE from the mammalian GI tract. Flagellated gram-negative anaerobes activate toll-like receptors (TLRs) through lipopolysaccharide (LPS) and flagellin components, stimulating host innate immunity, including production of host factors that antagonize VRE (). Specifically, TLR4 binding by LPS, and flagellin binding to TLR5, induce the production of the antimicrobial lectin RegIIIγ,Citation29-31 an important host defensin that contributes to the spatial barrier between gut microbes and the intestinal surface.Citation32
The complement of MGE in most VRE is highly variable, and the combination of pTEF2 and the integrated 33 Kb IS element is rare. We are therefore undertaking similar studies to determine the extent to which other VRE lineages are compromised in the ability to compete in the presence of native gut flora, and when found, to determine the underlying principles. These types of studies are important for determining the extent to which drug resistant strains retain their competitiveness.Citation33 With the introduction of ecological management as an important tool for treating and possibly preventing Clostridium difficile colitis,Citation34 it may well be possible to limit the first steps in infection – colonization and numeric expansion in the gut – by maintaining or supplementing the complement of antagonistic microbes in hospitalized patients, thereby preventing many if not most VRE hospital acquired infections.
Abbreviations
| GI | = | gastrointestinal |
| HAI | = | hospital acquired infection |
| IS | = | [element] insertion |
| MGE | = | mobile genetic element |
| PAI | = | pathogenicity island |
| VRE | = | Vancomycin resistant enterococci |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Portions of this work were supported by NIH grants AI083214, AI108710, and AI072360. AOG is the grateful recipient of support from the MBED training program (EY007145). The research described herein was supported by PHS grants AI072360 and AI108710, and the Harvard-wide Program on Antibiotic Resistance AI083214.
References
- Van Tyne D, Gilmore MS. Friend turned foe: evolution of enterococcal virulence and antibiotic resistance. Annu Rev Microbiol 2014; 68:337-56; PMID:25002090; http://dx.doi.org/10.1146/annurev-micro-091213-113003
- Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y. Molecular analysis of jejunal, ileal, caecal and recto-sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J Med Microbiol 2005; 54:1093-101; PMID:16192442; http://dx.doi.org/10.1099/jmm.0.45935-0
- Jett BD, Huycke MM, Gilmore MS. Virulence of enterococci. Clin Microbiol Rev 1994; 7:462-78; PMID:7834601
- Paulsen IT, Banerjei L, Myers GS, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, et al. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 2003; 299:2071-4; PMID:12663927; http://dx.doi.org/10.1126/science.1080613
- Ramsey M, Hartke A, Huycke M. The Physiology and Metabolism of Enterococci. In: Gilmore MS, Clewell DB, Ike Y, Shankar N, eds. Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. Boston, 2014; Boston: Massachusetts Eye and Ear Infirmary; 2014-. Available from: http://www.ncbi.nlm.nih.gov/books/NBK190432/
- Magill SS, Edwards JR, Bamberg W, Beldavs ZG, Dumyati G, Kainer MA, Lynfield R, Maloney M, McAllister-Hollod L, Nadle J, et al. Multistate point-prevalence survey of health care-associated infections. N Engl J Med 2014; 370:1198-208; PMID:24670166; http://dx.doi.org/10.1056/NEJMoa1306801
- Leclercq R, Derlot E, Duval J, Courvalin P. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med 1988; 319:157-61; PMID:2968517; http://dx.doi.org/10.1056/NEJM198807213190307
- Sahm DF, Kissinger J, Gilmore MS, Murray PR, Mulder R, Solliday J, Clarke B. In vitro susceptibility studies of vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother 1989; 33:1588-91; PMID:2554802; http://dx.doi.org/10.1128/AAC.33.9.1588
- Gilmore MS, Lebreton F, van Schaik W. Genomic transition of enterococci from gut commensals to leading causes of multidrug-resistant hospital infection in the antibiotic era. Curr Opin Microbiol 2013; 16:10-6; PMID:23395351; http://dx.doi.org/10.1016/j.mib.2013.01.006
- Cotter PD, Stanton C, Ross RP, Hill C. The impact of antibiotics on the gut microbiota as revealed by high throughput DNA sequencing. Discov Med 2012; 13:193-9; PMID:22463795
- Wu HJ, Wu E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012; 3:4-14; PMID:22356853; http://dx.doi.org/10.4161/gmic.19320
- Jernberg C, Lofmark S, Edlund C, Jansson JK. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J 2007; 1:56-66; PMID:18043614; http://dx.doi.org/10.1038/ismej.2007.3
- Frick JS, Autenrieth IB. The gut microflora and its variety of roles in health and disease. Curr Top Microbiol Immunol 2013; 358:273-89; PMID:22476557
- Mundy LM, Sahm DF, Gilmore M. Relationships between enterococcal virulence and antimicrobial resistance. Clin Microbiol Rev 2000; 13:513-22; PMID:11023953; http://dx.doi.org/10.1128/CMR.13.4.513-522.2000
- Donskey CJ, Chowdhry TK, Hecker MT, Hoyen CK, Hanrahan JA, Hujer AM, Hutton-Thomas RA, Whalen CC, Bonomo RA, Rice LB. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 2000; 343:1925-32; PMID:11136263; http://dx.doi.org/10.1056/NEJM200012283432604
- Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MR, Kamboj M, et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest 2010; 120:4332-41; PMID:21099116; http://dx.doi.org/10.1172/JCI43918
- Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, Gobourne A, Lee YJ, Dubin KA, Socci ND, Viale A, et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis 2012; 55:905-14; PMID:22718773; http://dx.doi.org/10.1093/cid/cis580
- Palmer KL, Gilmore MS. Multidrug-resistant enterococci lack CRISPR-cas. mBio 2010; 1:pii: e00227-10; PMID:21060735; http://dx.doi.org/10.1128/mBio.00227-10
- Lebreton F, van Schaik W, McGuire AM, Godfrey P, Griggs A, Mazumdar V, Corander J, Cheng L, Saif S, Young S, et al. Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. mBio 2013; 4:pii: e00534-13; http://dx.doi.org/10.1128/mBio.00534-13
- Huycke MM, Sahm DF, Gilmore MS. Multiple-drug resistant enterococci: the nature of the problem and an agenda for the future. Emerg Infect Dis 1998; 4:239-49; PMID:9621194; http://dx.doi.org/10.3201/eid0402.980211
- Gilmore MS, Rauch M, Ramsey MM, Himes PR, Varahan S, Manson JM, Lebreton F, Hancock LE. Pheromone killing of multidrug-resistant Enterococcus faecalis V583 by native commensal strains. Proc Natl Acad Sci U. S. A. 2015; 112:7273-8; PMID:26039987; http://dx.doi.org/10.1073/pnas.1500553112
- Brock TD, Peacher B, Pierson D. Survey of the Bacteriocines of Enterococci. J Bacteriol 1963; 86:702-7; PMID:14066464
- Manson JM, Hancock LE, Gilmore MS. Mechanism of chromosomal transfer of Enterococcus faecalis pathogenicity island, capsule, antimicrobial resistance, and other traits. Proc Natl Acad Sci U. S. A. 2010; 107:12269-74; PMID:20566881; http://dx.doi.org/10.1073/pnas.1000139107
- Hanin A, Sava I, Bao Y, Huebner J, Hartke A, Auffray Y, Sauvageot N. Screening of in vivo activated genes in Enterococcus faecalis during insect and mouse infections and growth in urine. PloS one 2010; 5:e11879; PMID:20686694; http://dx.doi.org/10.1371/journal.pone.0011879
- Dunny GM, Johnson CM. Regulatory circuits controlling enterococcal conjugation: lessons for functional genomics. Curr Opin Microbiol 2011; 14:174-80; PMID:21353627; http://dx.doi.org/10.1016/j.mib.2011.01.008
- Clewell DB. Bacterial sex pheromone-induced plasmid transfer. Cell 1993; 73:9-12; PMID:8462105; http://dx.doi.org/10.1016/0092-8674(93)90153-H
- Nakayama J, Abe Y, Ono Y, Isogai A, Suzuki A. Isolation and structure of the Enterococcus faecalis sex pheromone, cOB1, that induces conjugal transfer of the hemolysin/bacteriocin plasmids, pOB1 and pYI1. Biosci Biotechnol Biochem 1995; 59:703-5; PMID:7772836; http://dx.doi.org/10.1271/bbb.59.703
- Menard KL, Grossman AD. Selective pressures to maintain attachment site specificity of integrative and conjugative elements. PLoS genetics 2013; 9:e1003623; PMID:23874222; http://dx.doi.org/10.1371/journal.pgen.1003623
- Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated signals induce the bactericidal lectin RegIII gamma and protect mice against intestinal Listeria monocytogenes infection. J Exp Med 2007; 204:1891-900; PMID:17635956; http://dx.doi.org/10.1084/jem.20070563
- Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, Schnabl B, DeMatteo RP, Pamer EG. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature 2008; 455:804-7; PMID:18724361; http://dx.doi.org/10.1038/nature07250
- Pham TA, Clare S, Goulding D, Arasteh JM, Stares MD, Browne HP, Keane JA, Page AJ, Kumasaka N, Kane L, et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe 2014; 16:504-16; PMID:25263220; http://dx.doi.org/10.1016/j.chom.2014.08.017
- Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV. The antibacterial lectin RegIII gamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011; 334:255-8; PMID:21998396; http://dx.doi.org/10.1126/science.1209791
- Andersson DI, Hughes D. Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol Rev 2011; 35:901-11; PMID:21707669; http://dx.doi.org/10.1111/j.1574-6976.2011.00289.x
- van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 2013; 368:407-15; PMID:23323867; http://dx.doi.org/10.1056/NEJMoa1205037
- Van Tyne D, Martin MJ, Gilmore MS. Structure, function, and biology of the Enterococcus faecalis cytolysin. Toxins 2013; 5:895-911; PMID:23628786; http://dx.doi.org/10.3390/toxins5050895
- Pillar CM, Gilmore MS. Enterococcal virulence–pathogenicity island of E. Faecalis. Front Biosci 2004; 9:2335-46; PMID:15353291; http://dx.doi.org/10.2741/1400
- Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol 2013; 13:790-801; PMID:24096337; http://dx.doi.org/10.1038/nri3535