
ABSTRACT
Owing to their health benefits, dietary fermentable fibers, such as refined inulin, are increasingly fortified in processed foods to enhance their nutritional value. However, we previously demonstrated that when inulin was fed to Toll-like receptor 5 deficient (T5KO) mice susceptible to dysbiosis, a subset of them developed cholestasis and subsequently liver cancer in a gut microbiota-dependent manner. Therefore, we hypothesized that clearance of bacterial taxa, and thereby gut metabolites, involved in the onset and progression to liver cancer could abate the disease in these mice. Such a reshaping of microbiota by vancomycin treatment was sufficient to halt the development of liver cancer in inulin-fed T5KO mice; however, this intervention did not remedy disease penetrance for cholestatic liver injury and its sequelae, including hyperbilirubinemia, hypolipidemia, cholemia and liver fibrosis. Selective depletion of gut bacterial communities was observed in vancomycin-treated mice, including Gram-positive Lachnospiraceae and Ruminococcaceae belonging to the phylum Firmicutes, Bifidobacteria of the phylum Actinobacteria, which ferment fibers, and Clostridium cluster XIVa, which produce secondary bile acids. Lack of liver cancer in vancomycin-treated mice strongly correlated with the substantial loss of secondary bile acids in circulation. Although cholemia was unabated by vancomycin, the composition of serum bile acids shifted toward an abundance of hydrophilic primary bile acids, denoted by the increase in conjugated-to-unconjugated bile acid ratio. Taken together, the present study suggests that microbiotal regulation of bile acid metabolism is one of the critical mediators of fermentable fiber-induced liver cancer in dysbiotic mice.
Introduction
Liver cancer is the sixth most commonly diagnosed type of cancer and the fourth leading cause of cancer-related deaths worldwide.Citation1 While the rates for other cancers are declining, liver cancer is increasingly prevalent, accounting for 782,000 deaths with about 841,000 new cases reported every yearCitation1 and representing a major financial burden.Citation2 Though viral hepatitis is recognized as one known cause of hepatocellular carcinoma (HCC; a primary form of liver cancer), the prevalence of this HCC subtype does not account for the recent surge of non-viral HCC incidences. Many lines of evidence suggest that the etiology of HCC is multifactorial,Citation3 encompassing non-viral environmental factors such as poor dietary choices toward excessive intake of alcohol or foods rich in simple sugars and fats combined with a sedentary lifestyle.Citation4
Table 1. List of primers.
Recent advances in cancer research indicate that the gut microbiota represents a new environmental risk factor for HCC. Of note, the oncogenic properties of bacterial cell wall components, such as lipopolysaccharide and lipoteichoic acid, have been demonstrated in seminal studies.Citation5,Citation6 Liver cancer models driven either by hepatocarcinogen diethylnitrosamine (DEN) or overexpression of c-Myc have also identified secondary bile acids, such as deoxycholate and ω-muricholate, as oncometabolites that could promote HCC by inducing cellular senescenceCitation7 and/or impeding hepatic anti-tumor immune surveillance.Citation6,Citation8 Depleting Gram-positive bacteria using vancomycin was sufficient to confer protection against such a secondary bile acid-driven HCC in the DEN and c-Myc models,Citation6–Citation8 in agreement with the notion that major secondary bile acid producers are Gram-positive bacteria.
In our recent study,Citation9 we documented that a subset (~40%) of Toll-like receptor 5 deficient (T5KO) mice consistently developed hyperbilirubinemia and cholestatic liver injury within four weeks of feeding an open-source diet formulated to contain 7.5% refined inulin (a prebiotic, fermentable fiber). After six months, the cholestasis in these mice evolved into robust icteric HCC. These findings cautioned that highly-refined inulin may not operate in the same manner as their naturally-occurring and health-promoting counterparts in grains, fruits and vegetables. Rather, the onset of cholestasis in T5KO mice fed an inulin-containing diet (ICD) are more aligned with the pathology seen in humans who develop jaundice and liver disease following long-term intake of refined plant-derived or herbal supplements.Citation10-Citation15 Intriguingly, we found that the susceptibility of T5KO mice to ICD-induced HCC is dependent on their gut microbiota,Citation9 which we had previously characterized to be in a state of dysbiosis.Citation16,Citation17 Interventions that ablate the gut microbiota via broad-spectrum antibiotics or inhibit bacterial fermentation via β-acids (a fermentation inhibitor used to prevent spoilage in beer-making) were sufficient to prevent ICD-induced HCC.Citation9 However, the identities of the bacterial taxa and metabolites mediating ICD-induced HCC remain poorly understood.
Herein, we asked whether depleting specific group(s) of Gram-positive bacteria could mitigate the adverse effects of ICD in T5KO mice. We selected vancomycin for this study based on its efficacy to preferentially target Gram-positive bacteria. Though vancomycin therapy did not prevent hyperbilirubinemia in ICD-fed T5KO mice, it mitigated transaminitis and prevented liver disease from progressing to HCC. Key Gram-positive bacteria belonging to the phyla Firmicutes (i.e. Lachnospiraceae and Ruminococcaceae) and Actinobacteria (i.e. Bifidobacteria) involved in fermenting fibers and Clostridium cluster XIVa involved in producing secondary bile acids were notably ablated in vancomycin-given T5KO mice. This concurred with the loss of bacteria expressing butyryl-CoA: acetate transferase (BCoAT) and bile salt hydrolases in the feces. Taken together, our findings implicate that ICD-induced HCC is primarily driven by Gram-positive bacteria and substantiates the link between gut-liver axis in the etiology of this type of HCC.
Results
Vancomycin protects T5KO mice from inulin-induced HCC
Dietary supplements comprised of highly-refined botanical extracts are formulated to improve health; yet, a subset of individuals taking these supplements are reported to develop side effects,Citation10-Citation13 including jaundice and cholestasis that eventually leads to liver injury and failure.Citation14 T5KO mice exemplified these circumstances, whereby ~43% of mice (6 out of 14) fed ICD developed hyperbilirubinemia and liver injury as denoted by increased serum bilirubin ()) and alanine aminotransferase (ALT) activity ()), respectively. Based on their serum bilirubin levels, we segregated the mice into high (T5KO-HB) or normal (T5KO-NB) bilirubin groups. Consistent with our prior report,Citation9 serum α-fetoprotein (AFP; a clinical biomarker for HCC) was notably upregulated in T5KO-HB mice ()). Liver necropsy, following euthanasia at six months of ICD-feeding, confirmed the presence of multinodular tumors in every part of the liver ()). Tumor region was demarcated by hepatocytes exhibiting features of anisocytosis, mitotic figures and a trabecular pattern ()). The presence of bile duct hyperplasia was also noted in the tumor region ()).
Figure 1. Vancomycin blocks inulin-induced hepatocellular carcinoma. Inulin containing diet (ICD)-fed T5KO mice were maintained either on regular drinking water or water containing vancomycin (0.5 g/L). (a) total serum bilirubin. Based on serum bilirubin levels, mice were stratified into normal bilirubin (NB) or high bilirubin (HB, denoted by green dots present in red circle) groups. (b–c) serum levels of B) alanine transaminase (ALT), an enzyme predominantly present in liver and (c) alpha-fetoprotein, a tumor marker. (d) Representative macroscopic photographs of liver. Red arrows point to HCC nodules. Histological analysis (Hematoxylin and Eosin staining) of the liver: E i–ii) Dotted line demarcates the tumor from the non-tumor containing region. (e-iii) black arrow indicates the presence of bile ducts in the tumor region, and red arrows denote mitotic figures. (f) Collagen staining by picrosirius red. Data are representative of six mice per group. Error bars indicate mean ± SEM. ANOVA, *p < .05.
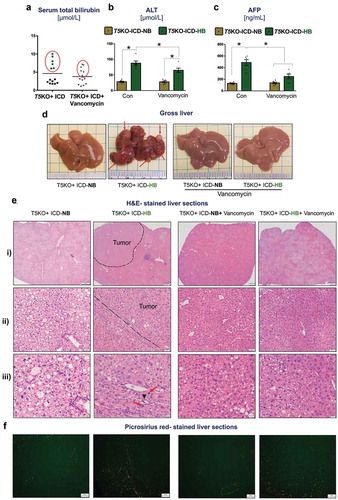
Vancomycin belongs to a class of first-generation glycopeptide antibiotics, which act by inhibiting the biosynthesis of N-acetylmuramic acid and N-acetylglucosamine polymers that form the cell wall of Gram-positive bacteria. To address the extent to which Gram-positive bacteria contributes to ICD-induced HCC, we treated T5KO mice with vancomycin starting at one week prior to feeding them ICD and continued the treatment up to six months. Intervention with vancomycin did not decrease the susceptibility of ICD-fed T5KO mice toward developing hyperbilirubinemia, whose disease penetrance remained at ~46% (6 out of 13 mice) ()). Nevertheless, such intervention reduced serum ALT and AFP by ~1.5-fold and ~2-fold, respectively, in T5KO-HB mice (). More importantly, the liver morphology of vancomycin-treated T5KO-HB mice was remarkably free of any discernible tumor nodules ()). Histological analysis on the livers of vancomycin-treated T5KO-HB mice affirmed the absence of frank HCC, though they appeared steatotic () and exhibited fibrosis ()). Intriguingly, vancomycin also mitigated the macrovesicular steatosis (large fat droplets/vacuoles) seen in control T5KO-NB mice (-iii)), which are known to develop fatty liver in a microbiota-dependent manner.Citation17 Of note, vancomycin treatment alone was sufficient to block inulin-induced development of HCC in a subset of T5KO mice.
Vancomycin partially reverts the metabolic abnormalities associated with ICD-induced HCC
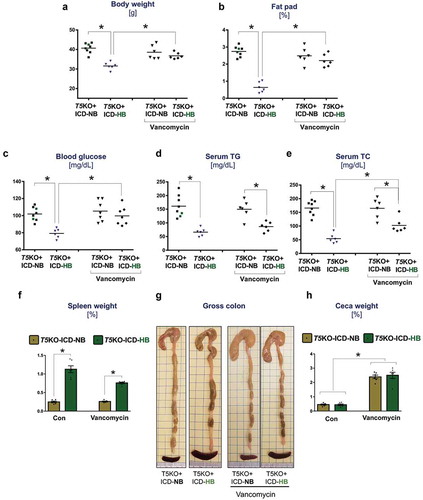
Aside from hyperbilirubinemia, other hallmark features that could distinguish cancer-prone T5KO mice were their low body weights, a deficit in abdominal adiposity, hypoglycemia, hypotriglyceridemia and hypocholesterolemia (–)) that are reminiscent of cancer-associated cachexia.Citation18 Unlike the vehicle-treated T5KO-HB mice, those which received vancomycin therapy displayed normalized body weights ()), fat pad weights ()), and blood glucose ()) that were comparable to their T5KO-NB counterparts. However, vancomycin-treated T5KO-HB mice remained hypotriglyceridemic and hypocholesterolemic, despite displaying mild improvements in these parameters when compared to vehicle-treated T5KO-HB mice (). Likewise, vancomycin could only reduce to some extent, but not normalize, the splenomegaly in T5KO-HB mice ()). Vancomycin treatment enlarged the ceca in both T5KO-HB and T5KO-NB mice (), which is expected for mice receiving antibiotics. Though T5KO mice are susceptible to develop spontaneous colitis,Citation19-Citation21 we did not observe signs of intestinal inflammation in any of the mice fed on ICD in this study (data not shown).
Figure 2. Vancomycin treatment improves body weight and metabolic markers of mice with hyperbilirubinemia. Dot plots represent (a) body weight, and (b) fat pad weight expressed as percent body weight. (c) Fasting (15 h) blood glucose levels. Lipids were analyzed in serum obtained from 5 h fasted mice. The serum concentration of (d) triglycerides (TGs), and (e) total cholesterol (TC). (f) Spleen weight normalized to body weight. (g) Images display gross morphology of ceca and colon. (h) Cecum weight normalized to body weight. Data are representative of six mice per group. Error bars indicate mean ± SEM. ANOVA, *p < .05.
Hepatoprotection conferred by vancomycin correlates with a substantial loss in SCFAs and major secondary bile acid-producing bacteria in the gut of ICD-fed T5KO mice
The above results suggest that the depletion of vancomycin-sensitive bacteria could be lowering ICD-induced HCC. In the vehicle-treated group, T5KO-HB mice displayed a ~ 2-fold increase in total fecal bacterial load when compared to T5KO-NB mice ()). However, such an increase was abrogated in T5KO-HB mice receiving vancomycin. Despite its specificity toward depleting Gram-positive bacteria, vancomycin therapy enriched Firmicutes (Gram-positive) and γ-Proteobacteria (Gram-negative), but depleted Bacteroidetes (Gram-negative) (–)). Comparatively, Clostridium cluster XIVa (Gram-positive) was enriched in vehicle-treated but absent in vancomycin-treated T5KO-HB mice ()). Gram-positive fermenting bacteria such as Bifidobacteria, Lachnospiraceae and Ruminococcaceae were likewise lacking in vancomycin-treated T5KO-HB mice (). Moreover, mice given vancomycin were also depleted of bacteria expressing butyryl-CoA: acetate transferase (BCoAT) ()), which is the major enzyme involved in the production of the short chain fatty acid (SCFAs), butyrate, from dietary fibers. In line, a profound reduction in cecal propionate and butyrate, along with moderate suppression of acetate production, was observed in vancomycin treated T5KO-HB mice ()).
Figure 3. Vancomycin suppresses secondary bile acid and SCFAs producers in inulin-fed mice. Bar graphs represent relative abundances of (a) total bacteria, (b) Bacteroidetes, (c) Firmicutes, (d) γ-Proteobacteria, (e) Clostridium cluster XIVa, (f) Bifidobacteria, (g) Lachnospiraceae and Ruminococcaceae. (h) Relative level of butyryl-coenzyme A (CoA) transferase (BCoAT). SCFAs were quantified in cecal content via 1 H NMR spectroscopy. (i) Bar diagram represents the level of SCFAs in cecal content. Relative gene expression of bile salt hydrolases (j) bsh1a, (k) bsh1b, and (l) bsh3 c. (m) Bile salt hydrolase (BSH) activity was measured in feces from indicated group of mice. Germ-free mice feces were used as negative control. BSH activity is expressed as mM of taurine released from the taurocholate per minute per g of feces. N.D. denotes not detected. Data are representative of six mice per group. Error bars indicate mean ± SEM. ANOVA, *p < .05.
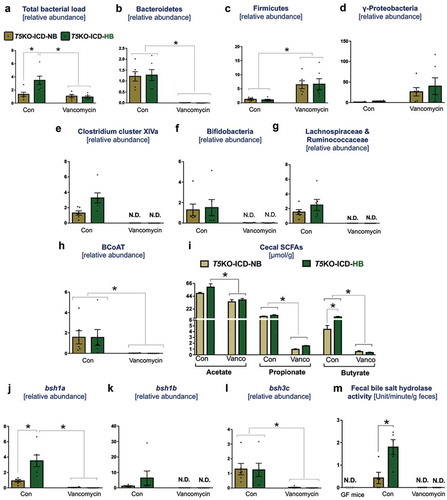
Next, to examine whether hepatoprotection was due to the loss of bile acid producers, we examined the abundance of bile salt hydrolases (BSH; bacterial enzymes involved in the deconjugation of primary bile acids) based on three major reference groups: bsh1a, bsh1b and bsh3c. The levels of bacteria expressing bsh1a, bsh1b and bsh3c, as well as fecal BSH activity, were undetectable in the feces of vancomycin-treated mice (–)), indicating that these bacteria are vancomycin-sensitive. On the flip side, T5KO-HB that developed HCC displayed a higher abundance of bsh1a-expressing bacteria and an increased fecal BSH activity compared to the non-HCC T5KO-NB mice.
Vancomycin depletes circulating secondary bile acids in ICD-fed T5KO mice, but does not correct cholemia
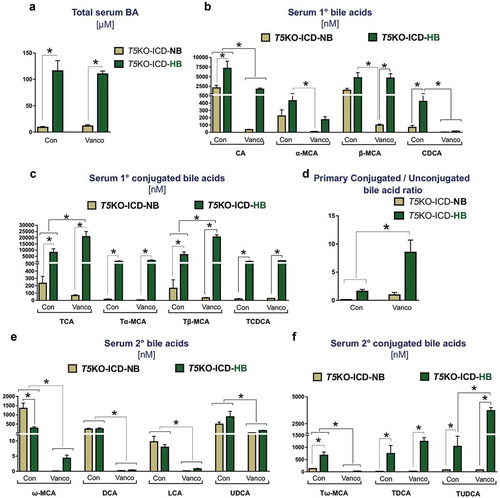
As we had previously reported,Citation9 the cholemia, which is the spillover of bile acids into systemic circulation, was invariably associated with hyperbilirubinemia in T5KO-HB mice with HCC but absent in T5KO-NB mice ()). Hence, we next asked whether the loss of secondary bile acid producers could mitigate cholemia of T5KO-HB mice. Cholemia of T5KO-HB mice was unabated by vancomycin treatment ()). Despite this observation, LC-MS based metabolomics revealed that mice maintained on vancomycin displayed a shift toward primary conjugated bile acids, as denoted by the increase in taurocholate and tauro-β-muricholate, and the decrease in unconjugated cholate and chenodeoxycholate in circulation (). The bile acid composition in vancomycin-treated mice was more hydrophilic than vehicle-treated mice, as evident by the higher conjugated-to-unconjugated bile acids ratio in mice given vancomycin ()). More profoundly, secondary bile acids – such as deoxycholate, lithocholate, and ω-muricholate – were substantially reduced in the vancomycin treated groupwith moderate reduction in ursodeoxycholate (). The level of tauroursodeoxycholate was not decreased by vancomycin treatment (). Collectively, these data suggest that the gut microbiota-bile acid metabolism axis is a significant intermediary factor promoting fermentable fiber-induced HCC in T5KO mice.
Figure 4. Bacterial-derived secondary bile acids are diminished upon vancomycin treatment. Bar graphs represent the serum concentration of (a) total bile acids, (b) primary unconjugated bile acids, (c) primary conjugated bile acids, (d) ratio of major primary conjugated to primary unconjugated bile acids, (e) secondary bile acids, and (f) secondary conjugated bile acids. Data are representative of six mice per group. Error bars indicate mean ± SEM. ANOVA, *p < .05. CA: cholate, α-MCA: alpha-muricholate, β-MCA: beta-muricholate, CDCA: chenodeoxycholate, TCA: taurocholate, Tα-MCA: tauro-alpha-muricholate, Tβ-MCA: tauro-beta-muricholate, TCDCA: taurochenodeoxycholate, ω-MCA: omega-muricholate, DCA: deoxycholate, LCA: lithocholate, UDCA: ursodeoxycholate, Tω-MCA: tauro-omega-muricholate, TDCA: taurodeoxycholate, TUDCA: tauroursodeoxycholate.
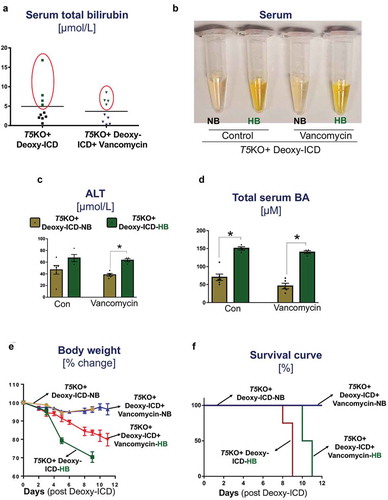
Supplementing ICD with deoxycholate potentiates body weight loss and morbidity in hyperbilirubinemic T5KO mice, irrespective of vancomycin treatment
To test whether secondary bile acid(s) are critical in promoting ICD-induced HCC, we next fed T5KO mice with ICD supplemented with 0.3% deoxycholate (Deoxy-ICD) while maintained on either regular water or vancomycin. As anticipated, a subset of T5KO mice displayed hyperbilirubinemia following one week of Deoxy-ICD feeding, irrespective of without (4 out of 10) or with (4 out of 9) vancomycin treatment (). Serum ALT and total bile acids were also elevated in these hyperbilirubinemic T5KO-HB mice (). Intriguingly, T5KO-HB mice without vancomycin lost more than 20% of body weight by day 8 of feeding ()). With vancomycin treatment, such body weight loss was delayed to day 10–11 ()). These T5KO-HB mice, in both groups, appeared moribund and had to be euthanized ()). In contrast, T5KO-NB mice that tolerated Deoxy-ICD maintained their body weight and displayed normal serum bilirubin levels, despite the increase in serum total bile acid levels to ~50 µM. While early termination of T5KO-HB mice on Deoxy-ICD diet prevented us from assessing for HCC parameters, such outcome nevertheless highlights that deoxycholate is likely a cholestatic agent for T5KO-HB mice.
Figure 5. Supplementing ICD diet with deoxycholate induces mortality in hyperbilirubinemic mice with or without vancomycin treatment. Vancomycin-pretreated and control T5KO mice were maintained on ICD supplemented with deoxycholate (Deoxy-ICD) up to 11 days. (a–d) Serum samples were collected post-one week of Deoxy-ICD feeding and analyzed for (a) total bilirubin. Based on the total bilirubin level, mice were re-grouped into normal bilirubin (NB) and high bilirubin (HB). (b) The image displays the appearance of serum color. (c–d) Serum levels of (c) ALT, and (d) total bile acids. (e–f) Line graphs display the (e) percent loss in body weight and (f) percent survival. Data represent the mean ± SEM of two pooled group experiments; n = 10 for water control and n = 9 for vancomycin groups. *p < .05.
Discussion
The mammalian gastrointestinal (GI) tract corrals trillions of bacteria that are collectively referred to as gut microbiota. This assemblage of gut microbes persists in a symbiotic relationship with their host by performing diverse metabolic functions and producing key nutrients that benefit the holobiont. Bacteria residing densely in the distal colon, for instance, largely depends on host-derived metabolites and undigested foods. For example, bacteria that express glucuronidases are capable of utilizing host-derived waste metabolites, such as bilirubin diglucuronide, as their carbon source. Other bacteria expressing BSH could acquire taurine or glycine as carbon sources from host-derived primary bile acids and generate secondary bile acids. These secondary bile acids can enter enterohepatic circulation and further serve as ligands for farnesoid X receptor (FXR) and Takeda G protein coupled receptor 5 (TGR5), whose signaling modulates cholesterol and bile acid homeostasis.Citation22 In addition, diverse bacteria ferment dietary fibers and generate large quantities of calorific short-chain fatty acids (SCFAs; e.g. acetate, butyrate and propionate). Since not all bacteria are capable of catalyzing similar reactions, the constituents of the gut microbiota actively participate in quorum-sensing and cross-feeding to meet their nutritional requirement.
While the production of secondary bile acids and microbial fermentation are often thought to be beneficial or benign to the host, such microbial activity could also be detrimental under certain circumstances such as during dysbiosis. Accumulation of secondary bile acids, e.g. deoxycholate known for its hydrophobicity and detergent-like properties, has been increasingly implicated in driving hepatic tumorigenesis by promoting cellular senescenceCitation7 and suppressing anti-tumor immunityCitation6,Citation7 in the liver. Another secondary bile acid, ω-muricholate, is shown to promote liver cancer by suppressing anti-tumor immune surveillance mediated by NKT cells.Citation8 In a similar vein, butyrate derived from bacterial fermentation could exert double-edged sword effects, preeminently described as the ‘butyrate paradox’,Citation23 in promoting or demoting cancer proliferation depending on the circumstances.Citation24-Citation26 Based on our previous work,Citation9 we posit that cholestasis and its sequelae could potentiate the pro-oncogenic property of butyrate. The adverse effects of such metabolites could be further amplified in the dysbiotic T5KO mice fed on a diet containing inulin, which is known to promote the growth of fermenters and secondary bile acid producers. Since the major fermenters and secondary bile acid producers in the GI tract are Gram-positive bacteria, we postulated that depleting specifically Gram-positive bacteria may confer a degree of protection to T5KO mice from ICD-induced HCC.
Outcomes from our vancomycin intervention study assert that ICD-induced HCC is largely dependent on Gram-positive bacteria and, by extension, dependent on their metabolites. Yet, it was unexpected for us to observe that vancomycin therapy could enrich Firmicutes (Gram-positive) but deplete Bacteroidetes (Gram-negative). This outcome may seem contentious, but not surprising as a prior study has reported similar occurrences in humans.Citation27 One possible explanation is that the Firmicutes thriving in vancomycin-treated mice could be intrinsically resistant to the antibiotic and the Bacteroidetes that was ablated could be dependent on the nutritional cross-feeding provided by vancomycin-sensitive bacteria. These findings at the phyla level, nevertheless, were unlikely to explain the hepato-protection seen in T5KO-HB mice given vancomycin. Hence, we focused our analyses more specifically on their activity, i.e. production of secondary bile acids, rather than specific taxa of bacteria. The observation that circulating secondary bile acids were depleted in T5KO-HB mice rescued from HCC by vancomycin suggests that secondary bile acid-producing bacteria are required for ICD-induced HCC. Other observations that substantiate this interpretation include: (i) the increase in taurine-conjugated primary bile acids in the serum metabolome due to the lack of bile acid deconjugation, (ii) the absence of bacteria expressing bsh groups 1a, 1b and 3c, and (iii) the loss of fecal BSH activity in T5KO-HB mice protected from HCC. Though these outcomes do not necessarily implicate the role of secondary bile acid producers in ICD-induced HCC, the correlation between their absence and the ensuing hepatoprotection was consistent with previous reports that studied a similar link in other models of HCC.Citation6-Citation8
Though vancomycin could prevent the manifestation of overt HCC at six months of ICD-feeding, the protection conferred was rather inadequate against another spectrum of ICD-induced pathologies. Most notable was its lack of effect on the ~40–46% incidences of hyperbilirubinemia in T5KO mice fed ICD. Cholemia in T5KO-HB mice was likewise unabated, though the bile acid composition shifted toward a larger pool of conjugated primary bile acids. Such an increase in the hydrophilic-to-hydrophobic bile acid ratio is a notable advantage nonetheless, since hydrophilic bile acids are less toxic, more water-soluble and are more readily excreted through the urinary or fecal routes. The level of ursodeoxycholate (UDCA) in the circulation was not affected by vancomycin since UDCA is a minor host-derived primary bile acid in rodents, unlike the UDCA in humans whose production requires microbial conversion from chenodeoxycholic acid.Citation28 Intriguingly, the level of the taurine-conjugated form of UDCA (TUDCA) was elevated in the circulation following vancomycin therapy. Considering that UDCA and TUDCA are known for their hydrophilicity and efficacy in treating cholestasis,Citation29 the elevation in TUDCA in vancomycin-treated mice may, in part, contribute to their resistance to ICD-induced HCC. We also note that the features of cancer-associated cachexia (i.e. low body weight and adiposity), but not sequela of cholestasis (i.e. hypolipidemia), could be ameliorated by vancomycin therapy. The overall dissimilarities between vehicle and vancomycin–treated T5KO-HB mice seem to illustrate a dichotomy between the two disease states induced by ICD feeding: cholestasis and the ensuing HCC. These findings collectively imply that bacterial families which belong to Gram-positive groups are required for HCC progression, but are not likely to be involved in initiating cholestasis or liver injury/fibrosis following ICD feeding. Further studies on other factors, such as host genetics or vancomycin-sensitive bacteria, are certainly warranted to fully understand the etiology and pathogenesis of the initial ICD-induced cholestasis.
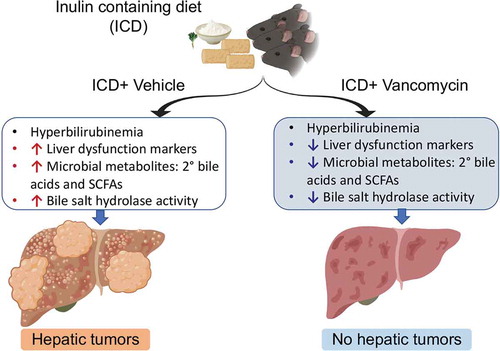
In summary, the present study sheds light on the contributory role of Gram-positive bacteria, specifically secondary bile acid producers, in promoting ICD-induced HCC in T5KO mice (). Notwithstanding the efficacy of vancomycin against Gram-positive bacteria including those with multi-drug resistance, vancomycin use in clinical settings is not recommended due to potential side effects and the risk of engendering vancomycin-resistant bacteria. Such concerns certainly limit the prospect of employing vancomycin-based interventions for treating human HCC. One thought is that instead of a simple generic depletion of gut bacteria using antibiotics, it is perhaps more ideal to inhibit their bile acid catabolism in the GI tract. This could be achieved by administering difructose anhydride III that is shown to decrease secondary bile acid generation, purportedly via inhibiting bacterial 7α-dehydroxylase.Citation7,Citation30 As an alternate means, bile salt hydrolase inhibitors could also be considered for restricting the generation of secondary bile acids.Citation31 Another viable alternative is to employ β-acids from hops (Humulus lupulus), which we had previously shown to be effective in inhibiting gut fermentation and thus prevent fermenting bacteria from generating SCFAs.Citation9 Collectively, our study suggests that selective targeting of certain bacterial species or their metabolism, rather than indiscriminately eradicating the entire microbiota with broad spectrum antibiotics, may be a fruitful approach to limit the generation of hepatotoxic metabolites in the gut.
Figure 6. A mechanistic model of vancomycin-mediated prevention of HCC. The image illustrates the causative factors that drive HCC and its remediation and reduction in HCC upon vancomycin treatment in a model of inulin-induced HCC.
Materials and methods
Mice
All animal studies described were conducted as per institutional animal care and use committee (IACUC) approvals obtained from the Pennsylvania State University and the University of Toledo. Toll-like receptor 5 (TLR5) deficient (T5KO) mice were originally generated by Dr. Shizuo Akira, Japan on C57BL/6 background. These mice were bred and maintained under specific pathogen-free conditions at the Pennsylvania State University and at the University of Toledo. Mice were housed in cages (n = 5 mice/cage) containing corncob bedding (Bed-O-Cob, The Andersons Co.) and nestlets (cat# CABFM00088, Ancare), fed ad libitum (LabDiet 5020 for breeders and LabDiet 5001 for weaned mice) and given regular, non-acidified drinking water. Mice were housed at 23°C under a 12-h light/dark phase cycle.
Diet
Inulin, a polyfructosan (Orafti® HP; source: chicory root; inulin content: 100%; degree of polymerization: > 23) was procured from Beneo (Tienen, Belgium). Inulin was incorporated into an open-source diet, i.e. inulin-containing diet (ICD: cat# D12081401) comprised of 7.5% inulin and 2.5% cellulose. Cellulose was retained in the diet to maintain roughage. Deoxycholate was procured from Sigma and supplemented into ICD (at 0.3% w/w) to formulate Deoxy-ICD diet (cat# D18112901). Diet was prepared by Research Diets (New Brunswick, NJ) and their composition is provided in detail in Singh et al..Citation9
Vancomycin treatment
Groups of experimental mice were administered with or without 0.5 g/L of vancomycin hydrochloride (DOT Scientific Inc.) in drinking water. Vancomycin treatment began one week prior to placing mice on ICD and thereafter maintained up to six months. Both experimental and control groups of mice were housed in a BSL2 facility as a precaution against, and to curtail the dissemination of, vancomycin-resistant bacteria that could potentially arise following long-term exposure to vancomycin.
Euthanasia and serum collection
At the end of six-month ICD-feeding, mice were euthanized via CO2 asphyxiation and blood was collected using a BD microtainer® (Becton, Dickinson) via cardiac puncture. Hemolysis-free sera were obtained after centrifugation and stored at −80ºC until further analysis.
Histochemical staining
Following euthanasia, a portion of liver tissues were submitted to the Penn State University Animal Diagnostic Laboratory for paraffin-embedded sectioning and hematoxylin and eosin (H&E) staining to describe liver tumors (HCC) and cells found in surrounding liver tissue. Histology images were captured from VS120 Virtual Slide Microscope (Olympus) through the OlyVIA software.
Picrosirius red staining
Picrosirius red staining was used to assess collagen accumulation in the liver. Briefly, deparaffinized liver (5 microns) sections were incubated with picrosirius red solution (0.1% sirius red in saturated picric acid) for one hour and quickly washed with acidified water (0.5% acetic acid). Collagen was selectively visualized as bright orange-red birefringent fibers and the image was acquired under a polarized light.
Blood glucose, serum lipids and transaminases measurement
One week prior to euthanasia, mice were fasted overnight (15 h), and blood glucose levels were measured using glucose test strips and a glucometer (Nova Max, Waltham, MA USA). Three days later, mice were bled retro-orbitally after 5 h fasting and hemolysis-free serum was collected using BD microtainer® (BD Biosciences) tubes. Serum cholesterol, triglycerides, alanine transaminase (ALT) and total bilirubin were measured by using biochemical kits from Randox Laboratories (Kearneysville, WV).
Enzyme-linked immunosorbent assay (ELISA)
Serum α-fetoprotein (AFP) was measured using the Duoset ELISA kits (R&D Systems; Minneapolis, MN) according to the manufacturer’s protocol.
Cecal short-chain fatty acid (SCFAs) analysis via NMR
1H NMR based-SCFAs quantitation analysis were performed as previously described.Citation32,Citation33 Briefly, cecal content samples (50 mg) were extracted with 0.6 ml of phosphate buffer (K2HPO4/NaH2PO4, 0.1 M, pH 7.4, 50% v/v D2O) containing 0.005% 3-(trimethysilyl) [2,2,3,3-2H4] propionate (TSP) as internal standard. Targeted analysis of SCFAs in cecal content was performed using a Bruker Avance NEO 600 MHz spectrometer equipped with an inverse cryogenic probe (Bruker Biospin, Germany) at 298 K. NMR spectra of cecal content samples were acquired for each employing the first increment of NOESY pulse sequence with a delay of 10 s added in the addition to recycle delay (RD) of 2 s. The quantification of SCFAs was calculated by NMR peak area against TSP using Chenomx (Chenomx Inc, Canada).
Serum total bile acid quantification
Total bile acids (TBA) in hemolysis-free sera was measured using a TBA assay kit (enzyme cycling method; Diazyme Laboratories) according to the manufacturer’s protocol.
Serum bile acid profiling
Targeted bile acid extraction and analysis by liquid chromatography-tandem mass spectrometry (LC MS/MS) (Waters Acquity -ABSciex 4000 Qtrap) was performed by West Coast Metabolomics Center (UC, Davis, https://metabolomics.ucdavis.edu/) to profile the composition of serum bile acids as previously described.Citation9
Analysis of bacterial composition and bile acid genes
Fecal samples were collected from control and vancomycin treated groups of T5KO mice. Total bacterial DNA was extracted from equal amounts of feces (~50 mg) using the QIAamp DNA Stool Mini Kit (QiagenInc, Valencia, CA).Citation34 The relative abundance of different groups of bacteria involved in secondary bile production was analyzed by quantitative PCR (Step One Plus Real-Time PCR System, Applied Biosystems) in a reaction mixture of fecal DNA, SYBR green master mix, and bacterial-specific primers (). The data were expressed as a relative fold-change in comparison to the control RNA polymerase beta subunit (rpoB) gene.
Fecal bile salt hydrolase (BSH) activity assay
Fecal BSH activity was determined using the previously described two-step procedure,Citation31,Citation35 but with slight modifications. Briefly, a 100 mg/ml fecal suspension was prepared in a buffer containing 0.6 M sodium chloride (pH 6.0), 0.1 M sodium phosphate (pH 6.0) and protease inhibitor (Sigma), and then subjected to three cycles of homogenization and sonication (3 x 10 seconds, 0.5 cycle, 50% amplitude). Next, samples were freeze-thawed three times to lyse fecal bacteria. Afterward, samples were centrifuged at 12,000 x g for 20 minutes at 4°C and the supernatants collected. The reaction set up for a 96 well flat bottom plate was as follows: 50 µl of supernatant, 38 µl of reaction buffer (0.1 M sodium-phosphate, pH 6.0) and 2 µl of 1 M dithiothreitol (DTT). The samples were preheated at 37°C for 5 minutes. Next, 10 µl of taurocholate (100 mM) was added to the samples for a total reaction volume of 100 µl. A baseline control without taurocholate was prepared for each individual sample. Samples were incubated at 37°C for 30 minutes. The reaction was stopped using 100 µl of 15% trichloroacetic acid and then centrifuged at 2,000 x g for 5 minutes at room temperature to remove the precipitate. Next, 50 µl of the supernatant was thoroughly mixed with 950 µl of ninhydrin reagent [0.25 mL of 1% wt/vol ninhydrin in 0.5 M sodium citrate buffer (pH 5.5), 0.6 ml of glycerol, and 0.1 ml of 0.5 M sodium citrate buffer (pH 5.5)] and then incubated in a water bath at 99°C for 10 minutes. After cooled on ice for 3 minutes, 100 µl of sample was transferred to a 96-well plate and read at an absorbance of 570 nM on a microplate reader. Germ-free feces were used as a negative control. A standard curve was generated using taurine (20 mM – 1 mM). BSH activity was calculated as mM of taurine released from taurocholate (the difference between reaction and baseline values; 1 mM equals to 1 Unit) and expressed as Unit per minute per gram of feces.
Statistical analysis
All data are presented as mean ± SEM. Statistical significance between two groups was calculated using unpaired, two-tailed t test. Data from more than two groups were compared using a one-way ANOVA followed by Tukey’s multiple comparison test. *p < .05 was considered statistically significant. All statistical analyzes were performed with the GraphPad Prism 7.0 program (GraphPad).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi:10.3322/caac.21492.
- Jinjuvadia R, Salami A, Lenhart A, Jinjuvadia K, Liangpunsakul S, Salgia R. Hepatocellular carcinoma: a decade of hospitalizations and financial burden in the United States. Am J Med Sci. 2017;354:362–369. doi:10.1016/j.amjms.2017.05.016.
- Blonski W, Kotlyar DS, Forde KA. Non-viral causes of hepatocellular carcinoma. World J Gastroenterol. 2010;16:3603–3615. doi:10.3748/wjg.v16.i29.3603.
- Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol. 2013;10:330–344. doi:10.1038/nrgastro.2013.41.
- Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, Caviglia J, Khiabanian H, Adeyemi A, Bataller R, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504–516. doi:10.1016/j.ccr.2012.02.007.
- Loo TM, Kamachi F, Watanabe Y, Yoshimoto S, Kanda H, Arai Y, Nakajima-Takagi Y, Iwama A, Koga T, Sugimoto Y, et al. Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov. 2017;7:522–538. doi:10.1158/2159-8290.CD-16-0932.
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi:10.1038/nature12347.
- Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360(6391):eaan5931. doi:10.1126/science.aan5931.
- Singh V, Yeoh BS, Chassaing B, Xiao X, Saha P, Aguilera Olvera R, Lapek JD, Zhang L, Wang W-B, Hao S, et al. Dysregulated microbial fermentation of soluble fiber induces cholestatic liver cancer. Cell. 2018;175:679–94 e22. doi:10.1016/j.cell.2018.09.004.
- Zhu J, Seo JE, Wang S, Ashby K, Ballard R, Yu D, Ning B, Agarwal R, Borlak J, Tong W, et al. The development of a database for herbal and dietary supplement induced liver toxicity. Int J Mol Sci. 2018;19(10):E2955. doi:10.3390/ijms19102955.
- Peterson BR, DeRoche TC, Huber AR, Shields WW. Cholestatic liver injury associated with dietary supplements: a report of five cases in active duty service members. Mil Med. 2013;178:e1168–71. doi:10.7205/MILMED-D-13-00228.
- Sharma T, Wong L, Tsai N, Wong RD. Hydroxycut((R)) (herbal weight loss supplement) induced hepatotoxicity: a case report and review of literature. Hawaii Med J. 2010;69:188–190.
- Cheng FK, Dunaway P. A case of supplement-induced hepatotoxicity. Case Rep Med. 2010;2010:262706. doi:10.1155/2010/262706.
- Navarro VJ, Khan I, Bjornsson E, Seeff LB, Serrano J, Hoofnagle JH. Liver injury from herbal and dietary supplements. Hepatology. 2017;65:363–373. doi:10.1002/hep.v65.1.
- Navarro VJ, Barnhart H, Bonkovsky HL, Davern T, Fontana RJ, Grant L, Reddy KR, Seeff LB, Serrano J, Sherker AH, et al. Liver injury from herbals and dietary supplements in the U.S. drug-induced liver injury network. Hepatology. 2014;60:1399–1408.
- Singh V, Chassaing B, Zhang L, San Yeoh B, Xiao X, Kumar M, Baker M, Cai J, Walker R, Borkowski K, et al. Microbiota-dependent hepatic lipogenesis mediated by stearoyl CoA desaturase 1 (SCD1) promotes metabolic syndrome in TLR5-deficient mice. Cell Metab. 2015;22:983–996. doi:10.1016/j.cmet.2015.09.028.
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi:10.1126/science.1179721.
- Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer-associated cachexia. Nat Rev Dis Primers. 2018;4:17105. doi:10.1038/nrdp.2017.105.
- Singh V, Yeoh BS, Carvalho F, Gewirtz AT, Vijay-Kumar M. Proneness of TLR5 deficient mice to develop colitis is microbiota dependent. Gut Microbes. 2015;6:279–283. doi:10.1080/19490976.2015.1060390.
- Carvalho FA, Koren O, Goodrich JK, Johansson ME, Nalbantoglu I, Aitken JD, Su Y, Chassaing B, Walters W, González A, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe. 2012;12:139–152. doi:10.1016/j.chom.2012.07.004.
- Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, Neish AS, Uematsu S, Akira S, Williams IR, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117:3909–3921. doi:10.1172/JCI33084.
- Wahlstrom A, Sayin SI, Marschall HU, Backhed F. intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24:41–50. doi:10.1016/j.cmet.2016.05.005.
- Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48:612–626. doi:10.1016/j.molcel.2012.08.033.
- Belcheva A, Irrazabal T, Robertson SJ, Streutker C, Maughan H, Rubino S, Moriyama E, Copeland J, Surendra A, Kumar S, et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell. 2014;158:288–299. doi:10.1016/j.cell.2014.04.051.
- Misikangas M, Tanayama H, Rajakangas J, Linden J, Pajari AM, Mutanen M. Inulin results in increased levels of beta-catenin and cyclin D1 as the adenomas increase in size from small to large in the Min/+ mouse. Br J Nutr. 2008;99:963–970. doi:10.1017/S0007114507853414.
- Pajari AM, Rajakangas J, Paivarinta E, Kosma VM, Rafter J, Mutanen M. Promotion of intestinal tumor formation by inulin is associated with an accumulation of cytosolic beta-catenin in Min mice. Int J Cancer. 2003;106:653–660. doi:10.1002/ijc.11270.
- Isaac S, Scher JU, Djukovic A, Jimenez N, Littman DR, Abramson SB, Pamer EG, Ubeda C. Short- and long-term effects of oral vancomycin on the human intestinal microbiota. J Antimicrob Chemother. 2017;72:128–136. doi:10.1093/jac/dkw383.
- Hay DW, Carey MC. Chemical species of lipids in bile. Hepatology. 1990;12:6S–14S. discussion S–6S.
- Goode EC, Rushbrook SM. A review of the medical treatment of primary sclerosing cholangitis in the 21st century. Ther Adv Chronic Dis. 2016;7:68–85. doi:10.1177/2040622315605821.
- Minamida K, Ohashi M, Hara H, Asano K, Tomita F. Effects of ingestion of difructose anhydride III (DFA III) and the DFA III-assimilating bacterium Ruminococcus productus on rat intestine. Biosci Biotechnol Biochem. 2006;70:332–339. doi:10.1271/bbb.70.332.
- Lin J, Negga R, Zeng X, Smith K. Effect of bile salt hydrolase inhibitors on a bile salt hydrolase from lactobacillus acidophilus. Pathogens. 2014;3:947–956. doi:10.3390/pathogens3040947.
- Tian Y, Nichols RG, Cai J, Patterson AD, Cantorna MT. Vitamin A deficiency in mice alters host and gut microbial metabolism leading to altered energy homeostasis. J Nutr Biochem. 2018;54:28–34. doi:10.1016/j.jnutbio.2017.10.011.
- Cai J, Zhang J, Tian Y, Zhang L, Hatzakis E, Krausz KW, Smith PB, Gonzalez FJ, Patterson AD. Orthogonal comparison of GC-MS and (1)H NMR spectroscopy for short chain fatty acid quantitation. Anal Chem. 2017;89:7900–7906. doi:10.1021/acs.analchem.7b00848.
- Singh V, Yeoh BS, Walker RE, Xiao X, Saha P, Golonka RM, Cai J, Bretin ACA, Cheng X, Liu Q, et al. Microbiota fermentation-NLRP3 axis shapes the impact of dietary fibres on intestinal inflammation. Gut. 1801-12;2019:68.
- Wang Z, Zeng X, Mo Y, Smith K, Guo Y, Lin J. Identification and characterization of a bile salt hydrolase from Lactobacillus salivarius for development of novel alternatives to antibiotic growth promoters. Appl Environ Microbiol. 2012;78:8795–8802. doi:10.1128/AEM.02519-12.
- Raetz M, Hwang SH, Wilhelm CL, Kirkland D, Benson A, Sturge CR, Mirpuri J, Vaishnava S, Hou B, DeFranco AL, et al. Parasite-induced TH1 cells and intestinal dysbiosis cooperate in IFN-gamma-dependent elimination of Paneth cells. Nat Immunol. 2013;14:136–142. doi:10.1038/ni.2508.
- Mullish BH, Pechlivanis A, Barker GF, Thursz MR, Marchesi JR, McDonald JAK. Functional microbiomics: evaluation of gut microbiota-bile acid metabolism interactions in health and disease. Methods. 2018;149:49–58. doi:10.1016/j.ymeth.2018.04.028.