
ABSTRACT
Helicobacter pylori-induced inflammation is the strongest known risk factor for gastric adenocarcinoma. Hypoxia-inducible factor-1 (HIF-1α) is a key transcriptional regulator of immunity and carcinogenesis. To examine the role of this mediator within the context of H. pylori-induced injury, we first demonstrated that HIF-1α levels were significantly increased in parallel with the severity of gastric lesions in humans. In interventional studies targeting HIF-1α, H. pylori-infected mice were treated ± dimethyloxalylglycine (DMOG), a prolyl hydroxylase inhibitor that stabilizes HIF-1α. H. pylori significantly increased proinflammatory chemokines/cytokines and inflammation in vehicle-treated mice; however, this was significantly attenuated in DMOG-treated mice. DMOG treatment also significantly decreased function of the H. pylori type IV secretion system (T4SS) in vivo and significantly reduced T4SS-mediated NF-κB activation and IL-8 induction in vitro. These results suggest that prolyl hydroxylase inhibition protects against H. pylori-mediated pathologic responses, and is mediated, in part, via attenuation of H. pylori cag-mediated virulence and suppression of host proinflammatory responses.
Introduction
Helicobacter pylori-induced inflammation and injury is the strongest known risk factor for gastric adenocarcinoma, the fourth leading cause of cancer-related mortality worldwide and which accounts for > 800,000 deaths annually.Citation1 Heightened risk for H. pylori-mediated pathologic outcomes is orchestrated by complex interactions between H. pylori virulence determinants, host immune responses, and the exposome. One H. pylori strain-specific virulence determinant that augments disease risk is the cag pathogenicity island, and strains harboring this constituent induce more severe gastric injury. The cag island encodes a type IV secretion system (T4SS), which translocates the effector protein CagA into host gastric epithelial cells. Intracellular CagA can undergo tyrosine-phosphorylation or remain unphosphorylated; in either form, CagA aberrantly activates numerous signaling pathways which can induce proinflammatory responses.Citation2–4 However, only a subset of persons infected by cag-positive strains ever develop cancer,Citation5 underscoring the importance of defining precise interactions that increase gastric cancer risk.
Environmental conditions also accentuate the risk for H. pylori-mediated inflammation and carcinogenesis. Our laboratory previously demonstrated that iron deficiency enhances the ability of H. pylori to induce gastric carcinogenesis in rodent models through augmentation of H. pylori virulence and alterations in host responses that drive inflammation.Citation6,Citation7 Another environmental condition that contributes to H. pylori-induced gastric carcinogenesis is oxygen availability, and populations residing at high altitudes are at heightened risk for gastric cancer.Citation8 Both oxygen and iron regulate intracellular levels of hypoxia-inducible factors.Citation9 Under normoxia, oxygen-dependent prolyl hydroxylases (PHD) are active within the cell and can hydroxylate HIF-1α leading to ubiquitin-mediated proteasomal degradation. Under hypoxic conditions, hydroxylation is inhibited due to inactivation of PHD, and HIF-1α is stabilized and functions as a central mediator of cellular adaptation to hypoxic conditions. Several other factors can induce HIF-1α via regulating reactive oxygen species (ROS) or kinases, and gastric epithelial ROS, whether endogenous or induced by H. pylori, enhance HIF-1α expression in gastric mucosa under normoxic conditions.Citation10 Importantly, HIFs have emerged as major transcriptional regulators of immunityCitation11 and are involved in cancer progression.Citation12,Citation13 Thus, we sought to investigate the role of HIF-1α in the development of gastric inflammation and injury within the context of H. pylori infection. To address this, we utilized in vivo, ex vivo, and in vitro models of HIF-1α stabilization via DMOG (dimethyloxalylglycine), a cell-permeable inhibitor of PHD, and demonstrate that DMOG is protective in H. pylori-induced injury and inhibits the cag type IV secretion system-mediated virulence potential of H. pylori and corresponding host proinflammatory immune responses.
Results
HIF-1α expression is associated with more advanced disease in humans
HIFs have been shown to play an important role in the progression to cancer. Thus, to address this in a human population at heightened risk for inflammation-driven gastric cancer, we assessed HIF-1α expression levels in gastric tissue from patients with normal gastric mucosa, non-atrophic gastritis, multifocal atrophic gastritis, intestinal metaplasia, or gastric cancer (Supplementary Table S1, ()). HIF-1α expression levels paralleled the severity of gastric disease, with the highest levels among patients with gastric cancer ().
Figure 1. HIF-1α is associated with more advanced disease in humans. HIF-1α IHC was quantified in patients with normal gastric mucosa (N = 6), non-atrophic gastritis (NAG, N = 8), multifocal atrophic gastritis (MAG, N = 10), intestinal metaplasia (IM, N = 13), and gastric cancer (GC, N = 10) (a-b). Representative IHC images are shown at 200X and scale bars represent 100 µm (a). AGS cells were co-cultured with H. pylori clinical isolates from low-risk (N = 9) or high-risk (N = 9) patients and HIF-1α expression was assessed by qRT-PCR (c). ANOVAs with Sidak multiple comparisons test (b-c) were used for statistical analyses and standard error of the mean is shown. ****, P < .0001; ***, P < .001; **, P < .01; *, P < .05.
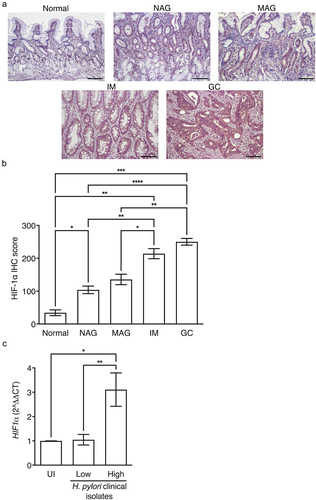
We next assessed the ability of H. pylori clinical strains isolated from patients from low versus high gastric cancer risk regions in Colombia to induce HIF-1α. The majority of patients from the low gastric cancer risk region (Pacific coastal town of Tumaco, Colombia) had a histologic diagnosis of non-atrophic gastritis, while the majority of patients from the high gastric cancer risk region (Andean mountain town of Túquerres, Colombia) had more advanced histologic diagnoses of intestinal metaplasia (Supplementary Table 2). Despite the high prevalence of H. pylori infection in each region (>90%), patients from the high-risk mountain region harbor a 25-fold increase in cancer rate compared to the low-risk coastal region.Citation14,Citation15 All H. pylori strains isolated from the low-risk and high-risk patients were urease-, catalase-, and oxidase-positive, and the majority of H. pylori strains isolated from the low-risk and high-risk patients were cagA+, vacA s1m1, sabA+, and babA+ (Supplementary Table 2). However, the MLST classifications differed between strains isolated from the low-risk versus high-risk patients,Citation16 whereby H. pylori strains isolated from low-risk patients were hspWAfrica types, while H. pylori strains isolated from high-risk patients were hpEurope types (Supplementary Table 2). H. pylori clinical strains were co-cultured with gastric epithelial cells and HIF-1α expression was assessed by qRT-PCR. H. pylori strains isolated from high-risk patients induced significantly higher levels of HIF-1α, compared to strains isolated from low-risk patients (), indicating that H. pylori strains infecting high-risk mountainous populations may adapt to hypoxia, which could alter H. pylori virulence.
DMOG treatment paradoxically reduces H. pylori-induced injury in vivo
We next conducted interventional studies targeting HIF-1α using a C57BL/6 mouse model of H. pylori-induced inflammation and injury. To first assess the expression of HIF-1α in this model, immunohistochemistry (IHC) was performed on gastric tissue sections from uninfected and H. pylori-infected C57BL/6 mice. HIF-1α was quantified in both the gastric epithelium and gastric inflammatory infiltrates. Although there were no significant differences in HIF-1α expression in epithelial cells following infection, H. pylori significantly increased levels of HIF-1α in foci of inflammation (Supplementary Figure S1A-C). To then investigate the functional role of HIF-1α in H. pylori-induced inflammation and injury, C57BL/6 mice were treated with or without DMOG, a cell-permeable inhibitor of PHD that stabilizes HIF-1α and then challenged with or without H. pylori. To first assess the direct effects of DMOG in vivo, we quantified levels of HIF-1α as well as carbonic anhydrase IX (CA9), a direct downstream target of PHD, by IHC. DMOG treatment significantly increased levels of HIF-1α in inflammatory infiltrates of H. pylori-infected mice (Supplementary Figure S1D) and significantly increased levels of CA9 in the gastric corpus (Supplementary Figure S1E), indicating that DMOG directly leads to HIF-1α stabilization and activation of HIF-1α downstream targets.
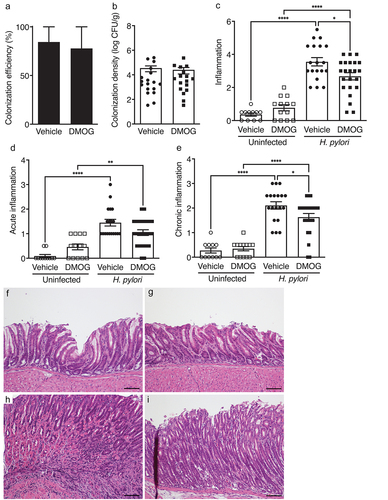
Having established the effectiveness of DMOG treatment, H. pylori colonization was assessed. DMOG treatment had no effect on H. pylori colonization efficiency () or colonization density () compared to vehicle-treated controls. We hypothesized that stabilization of HIF-1α would exacerbate H. pylori-induced disease in vivo; however, DMOG treatment reciprocally reduced H. pylori-induced inflammation (). When acute and chronic inflammation were independently assessed, DMOG treatment reduced acute inflammation, but this was not statistically significant; however, DMOG significantly reduced levels of chronic inflammation (). Collectively, these findings demonstrate that treatment with DMOG is protective in an in vivo model of H. pylori-induced inflammation and injury.
Figure 2. DMOG reduces H. pylori-induced inflammation in vivo. mice were treated with vehicle (N = 30) or DMOG (N = 36) and then challenged with or without H. pylori. Colonization efficiency (a) and colonization density (b) are shown. Total gastric inflammation (c), acute inflammation (d), and chronic inflammation (e) were quantified and representative H&E images from uninfected mice treated with vehicle (f) or DMOG (g) and H. pylori-infected mice treated with vehicle (h) or DMOG (i) are shown at 200X and scale bars represent 100 µm. Open symbols: uninfected mice; closed symbols: H. pylori-infected mice. Circles: vehicle-treated; squares: DMOG-treated. Unpaired parametric t-test (b) and one-way ordinary ANOVAs with Sidak multiple comparison test (c-e) were used for statistical analyses and standard error of the mean is shown. ****, P < .0001; **, P < .01; *, P < .05.
DMOG treatment reduces proinflammatory immune responses and M1 macrophage polarization markers in vivo
To define mechanisms by which DMOG paradoxically elicits a protective effect in H. pylori-induced injury, we next assessed chemokine and cytokine expression in gastric tissue from uninfected and H. pylori-infected mice treated with vehicle or DMOG ( and Supplementary Figure S2). H. pylori infection induced significant increases in the levels of several proinflammatory chemokines and cytokines, and a subset of these inflammatory mediators were significantly attenuated among infected mice treated with DMOG. H. pylori infection, regardless of treatment group, resulted in significant increases in gastric mucosal levels of G-CSF, IP-10, and IL-17 (), compared to uninfected controls. H. pylori infection of vehicle-treated mice also resulted in significant increases in levels of the chemokines KC, MIP-1β, and RANTES () and the proinflammatory cytokines IFN-γ, IL-1β, IL-6, IL-7, TNF-α, and IL-12 (), when compared to uninfected, vehicle-treated mice. However, these chemokine and cytokine proinflammatory responses were significantly attenuated in DMOG-treated mice infected with H. pylori ().
Figure 3. DMOG treatment reduces proinflammatory immune responses and M1 macrophage polarization markers in vivo. Chemokines and cytokines were analyzed in gastric tissue from mice treated with vehicle or DMOG and then challenged with or without H. pylori. Levels of G-CSF (a), IP-10 (b), and IL-17 (c) were increased with H. pylori infection. H. pylori-induced chemokines KC (d), MIP-1β (e), and RANTES (f) as well as proinflammatory cytokines INF-γ (g), IL-1β (h), IL-6 (i), IL-7 (j), TNF-α (k), IL-12 (p40) (l), and IL-12 (p70) (m) were significantly attenuated by DMOG treatment. Open symbols: uninfected mice; closed symbols: H. pylori-infected mice. Circles: vehicle-treated; squares: DMOG-treated. ANOVAs with Sidak multiple comparisons test were used for statistical analyses. ****, P < .0001; ***, P < .001; **, P < .01; *, P < .05.
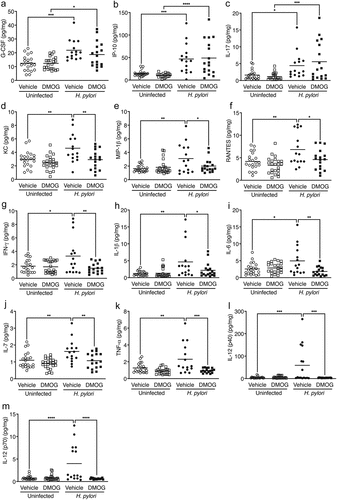
To further investigate the mechanisms underpinning this phenotype, we next assessed M1 macrophage polarization markers within the context of H. pylori infection and DMOG treatment in vivo. It has been previously demonstrated that DMOG alters macrophage polarization via reductions in levels of the proinflammatory cytokines IL-1β, IL-6, and TNF-α which drive M1 macrophage polarization.Citation17–19 In this model of H. pylori-induced gastric injury, in addition to reductions in levels of the previously identified M1 macrophage markers IL-1β, IL-6, and TNF-α (), DMOG also significantly reduced the M1 macrophage markers IFN-γ, IL-7, IL-12(p40), and IL-12(p70) ( and 3l-m). Collectively, these data demonstrate that DMOG treatment significantly reduces proinflammatory immune responses induced by H. pylori, and this is likely mediated through reduced M1 macrophage polarization.
Since HIF-1α is an important regulator of immunity and is highly expressed in immune cells, we next employed a novel and enriched primary ex vivo model system using cells isolated from a mouse model that is prone to gastric carcinogenesis, INS-GAS mice. To first validate the expression pattern of HIF-1α seen in the C57BL/6 model, IHC was performed on gastric tissue sections from uninfected and H. pylori-infected INS-GAS mice and HIF-1α was quantified. As observed in our C57BL/6 mouse model, infection with H. pylori significantly increased levels of HIF-1α in foci of inflammation (Supplementary Figure S3A-C). Having validated HIF-1α expression patterns in INS-GAS mice, gastric organoids were generated from these mice and seeded in the upper chamber of a transwell system to form 2D monolayers, while autologous bone marrow-derived macrophages and splenocytes treated with anti-CD3 and anti-CD28, to induce T cell activation, were placed in the lower chamber. Epithelial gastric organoid monolayers were then infected with H. pylori and chemokine and cytokine expression was quantified from inflammatory macrophage/T cell lysates. Consistent with our in vivo findings (), H. pylori significantly increased the proinflammatory chemokines Kc and Rantes (Supplementary Figure S3D-E) as well as proinflammatory cytokines Il1β, Il17, and Tnfα (Supplementary Figure S3F-H). Further, H. pylori also significant increased levels of Il9 and Il22 (Supplementary Figure S3I-J), cytokines that are regulated by HIF-1α and which contributes to T cell differentiation and immunity.Citation20–22 Collectively, these data validate our in vivo findings in another mouse model, but also reveal the important contribution of immune cell components in HIF-1α-mediated chemokine and cytokine induction.
DMOG treatment attenuates H. pylori cag T4SS function and the subsequent induction of downstream proinflammatory responses
To next assess direct effects of DMOG on H. pylori virulence, gastric epithelial cells were treated with vehicle or DMOG and then challenged with H. pylori. Concordant with the finding that HIF-1α is more robustly expressed in inflammatory immune infiltrates in vivo (Supplementary Figure 1A-C), HIF-1α expression was stabilized by DMOG in gastric epithelial cells in vitro, and was augmented with H. pylori infection (). However, consistent with the in vivo suppressive phenotype, DMOG significantly reduced H. pylori-induced NF-κB activation () and IL-8 induction (). To confirm that DMOG did not exert adverse effects on H. pylori growth and viability, H. pylori were grown in the presence or absence of DMOG. There were no effects of DMOG on H. pylori growth or viability, compared to control ().
Figure 4. DMOG attenuates H. pylori cag T4SS function and induction of proinflammatory responses. Levels of HIF-1α were assessed by Western blot analyses (a), NF-κB activation by luciferase assay (b), and IL-8 by ELISA (c) in AGS cells treated with vehicle or DMOG and then challenged with or without H. pylori. H. pylori was grown in Brucella broth (BB) with vehicle or DMOG and bacterial growth was measured by OD600 (d). In vivo-adapted H. pylori were isolated from infected mice treated with vehicle or DMOG and co-cultured with AGS cells to assess CagA translocation by Western blot analysis (e-f) and IL-8 by ELISA (g). The percentage of H. pylori that retained T4SS function (e) and CagA translocation (f) were determined. ANOVAs with Sidak multiple comparisons test were used for statistical analyses and standard error of the mean is shown. ****, P < .0001; ***, P < .001; **, P < .01; *, P < .05.
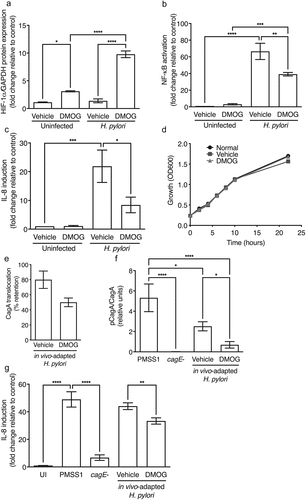
To increase the depth of our mechanistic analyses, we next assessed the effects of in vivo-DMOG-adaptation on the virulence potential of H. pylori isolates. In vivo-adapted H. pylori strains were isolated from mice treated with vehicle or DMOG, minimally passaged, and then co-cultured with gastric epithelial cells to assess translocation of the effector protein CagA and induction of the downstream proinflammatory chemokine IL-8. The input parental H. pylori strain PMSS1 and the PMSS1 cagE− isogenic mutant were used as positive and negative controls, respectively, as the parental strain PMSS1 harbors the ability to translocate CagA and induce IL-8, while loss of cagE prevents CagA translocation and partially attenuates IL-8 induction. In vivo-adaptation to DMOG resulted in a marked reduction in the ability of output H. pylori strains to translocate CagA, whereby 50% of isolates exposed to DMOG completely lost the ability to translocate CagA, compared to only 20% of H. pylori isolates in vivo-adapted to vehicle (). Among strains that maintained the ability to translocate CagA, in vivo-adaptation to DMOG resulted in a significant reduction in levels of CagA translocation compared to isolates in vivo-adapted to vehicle (). Loss of CagA translocation was paralleled by significant reductions in IL-8 secretion (). These findings suggest that DMOG, while increasing the stabilization of HIF-1α, counterbalances this effect via a concomitant suppressive effect on H. pylori cag T4SS function and virulence, which may attenuate inflammation and facilitate persistence.
Discussion
Hypoxia is a well-recognized condition affecting the development and progression of tumors via exerting an important role in therapeutic resistance, recurrence, and metastatic potential. HIF-1α, which is induced under hypoxic conditions, is an important transcriptional regulator of a number of signaling pathways that mediate the development and progression to cancer. In gastric cancer, HIF-1α has been shown to inhibit apoptosis, while promoting proliferation, angiogenesis, metastasis, and stemness of gastric cancer cells.Citation23 Further, HIF-1α expression is associated with increased invasion and metastasis and reduced overall survival in gastric cancer patients.Citation12 Recent studies have also described non-transcriptional functions for HIF-1α that contrast with its canonical roles in carcinogenesis, via contributions to cell cycle arrestCitation24,Citation25 and inhibition of DNA replication.Citation26
Prolyl hydroxylases (PHDs) regulate the stability of hypoxia-inducible factors, and hydroxylase inhibitors, such as DMOG, result in the stabilization of HIF-1α. However, published data have identified an important paradox as DMOG also exerts a protective effect in certain models of inflammation. In the current study, we demonstrate that DMOG is protective against H. pylori-induced gastric inflammation and injury by attenuating host proinflammatory immune responses and M1 macrophage responses. Specifically, our findings demonstrate that DMOG decreases production of a subset of chemokines that are important in the recruitment and infiltration of innate immune components, particularly neutrophils. Further, DMOG significantly attenuates the induction of several potent Th1 proinflammatory cytokines, including IL-1β, IL-6, and TNF-α, all of which signal via type I cytokine receptors that are structurally divergent from other cytokine receptor types. In addition to dampening Th1 proinflammatory cytokine responses, DMOG also significantly attenuates the secreted M1 macrophage markers IFN-γ, IL-1β, IL-6, IL-12, and TNF-α, which in turn further dampens Th1 adaptive immune responses to H. pylori infection.
In addition to suppressive effects on the proinflammatory immune response, our findings also demonstrate that DMOG can attenuate H. pylori virulence mechanisms. However, although we have demonstrated that DMOG treatment significantly attenuates cag type IV secretion system function and concomitant CagA translocation, this study has not addressed the role of other important H. pylori cag T4SS effectors, adhesins, or other well characterized virulence factors.
Consistent with our results, DMOG is also protective in dextran-sodium sulfate (DSS)-induced colitis and attenuates production of the DSS-induced proinflammatory cytokines IL-1β, IL-6, IL-12, and TNF-α.Citation27 In addition, concordant with our findings, several studies have demonstrated DMOG-mediated protection in a variety of models through alterations in macrophage polarization, with decreases in the M1 macrophage markers IL-1β, IL-6, and TNF-α and/or increases in the M2 macrophage markers IL-4, IL-10, Arg-1.Citation17–19 In other models of microbial-induced injury, DMOG attenuates Clostridium difficile toxin-induced injury via decreasing proinflammatory chemokine and cytokine production,Citation28 and significantly reduced mortality in a Pseudomonas aeruginosa-mediated model of pneumonia.Citation29 DMOG protection has also been reported to be mediated through enhanced mucosal barrier function leading to decreased membrane permeability;Citation30–32 we have not yet investigated the role of DMOG in enhanced barrier function in our model of H. pylori-induced gastric inflammation and injury, but our findings provide a framework for these studies in the future. Collectively, these studies demonstrate the importance of PHD inhibition in reducing the severity of disease in both chemically- and microbially-induced models of inflammation and injury through a variety of different mechanisms.
HIF-1α is induced by certain bacterial pathogens,Citation33,Citation34 while other bacterial pathogens are able to interfere with HIF-1α activation.Citation35,Citation36 Given the duality and complex role of HIF-1α in regulating inflammation, carcinogenesis, and immune-mediated bactericidal activity,Citation37 the question of whether HIF-1α is beneficial or detrimental for H. pylori-induced disease is central. A prior study addressed this by demonstrating that myeloid HIF-1α contributes to proinflammatory responses induced by H. pylori, but is protective against H. pylori-mediated gastritis in vivo,Citation38 emphasizing the complex interplay of the innate immune and inflammatory phenotypes in driving pathologic responses.
Collectively, these new data highlight the importance of HIF-1α in populations at high risk for gastric cancer, but also demonstrate that use of a PHD inhibitor, DMOG, and consequent stabilization of HIF-1α is protective in H. pylori-induced gastric inflammation and injury. Importantly, hydroxylase inhibitors, such as Roxadustat, have been clinically approved for other therapiesCitation39 and could potentially be repurposed. Thus, it is of paramount importance to more fully dissect the distinct cell-specific roles of HIF-1α in regulating host inflammatory responses as well as signaling pathways related to carcinogenesis as a means to identify actionable targets that can limit H. pylori-induced disease.
Methods
Murine model of H. pylori-induced injury
C57BL/6NHsd (Envigo #4405) mice and transgenic hypergastrinemic INS-GAS+/+ mice on a FVB/N backgroundCitation40–42 were bred and housed in the Vanderbilt University Medical Center animal care facilities. C57BL/6 mice were subjected to intraperitoneal injection of vehicle (PBS) or DMOG (8 mg, Cayman Chemicals #71210), every 48 hours for seven days prior to H. pylori challenge. Mice were orogastrically challenged with Brucella broth (BD Biosciences #211088) or with H. pylori strain PMSS1 and mice were euthanized eight weeks post-challenge, as previously described.Citation7
Histopathology
Gastric tissue was fixed, paraffin-embedded, and stained with hematoxylin and eosin (H&E). A pathologist assessed indices of gastric inflammation and injury as previously described.Citation7 Severity of acute and chronic inflammation was graded 0–3 in both the gastric antrum and corpus for a cumulative score of 0–12.
Chemokine and cytokine multiplex bead array
Gastric tissue was lysed (CelLytic MT Cell Lysis Reagent, Sigma #C3228) and lysates were diluted 1:3 and mixed with magnetic beads according to the manufacturer’s instructions (MILLIPLEX Cytokine/Chemokine Magnetic Bead Panel, Millipore MCYTOMAG-70K-PMX) and as previously described.Citation7 Data were analyzed via Millipore software platform and standardized to protein concentrations.
Human gastric epithelial cell culture
AGS human gastric epithelial cells (ATCC CRL-1739, mycoplasma-negative) were grown in RPMI 1640 with L-glutamine (Corning #10–040-CV) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals #S11150), and HEPES buffer (1 mM, Corning #25–060-Cl) at 37°C with 5% CO2, as previously described.Citation43 AGS cells were co-cultured with H. pylori at a multiplicity of infection (MOI) of 100:1 for six hours unless otherwise noted.
Western blot analysis
Protein lysates from AGS:H. pylori co-cultures were separated by SDS-PAGE and transferred to PVDF membranes (Thermo Scientific #88518). Levels of total CagA (anti-CagA antibody, Austral Biologicals #HPP-5003-9) and phosphorylated CagA (anti-pY99 antibody, Santa Cruz #sc-7020) were determined and quantified using ImageJ 1.50i, as previously described.Citation7
NF-κB activation assay
H. pylori were co-cultured with NF-κB luciferase gastric epithelial (AGS) reporter cells at a MOI of 10:1 for four hours, and NF-κB activation was measured using a Steady-Glo luciferase assay substrate according to the manufacturer’s instructions (Promega #E2510), as previously described.Citation6
IL-8 ELISA
Levels of IL-8 were quantified in AGS:H. pylori co-culture supernatants by Quantikine® IL-8 ELISA (R&D Systems #D8000C) according to the manufacturer’s instructions. Data were analyzed with Gen5 software (Synergy4, BioTek), as previously described.Citation7
Statistical analysis
Mean values are shown with standard error of the mean (SEM) for experiments performed and replicated on at least three independent occasions. Unpaired parametric t-tests and one-way ordinary ANOVAs with Sidak multiple comparison test were used for statistical analyses. When data did not assume normal Gaussian distribution, either a nonparametric Mann Whitney t test or a Kruskal-Wallis ANOVA test with Dunn’s multiple comparisons test were used. All analyses were performed using GraphPad Prism. P < .05 was considered statistically significant (****, P < .0001; ***, P < .001; **, P < .01; *, P < .05).
Ethics
All animal and human studies were conducted in accordance with the Declaration of Helsinki principles and have been approved by the VUMC Institutional Animal Care and Use Committee (IACUC) and the Institutional Review Board (IRB), respectively. The IRB of Louisiana State University Health Sciences Center, the Institutional Review Committee of Memorial Medical Center in New Orleans, Louisiana, the Committees on Ethics of Universidad del Valle and Hospital Departamental de Nariño in Colombia, the Committees on Ethics of Pontificia Universidad Catolica de Chile, and the IRB of Vanderbilt University Medical Center approved these protocols.
Author contributions
JMN conceived, designed and performed experiments, collected, analyzed, and interpreted data, and drafted the manuscript. MPB performed IHC experiments, reviewed pathology, and quantified IHC data. JRG assisted with animal experiments. AGD performed and optimized IHC experiments. GS performed gastric organoid and immune cell experiments. KA performed genotyping of H. pylori clinical strains. MGH performed Western blot experiments and analyzed data. JCR acquired human gastric tissue samples for IHC analysis. CTT conceived and designed experiments. RMP conceived and designed experiments and drafted the manuscript. All authors revised the manuscript and approved the final version of the manuscript.
Supplemental Material
Download Zip (14.4 MB)Acknowledgments
We would like to acknowledge the following funding support through the National Institutes of Health: R01CA077955 (RMP), R01DK058587 (RMP), P01CA116087 (RMP), and P30DK058404 (RMP) as well as ANID FONDAP 152220002 (JCR) and the Science Foundation Ireland (CTT). We also acknowledge the following resources at Vanderbilt University Medical Center: Animal Care and Use Program, Division of Animal Care, Translational Pathology Shared Resource Core (NCI/NIH Cancer Center Support Grant P30CA68485), and Digestive Disease Research Center Tissue Morphology Core.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available in figshare at https://doi.org/10.6084/m9.figshare.22154753.v2, Noto, Jennifer (2023): MILLIPLEX Cytokine/Chemokine Magnetic Bead Panel. figshare. Dataset. https://doi.org/10.6084/m9.figshare.22154753.v2.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2263936
Additional information
Funding
References
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–13. doi:10.3322/caac.21660.
- Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Sci. 2000;287:1497–1500. doi:10.1126/science.287.5457.1497.
- Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Sci. 2003;300(5624):1430–1434. doi:10.1126/science.1081919.
- Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, Neish AS, Collier-Hyams L, Perez-Perez GI, Hatakeyama M, et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA. 2005;102:10646–10651. doi:10.1073/pnas.0504927102.
- Blaser MJ, Berg DE. Helicobacter pylori genetic diversity and risk of human disease. J Clin Invest. 2001;107(7):767–773. doi:10.1172/JCI12672.
- Noto JM, Gaddy JA, Lee JY, Piazuelo MB, Friedman DB, Colvin DC, Romero-Gallo J, Suarez G, Loh J, Slaughter JC, et al. Iron deficiency accelerates Helicobacter pylori-induced carcinogenesis in rodents and humans. J Clin Invest. 2013;123:479–492. doi:10.1172/JCI64373.
- Noto JM, Piazuelo MB, Shah SC, Romero-Gallo J, Hart JL, Di C, Carmichael JD, Delgado AG, Halvorson AE, Greevy RA, et al. Iron deficiency linked to altered bile acid metabolism promotes Helicobacter pylori-induced inflammation-driven gastric carcinogenesis. J Clin Invest. 2022;132:e147822. doi:10.1172/JCI147822.
- Torres J, Correa P, Ferreccio C, Hernandez-Suarez G, Herrero R, Cavazza-Porro M, Dominguez R, Morgan D. Gastric cancer incidence and mortality is associated with altitude in the mountainous regions of Pacific Latin America. Cancer Causes Control. 2013;24(2):249–256. doi:10.1007/s10552-012-0114-8.
- Hirota K. An intimate crosstalk between iron homeostasis and oxygen metabolism regulated by the hypoxia-inducible factors (HIFs). Free Radic Biol Med. 2019;133:118–129. doi:10.1016/j.freeradbiomed.2018.07.018.
- Park JH, Kim TY, Jong HS, Kim TY, Chun YS, Park JW, Lee CT, Jung HC, Kim NK, Bang YJ. Gastric epithelial reactive oxygen species prevent normoxic degradation of hypoxia-inducible factor-1alpha in gastric cancer cells. Clin Cancer Res. 2003;9:433–440.
- McGettrick AF, O’Neill LAJ. The role of HIF in immunity and inflammation. Cell Metab. 2020;32(4):524–536. doi:10.1016/j.cmet.2020.08.002.
- Lin S, Ma R, Zheng XY, Yu H, Liang X, Lin H, Cai XJ. Meta-analysis of immunohistochemical expression of hypoxia inducible factor-1alpha as a prognostic role in gastric cancer. World J Gastroenterol. 2014;20:1107–1113. doi:10.3748/wjg.v20.i4.1107.
- Cowman SJ, Koh MY. Revisiting the HIF switch in the tumor and its immune microenvironment. Trends Cancer. 2022;8(1):28–42. doi:10.1016/j.trecan.2021.10.004.
- Correa P, Cuello C, Duque E, Burbano LC, Garcia FT, Bolanos O, Brown C, Haenszel W. Gastric cancer in Colombia. III. Natural history of precursor lesions. J Natl Cancer Inst. 1976;57:1027–1035. doi:10.1093/jnci/57.5.1027.
- Cuello C, Correa P, Haenszel W, Gordillo G, Brown C, Archer M, Tannenbaum S. Gastric cancer in Colombia. I. Cancer risk and suspect environmental agents. J Natl Cancer Inst. 1976;57(5):1015–1020. doi:10.1093/jnci/57.5.1015.
- de Sablet T, Piazuelo MB, Shaffer CL, Schneider BG, Asim M, Chaturvedi R, Bravo LE, Sicinschi LA, Delgado AG, Mera RM, et al. Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut. 2011;60:1189–1195. doi:10.1136/gut.2010.234468.
- Strowitzki MJ, Kimmer G, Wehrmann J, Ritter AS, Radhakrishnan P, Opitz VM, Tuffs C, Biller M, Kugler J, Keppler U, et al. Inhibition of HIF-prolyl hydroxylases improves healing of intestinal anastomoses. JCI Insight. 2021;6(8):e13919. doi:10.1172/jci.insight.139191.
- Chen MH, Wang YH, Sun BJ, Yu LM, Chen QQ, Han XX, Liu YH. HIF-1alpha activator DMOG inhibits alveolar bone resorption in murine periodontitis by regulating macrophage polarization. Int Immunopharmacol. 2021;99:107901. doi:10.1016/j.intimp.2021.107901.
- Hams E, Saunders SP, Cummins EP, O’Connor A, Tambuwala MT, Gallagher WM, Byrne A, Campos-Torres A, Moynagh PM, Jobin C, et al. The hydroxylase inhibitor dimethyloxallyl glycine attenuates endotoxic shock via alternative activation of macrophages and IL-10 production by B1 cells. Shock. 2011;36(3):295–302. doi:10.1097/SHK.0b013e318225ad7e.
- Roy S, Rizvi ZA, Clarke AJ, Macdonald F, Pandey A, Zaiss DMW, Simon AK, Awasthi A. EGFR-HIF1alpha signaling positively regulates the differentiation of IL-9 producing T helper cells. Nat Commun. 2021;12:3182. doi:10.1038/s41467-021-23042-x.
- Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, Sun J, Pan F, Zhou J, Zhang W, et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat Commun. 2020;11(1):4457. doi:10.1038/s41467-020-18262-6.
- Fachi JL, Pral LP, Dos Santos JAC, Codo AC, de Oliveira S, Felipe JS, Zambom FFF, Camara NOS, Vieira P, Colonna M, et al. Hypoxia enhances ILC3 responses through HIF-1alpha-dependent mechanism. Mucosal Immunol. 2021;14:828–841. doi:10.1038/s41385-020-00371-6.
- Li M, Li G, Yang X, Yin W, Lv G, Wang S. HIF in gastric cancer: regulation and therapeutic target. Molecules. 2022;27(15):4893. doi:10.3390/molecules27154893.
- Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol. 2003;23:359–369. doi:10.1128/MCB.23.1.359-369.2003.
- Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23:1949–1956. doi:10.1038/sj.emboj.7600196.
- Hubbi ME, Kshitiz Gilkes DM, Rey S, Wong CC, Luo W, Kim DH, Dang CV, Levchenko A, Semenza GL, Semenza GL. A nontranscriptional role for HIF-1α as a direct inhibitor of DNA replication. Sci Signal. 2013;6:ra10. doi:10.1126/scisignal.2003417.
- Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, Taylor CT. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134:156–165. doi:10.1053/j.gastro.2007.10.012.
- Hirota SA, Fines K, Ng J, Traboulsi D, Lee J, Ihara E, Li Y, Willmore WG, Chung D, Scully MM, et al. Hypoxia-inducible factor signaling provides protection in clostridium difficile-induced intestinal injury. Gastroenterology. 2010;139:259–269 e253. doi:10.1053/j.gastro.2010.03.045.
- Schaible B, McClean S, Selfridge A, Broquet A, Asehnoune K, Taylor CT, Schaffer K, Karhausen J. Hypoxia modulates infection of epithelial cells by Pseudomonas aeruginosa. PLoS One. 2013;8(2):e56491. doi:10.1371/journal.pone.0056491.
- Lei X, Teng W, Fan Y, Zhu Y, Yao L, Li Y, Zhu S, Pileggi M. The protective effects of HIF-1α activation on sepsis induced intestinal mucosal barrier injury in rats model of sepsis. PLoS One. 2022;17:e0268445. doi:10.1371/journal.pone.0268445.
- Kim YI, Yi EJ, Kim YD, Lee AR, Chung J, Ha HC, Cho JM, Kim SR, Ko HJ, Cheon JH, et al. Local stabilization of hypoxia-inducible factor-1alpha controls intestinal inflammation via enhanced gut barrier function and immune regulation. Front Immunol. 2020;11:609689. doi:10.3389/fimmu.2020.609689.
- Harnoss JM, Gebhardt JM, Radhakrishnan P, Leowardi C, Burmeister J, Halligan DN, Yuan S, Kennel KB, Strowitzki MJ, Schaible A, et al. Prolyl hydroxylase inhibition mitigates pouchitis. Inflamm Bowel Dis. 2020;26:192–205. doi:10.1093/ibd/izz218.
- Cane G, Ginouves A, Marchetti S, Busca R, Pouyssegur J, Berra E, Hofman P, Vouret-Craviari V. HIF-1alpha mediates the induction of IL-8 and VEGF expression on infection with Afa/Dr diffusely adhering E. coli and promotes EMT-like behaviour. Cell Microbiol. 2010;12:640–653. doi:10.1111/j.1462-5822.2009.01422.x.
- Kempf VA, Lebiedziejewski M, Alitalo K, Walzlein JH, Ehehalt U, Fiebig J, Huber S, Schutt B, Sander CA, Muller S, et al. Activation of hypoxia-inducible factor-1 in bacillary angiomatosis: evidence for a role of hypoxia-inducible factor-1 in bacterial infections. Circulation. 2005;111:1054–1062. doi:10.1161/01.CIR.0000155608.07691.B7.
- Patel NJ, Zaborina O, Wu L, Wang Y, Wolfgeher DJ, Valuckaite V, Ciancio MJ, Kohler JE, Shevchenko O, Colgan SP, et al. Recognition of intestinal epithelial HIF-1alpha activation by Pseudomonas aeruginosa. Am J Physiol Gastrointest Liver Physiol. 2007;292:G134–142. doi:10.1152/ajpgi.00276.2006.
- Rupp J, Gieffers J, Klinger M, van Zandbergen G, Wrase R, Maass M, Solbach W, Deiwick J, Hellwig-Burgel T. Chlamydia pneumoniae directly interferes with HIF-1alpha stabilization in human host cells. Cell Microbiol. 2007;9:2181–2191. doi:10.1111/j.1462-5822.2007.00948.x.
- Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–1815. doi:10.1172/JCI23865.
- Matak P, Heinis M, Mathieu JR, Corriden R, Cuvellier S, Delga S, Mounier R, Rouquette A, Raymond J, Lamarque D, et al. Myeloid HIF-1 is protective in Helicobacter pylori-mediated gastritis. J Immunol. 2015;194:3259–3266. doi:10.4049/jimmunol.1401260.
- Zhu X, Jiang L, Wei X, Long M, Du Y. Roxadustat: not just for anemia. Front Pharmacol. 2022;13:971795. doi:10.3389/fphar.2022.971795.
- Wang TC, Koh TJ, Varro A, Cahill RJ, Dangler CA, Fox JG, Dockray GJ. Processing and proliferative effects of human progastrin in transgenic mice. J Clin Invest. 1996;98(8):1918–1929. doi:10.1172/JCI118993.
- Wang TC, Dangler CA, Chen D, Goldenring JR, Koh T, Raychowdhury R, Coffey RJ, Ito S, Varro A, Dockray GJ, et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterol. 2000;118(1):36–47. doi:10.1016/S0016-5085(00)70412-4.
- Fox JG, Wang TC, Rogers AB, Poutahidis T, Ge Z, Taylor N, Dangler CA, Israel DA, Krishna U, Gaus K, et al. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterol. 2003;124(7):1879–1890. doi:10.1016/S0016-5085(03)00406-2.
- Noto JM, Rose KL, Hachey AJ, Delgado AG, Romero-Gallo J, Wroblewski LE, Schneider BG, Shah SC, Cover TL, Wilson KT, et al. Carcinogenic Helicobacter pylori strains selectively dysregulate the in vivo gastric proteome, which may be associated with stomach cancer progression. Molecular & Cellular Proteomics: MCP. 2019;18(2):352–371. doi:10.1074/mcp.RA118.001181.