
Abstract
Fluorescent derivatives of actin and actin-binding domains are powerful tools for studying actin filament architecture and dynamics in live cells. Growing evidence, however, indicates that these probes are biased, and their cellular distribution does not accurately reflect that of the cytoskeleton. To understand the strengths and weaknesses of commonly used live-cell probes—fluorescent protein fusions of actin, Lifeact, F-tractin, and actin-binding domains from utrophin—we compared their distributions in cells derived from various model organisms. We focused on five actin networks: the peripheral cortex, lamellipodial and lamellar networks, filopodial bundles, and stress fibers. Using phalloidin as a standard, we identified consistent biases in the distribution of each probe. The localization of F-tractin is the most similar to that of phalloidin but induces organism-specific changes in cell morphology. Both Lifeact and GFP-actin concentrate in lamellipodial actin networks but are excluded from lamellar networks and filopodia. In contrast, the full utrophin actin-binding domain (Utr261) binds filaments of the lamellum but only weakly localizes to lamellipodia, while a shorter variant (Utr230) is restricted to the most stable subpopulations of actin filaments: cortical networks and stress fibers. In some cells, Utr230 also detects Golgi-associated filaments, previously detected by immunofluorescence but not visible by phalloidin staining. Consistent with its localization, Utr230 exhibits slow rates of fluorescence recovery after photobleaching (FRAP) compared to F-tractin, Utr261 and Lifeact, suggesting that it may be more useful for FRAP- and photo-activation-based studies of actin network dynamics.
Abbreviations
| FRAP | = | fluorescence recovery after photobleaching |
Introduction
Several tools are now available to visualize actin filaments in cultured cells, isolated tissues, and whole organisms. The oldest and most widely used of these actin probes are fluorescent derivatives of the filament-binding toxin phalloidin, which is generally assumed to provide the most complete and accurate picture of the actin cytoskeleton. Phalloidin is relatively small and its interaction with actin filaments is generally insensitive to the presence of actin binding proteins. Some derivatives of phalloidin also undergo a fluorescence enhancement upon binding to actin, resulting in extremely high signal to noise ratios.Citation1 Unfortunately, phalloidin does not pass readily through lipid bilayers and must be microinjected to image actin filaments in live cells. In addition, phalloidin binding stabilizes actin filaments, perturbing their normal dynamics.
Genetically encoded actin reporters are much more useful for live cell imaging. These reporters fall into two classes: (i) fluorescent protein-tagged actin and (ii) fluorescent derivatives of peptides and protein domains that bind actin filaments. The second class includes tandem calponin homology domains from the filament-crosslinking protein utrophin (UtrCH or Utr261); an actin-binding peptide from yeast ABP140, called Lifeact; and an actin-binding peptide from rat neuronal inositol 1,4,5-triphosphate 3-kinase A (ITPKA), called F-tractin.Citation2,3,4
Experimental limitations have been previously described for each of these live-cell actin reporters. The contractile ring in yeast, for example, cannot be detected by overexpression of fluorescent protein-tagged actin.Citation5 The actin-binding peptide Lifeact is excluded from actin-rich membrane protrusions in the limb mesenchymal cells of chick embryos and fails to bind cofilin-actin filaments induced by heat shock in STHdh cells.Citation6,7 A truncation of the utrophin actin-binding domain (Utr230), in contrast, uniquely labels short actin filaments in mammalian somatic nuclei.Citation8
One explanation for the exclusion of actin reporters from specific filament structures is an incompatibility between the reporter and the mechanism driving filament assembly. For example, it has been shown that tagged actin is a poor substrate for formin-family actin nucleators.Citation5 Endogenous filament-binding proteins may also occlude the binding sites of filament-binding reporters (and vice-versa). Alternately, reporter binding may be sensitive to the kinetics of filament turnover and filament lifetimes. Thus, the careful choice of an appropriate actin probe is critical to the design of any in vivo study of actin filaments.
To more systematically characterize the bias in live-cell actin probes, and to provide a rational basis for selecting the most appropriate reporter for a given application, we compared the localization and dynamics of the most commonly used reporters in cell lines derived from multiple organisms (Homo sapiens, Mus musculus, Xenopus laevis and Drosophila melanogaster) routinely used in studies of cytoskeletal dynamics. We chose cells that: (i) contain well-characterized actin structures; (ii) derive from widely used model organisms; and (iii) have previously been used to study live-cell actin dynamics. Within each cell line, we compared the distribution of each live-cell actin probe to that of a fluorescent phalloidin derivative. To compare the dynamics of reporter binding to the rate of actin filament turnover, we also performed fluorescence recovery after photobleaching (FRAP) on labeled probes bound to relatively stable actin-based structures.
We uncovered several significant biases in commonly used live-cell actin reporters that are consistent across species. The most commonly used actin reporters, Lifeact and Utr261, have the most limited distributions, likely governed by filament regulatory mechanisms. Poor Lifeact binding and GFP-actin incorporation are common among formin-generated filaments, whereas Utr261 is excluded from multiple models of the lamellipod, the lone Arp2/3-generated actin filament structure included in our analysis. In contrast, the localization of the truncated variant of the utrophin actin-binding domain, Utr230, appears to be restricted to highly stable actin filaments. In addition to being the only reporter to bind punctate nuclear actin filaments in somatic cells,Citation8 Utr230 also detects Golgi-associated filaments of D. melanogaster S2 cells, both of which had been previously undetectable by other actin visualization techniques. Surprisingly, neither of the filament structures bound by Utr230 can be detected by phalloidin staining, suggesting that even the binding of phalloidin is limited to a subset of actin filament architectures. Finally, consistent with results from Drosophila oocytes,Citation9 we find that, of all the live cell actin reporters tested, F-tractin most closely reproduces the distribution observed with phalloidin.
Results
Cell lines and actin probes
We studied the localization and dynamics of actin probes in four cell lines derived from different organisms: macrophage-like S2 cells from D. melanogaster, XTC fibroblasts from X. laevis, U2-OS bone epithelial cells from H. sapiens, and B16-F10 skin epithelial cells from M. musculus. In each cell type we expressed five different commonly used, genetically-encoded actin reporters fused to eGFP, in addition to expression of eGFP alone for use as a control (). For mouse B16-F10, human U2-OS and D. melanogaster S2 cells, we created stable cell lines. Due to a lack of well-established protocols and to technical difficulties associated with selecting stable lines, we employed transient transfection to express actin reporters in X. laevis XTC cells.
Table 1. Actin reporter construct generation
Localization of actin probes in spreading D. melanogaster S2 cells plated on ConA
When plated on the lectin Concanavalin A (ConA), S2 cells adhere strongly and spread radially, generating a thin, continuous membrane protrusion around the cell periphery. Radial protrusion is driven by assembly of actin filaments that form dynamic networks similar to those found at the leading edge of many different polarized, motile cells.Citation10 The most dynamic of these networks, formed close to the plasma membrane by the nucleation and filament branching activity of the Arp2/3 complex, is usually called the lamellipod. Closer to the cell body lies a less dense and slower moving network, often called the lamellum.Citation11
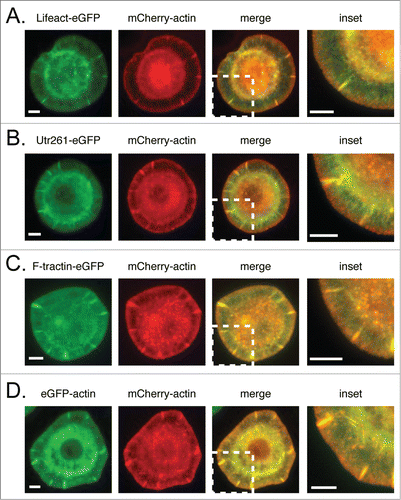
In cells plated on ConA, mCherry-actin concentrates in membrane-proximal actin networks more strongly than either Lifeact-eGFP or Utr261-eGFP, neither of which exhibits the same high concentration near the membrane as mCherry-actin ( and ). Differences between the localization of F-tractin-eGFP and mCherry-actin, in contrast, are relatively minor (). Comparing eGFP- and mCherry-labeled actin in the same cell () demonstrates that these localization differences are not caused by differences between fluorescent proteins, but reflect differential interactions with actin filaments.
Figure 1. Comparison of mCherry-actin localization with eGFP-tragged actin reporters. mCherry-actin and eGFP reporter localization in fixed S2 cells with corresponding linescans in the lamellum and lamellipod for (A–B) F-tractin-eGFP, (C–D) Lifeact-eGFP, (E–F) Utr261-eGFP, (G–H) eGFP actin (control). Scale bars indicate 5 microns.
To determine whether differences in reporter localization reflect biases against specific actin networks, we compared each eGFP-labeled probe to a fluorescent derivative of phalloidin in the same cell. To perform this comparison, we fixed probe-expressing cells with paraformaldehye to preserve the actin cytoskeleton and then stained them with Alexa 568-phalloidin. We then collected a z-series of widefield fluorescence images and chose the closest focal plane to the coverslip for our analysis. We based our conclusions on analysis of at least 20 cells from each probe-expressing cell line.
All the images of live-cell actin probes had lower contrast than the corresponding phalloidin images (). This difference in contrast is due, in part, to fluorescence enhancement of phalloidin derivatives upon binding actin filaments, but it is primarily related to the existence of soluble pools of unbound probe in the cell. The contribution of this pool of unbound probe to our images complicated analysis and so we normalized and compared the live-cell probe and phalloidin images in two different ways. We normalized the images to have either the same maximum fluorescence intensity or the same total integrated fluorescence, and we compared pairs of images by either subtracting one from the other or taking their ratios, respectively. In this work we describe only the biases that were consistently observed by both of these methods and in the majority of analyzed cells.
Figure 2. Comparison of live-cell actin probes and phalloidin in Drosophila S2 cells on ConA. Comparison of Alexa 564 phalloidin localization and eGFP actin reporters in fixed S2 cells on ConA stably expressing (A) eGFP, (B) eGFP-actin, (C) Utr230-eGFP, (D) Utr261-eGFP, (E) F-tractin-eGFP and (F) Lifeact-eGFP. Scale bars indicate 5 microns.
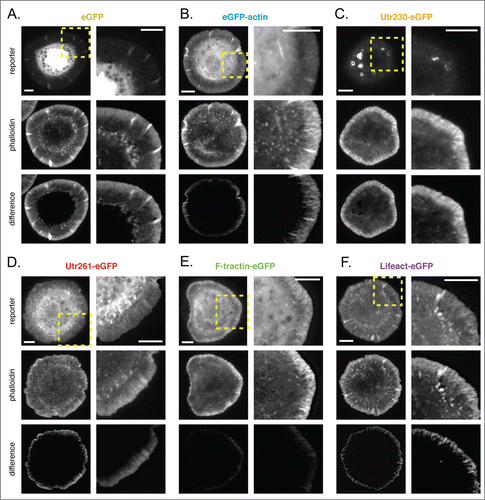
Subtracting images of soluble eGFP from images of fluorescent phalloidin highlights the peripheral actin networks of the lamellipod and lamellum, previously characterized in spreading S2 cells (; ratiometric comparison in Fig. S1A; additional subtraction results in Fig. S2A). In addition to diffuse staining near the membrane, phalloidin also reveals filamentous structures that project radially from the plasma membrane toward the cell body. Given the resolution of our imaging and actin density near the plasma membrane, these structures are almost certainly actin bundles rather than single filaments.
GFP-actin localizes poorly to filamentous structures in the periphery and cell body. The eGFP-actin probe accumulates in the cell periphery but its concentration does not rise as steeply as that of phalloidin and it does not display the same pattern of radial, filamentous structures (; Fig. S1B; Fig. S2B). This is particularly obvious in the difference images generated by subtracting the normalized eGFP-actin intensity from that of phalloidin (, bottom).
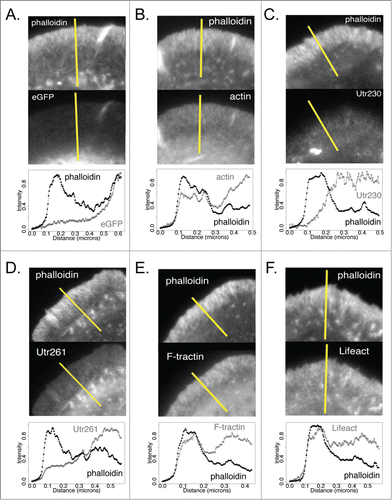
Interestingly, F-tractin-eGFP more closely reproduces the pattern of phalloidin staining in the peripheral actin networks of S2 cells compared to eGFP-actin. The F-tractin probe reveals the same radial, filamentous actin structures as phalloidin (; Fig. S1E; Fig. S2E) and its concentration rises more sharply near the plasma membrane than eGFP-actin (). Subtracting a normalized F-tractin image from its corresponding phalloidin image reveals almost no systematic difference in localization near the leading edge (, bottom).
Probes based on either the actin-binding domain of utrophin or the Lifeact peptide fail to recognize membrane-proximal, lamellipodial actin networks in spreading S2 cells. The longer utrophin-based probe, Utr261-GFP, reveals radial filamentous structures in the cell periphery but, unlike both phalloidin and GFP-actin, this probe does not display a steep rise in accumulation near the plasma membrane. This is clear in difference images in which the Utr261 signal is subtracted from the phalloidin signal (, bottom; Fig. S1D; Fig. S2D) as well as in linescans of Utr261-eGFP fluorescence, which reveal that the probe concentration remains almost constant from the cell edge inward to the cell body (). The Lifeact-based probe has a similar distribution to that of Utr260 but accumulates slightly more near the plasma membrane (; ; Fig. S1F; Fig. S2F), in the region occupied by lamellipodial actin networks.
Figure 3. Intensity plot profiles of actin probes and phalloidin in DrosophilaS2 cells on ConA Comparison of intensity profiles across lines (shown in yellow) for Alexa 564 phalloidin (top) and eGFP actin reporters (bottom) in fixed S2 cells on ConA. (A) eGFP, (B) eGFP-actin, (C) Utr230-eGFP, (D) Utr261-eGFP, (E) F-tractin-eGFP and (F) Lifeact-eGFP.
Finally, a truncated version of the actin binding domain from utrophin (residues 1-230), Utr230-eGFP, is entirely absent from lamellar and lamellipodial actin networks in spreading S2 cells but localizes to ring-shaped structures in the cell body that are not detected by phalloidin (; Fig. S1C; Fig. S2C). Interestingly, we recently characterized these structures as elements of the Golgi apparatus.Citation12 Some actin-binding proteins have previously been shown to localize to the Golgi apparatus, and several studies have reported the presence of filamentous actin associated with this organelle.Citation13,14 If the localization of our Utr230 probe in S2 cells is driven by its interaction with actin, this probe might prove useful in studying the function and dynamics of Golgi-associated cytoskeletal structures.
Localization of actin probes in non-spreading D. melanogaster S2 cells plated on poly-D-lysine
Drosophila S2 cells adhere more weakly to surfaces coated with poly-D-lysine (PDL) than to surfaces coated with ConA. S2 cells do not spread on PDL-coated surfaces but remain more-or-less spherical, occasionally blebbing or projecting actin-rich filopodia. These non-spreading cells are well suited to visualizing three actin-based structures: (1) the thin, membrane-proximal cell cortex; (2) dynamic filopodial bundles; and (3) smaller rod-shaped or globular structures within the cell body. All three of these structures appear in difference images generated by subtracting normalized soluble eGFP signal from that of fluorescent phalloidin (; additional difference images shown in Fig. S3A).
Figure 4. Comparison of live-cell actin probes and phalloidin in DrosophilaS2 cells on PDL. Comparison of Alexa 564 phalloidin localization and eGFP actin reporters in fixed S2 cells on PDL stably expressing (A) eGFP, (B) eGFP-actin, (C) Utr230-eGFP, (D) Utr261-eGFP, (E) F-tractin-eGFP and (F) Lifeact-eGFP. Scale bars indicate 5 microns.
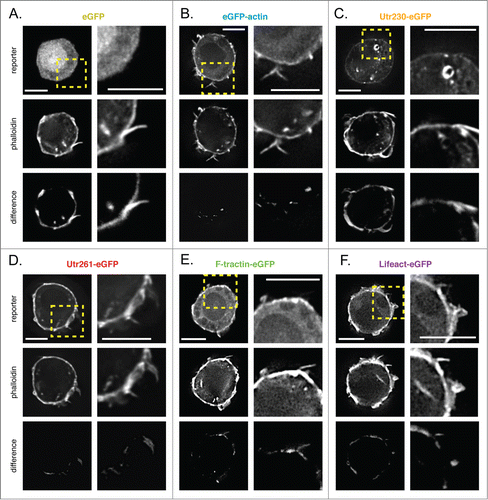
eGFP-actin accumulates in the cortex and in filopodia but fails to recognize actin structures in the cell body (; Fig. S3B). Subtracting normalized eGFP-actin from phalloidin images reveals very little difference in localization near the plasma membrane of these cells (, bottom).
Utr261-, F-tractin-, and Lifeact-eGFP accumulate to a similar degree in cortical actin networks and are all partially excluded from filopodial actin bundles (; Fig. S3D–F; Fig. S4). Soluble eGFP is also absent from filopodia in S2 cells (; Fig. S4), so it is likely that these actin filament-binding probes simply fail to diffuse into short-lived and rapidly growing filopodia. Of all the live cell probes, only Utr261-eGFP recognizes the globular, phalloidin-stained structures found in the cell body.
As in spreading S2 cells, the Utr230-eGFP probe localizes to the Golgi membranes in the cell body of rounded S2 cells. Otherwise, this probe is excluded from all other actin filament-containing structures, except for a slight accumulation in the cortex (; Fig. S3C).
Comparison of live-cell actin probes in XTC cells from X. laevis
XTC cells are a mesoderm-derived cell line from the African clawed frog, X. laevis. When plated on poly-L-lysine (PLL), XTC cells adhere tightly and spread by radial extension of a thin, actin-rich protrusion. The morphology and dynamics of spreading XTC cells are similar to those of D. melanogaster S2 cells plated ConA-coated surfaces. The architecture and dynamics of the peripheral actin networks in spreading XTC cells are characteristic of the lamellipodial and lamellar networks that underlie membrane protrusions in many organisms. These cells also produce dynamic filopodial protrusions. Difference images generated by subtracting normalized images of soluble eGFP from phalloidin highlight a broad peripheral actin network, containing well-defined radial filament bundles that occasionally project beyond the plasma membrane to form filopodia (; additional difference images shown in Fig. S5A).
Figure 5. Comparison of live-cell actin probes and phalloidin in XenopusXTC cells on PLL. Comparison of Alexa 564 phalloidin localization and eGFP actin reporters in fixed XTC cells transiently expressing (A) eGFP, (B) eGFP-actin, (C) Utr230-eGFP, (D) Utr261-eGFP, (E) F-tractin-eGFP and (F) Lifeact-eGFP. Scale bars indicate 5 microns.
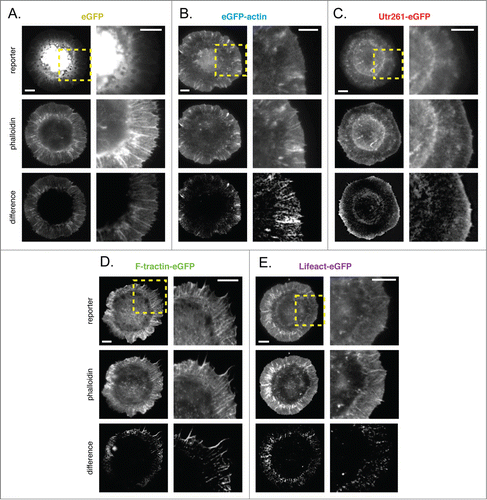
As in S2 cells, eGFP-actin is enriched near the plasma membrane of XTC cells but only weakly incorporated into radial filament bundles (; Fig. S5B). Also as in S2 cells, Utr261-eGFP is conspicuously absent from the most membrane-proximal regions of the actin network, the region enriched in Arp2/3-generated lamellipodial actin networks. The Utr261-based probe accumulates in a zone more distal from the cell edge and closer to the cell body, a zone characterized by slower-moving lamellar actin networks. Similar to eGFP-actin, Utr261 does not incorporate well into radial filament bundles in the cell periphery (; Fig. S5C). The F-tractin- and Lifeact-based probes accumulate in both lamellar and membrane-proximal lamellipodial actin networks. These probes, however, fail to accumulate to the same extent as phalloidin in the densest radial filament bundles as well as some filopodia (; Fig. S5D–E). Importantly, expression of F-tractin-eGFP induces an aberrant morphology in XTC cells, producing dense, radial actin bundles as well as significantly more and significantly longer filopodia than untransfected cells or cells expressing other reporters (Fig. S6). This effect makes F-tractin problematic for imaging actin dynamics in XTC cells.
Localization of Utr230-eGFP in XTC cells could not be determined as we could not generate cells expressing significant amounts of this probe. We detected no fluorescence in XTC cells transiently transfected with Utr230-eGFP, and we were unable to select cell lines stably expressing Utr230-based probes. Bacterially expressed Utr230 has poor solubility (unpublished data), so it is possible that the construct is quickly degraded when not bound to filaments.
Comparison of live-cell actin probes in mouse B16-F10 cells
Mouse B16-F10 cells are adherent and, when plated on laminin-coated substrates, they adopt a predominantly epithelial morphology. When plated at relatively low densities, many B16-F10 cells appear polarized, with asymmetric membrane protrusions similar to those observed in more rapidly migrating cells. Membrane protrusions ruffle continually at the periphery of B16-F10 cells and are more highly dynamic than those produced by either XTC or S2 cells. Subtracting normalized soluble eGFP images from images of phallodin-staining highlights several actin structures: a thin, membrane-proximal lamellipodial network, radial filament bundles that sometimes project into filopodia-like protrusions, and faint transverse actin ‘arcs’ adjacent to the cell body (; additional difference images shown in Figure S7A). Phalloidin also recognizes ventral stress fibers in the cell body but these structures are obliterated by subtraction of the soluble eGFP signal, which is highest in the cell body.
Figure 6. Comparison of live-cell actin probes and phalloidin in mouse B16-F10 cells on laminin. Comparison of Alexa 564 phalloidin localization and eGFP actin reporters in fixed B16-F10 cells stably expressing (A) eGFP, (B) eGFP-actin, (C) Utr230-eGFP, (D) Utr261-eGFP, (E) F-tractin-eGFP and (F) Lifeact-eGFP.
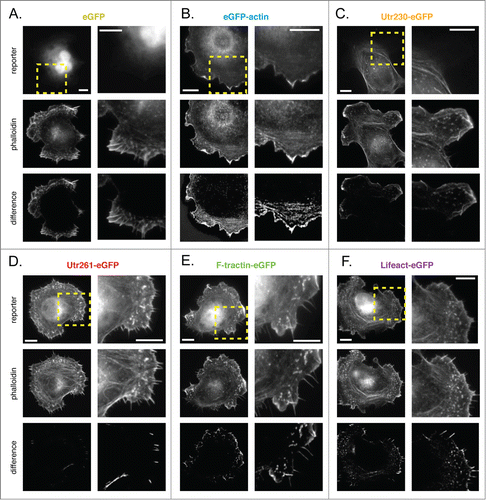
eGFP-actin is enriched in lamellipodial networks adjacent to the membrane of B16-F10 cells, but does not incorporate well into radial filament bundles or transverse actin arcs (; Fig. S7B). As in XTC and S2 cells, Utr230-eGFP is entirely excluded from the membrane-proximal lamellipodial network of B16-F10. This probe also fails to accumulate in radial filament bundles and filopodia, but does label transverse actin arcs as well as ventral stress fibers in the cell body (; Fig. S7C).
In B16-F10 cells the localizations of Utr261, F-tractin, and Lifeact all more-or-less match that of phalloidin in lamellipodia and stress fibers (; Fig. S7D–F). All three of these constructs are, however, excluded from filopodia and radial lamellar filament bundles. As in the filopodia of XTC and S2 cells, it is unclear whether exclusion results from binding preferences or restricted diffusion.
Comparison of live-cell actin probes in human U2-OS cells
U2-OS cells are derived from a human osteosarcoma and, when plated on fibronectin, have a typical adherent, epithelial morphology. The major actin structures revealed by phalloidin are ventral stress fibers running through the cell body and, in some cells, a membrane-proximal actin cortex (; additional difference images shown in Fig. S8A).
Figure 7. Comparison of live-cell actin probes and phalloidin in human U2-OS cells on fibronectin. Comparison of Alexa 564 phalloidin localization and eGFP actin reporters in fixed U2-OS cells stably expressing (A) eGFP, (B) eGFP-actin, (C) Utr230-eGFP, (D) Utr261-eGFP, (E) F-tractin-eGFP and (F) Lifeact-eGFP. Scale bars indicate 5 microns.
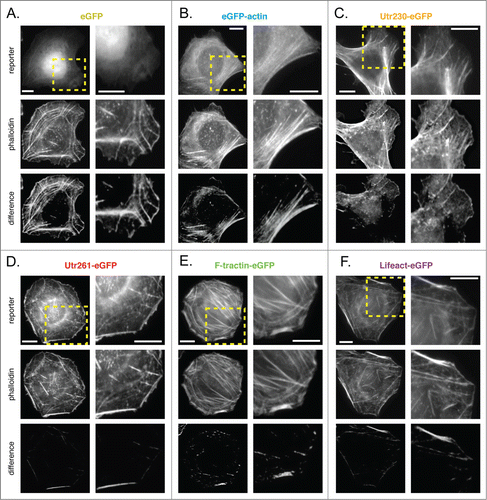
As in B16-F10 cells, eGFP-actin does not incorporate well into stress fibers but accumulates at the cell periphery (; Fig. S8B). Utr261-eGFP, F-tractin-eGFP and Lifeact-eGFP all detect cortical actin networks and stress fibers to the same degree as phalloidin. The major exceptions to this rule are the densest stress fiber bundles (; Fig. S8D-F). Most likely, filaments buried inside the bundles are more accessible to the small molecule phalloidin but inaccessible to the much larger eGFP-fusion reporters. As in other cell types, Utr230-eGFP localizes to stress fibers, but is wholly excluded from cortical actin and other short actin filaments throughout the cell body (; Fig. S8C).
FRAP analysis suggest that probe preferences are related to actin network dynamics.
To assess the suitability of live-cell actin probes for fluorescence recovery studies of actin dynamics, and to determine whether biases in a probe's localization correlate with the lifetime of its interaction with an actin filament, we photobleached probes associated with lamellipodia in B16-F10 and stress fibers in U2-OS cells. We chose these structures because they have different rates of filament turnover and because they were recognized by most of our live-cell actin probes (Movies S1-S9). We quantified recovery of fluorescence in photobleached spots and then fit normalized recovery curves using single, double, and triple exponentials (). From fitting, we computed the fractions of freely diffusing and filament-bound probe and the halftime (t1/2) for recovery of each fraction ().
Table 2. In vivo dynamics of actin reporters
Figure 8. FRAP of actin reporters at U2-OS cell stress fibers and B16-F10 lamellipodia. (A) Normalized FRAP recovery curves for actin reporters on U2-OS cell stress fibers. Color scheme follows: cyan, eGFP-actin; red, Utr261-eGFP; purple, Lifeact-eGFP; green, F-tractin-eGFP; orange, Utr230-eGFP. (B) Inset from (A) showing early recovery of Utr261-eGFP, F-tractin-eGFP and Lifeact-eGFP. (C) Normalized FRAP recovery curves for actin reporters in the mouse B16-F10 lamellipod. (D) Inset from (C) showing early recovery of Utr261-eGFP, F-tractin-eGFP and Lifeact-eGFP.
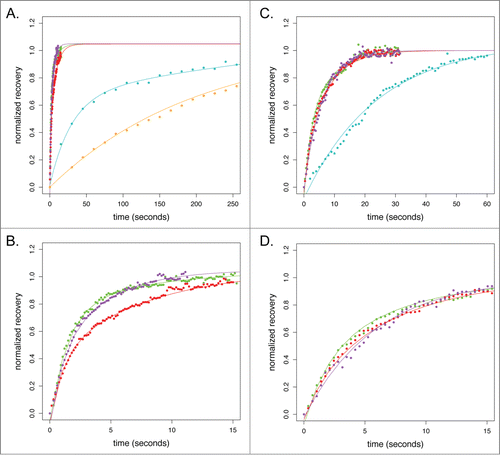
The t1/2 for recovery of eGFP-actin in mouse lamellipodia is 35 seconds while t1/2 for recovery in human stress fibers is 341 seconds. We assume that the recovery of eGFP-actin most accurately reflects turnover of filaments within each structure and our measured recovery rates are in good agreement with previous studies ( and ; ).Citation15,16 Similar recovery times for the other live-cell probes would suggest that their dissociation is slower than the turnover rate of the filaments to which they are bound. Significantly faster recovery rates would suggest that probe dissociation is too fast for this probe to be useful in FRAP studies of filament dynamics. Significantly slower rates of recovery would suggest that binding of the probe slows the rate of filament turnover.
Although actin filaments in human stress fibers turn over ten times more slowly than filaments in murine lamellipodia, the t1/2 for recovery of filament-bound Lifeact, Utr261, and F-tractin in both structures is <10 seconds. These fast dynamics suggest that fluorescence recovery depends almost entirely on probe binding kinetics rather than actin filament turnover (; ). These probes, therefore, are not suitable for FRAP studies.
Surprisingly, the recovery rate of stress fiber-associated Utr230-eGFP (t½ of 217 seconds) is much closer to that measured using eGFP-actin (; ). Since the recovery curves could only be fit with a single exponential and the contribution of the freely diffusing pool of Utr230 could not be detected, this value more likely represents the lower limit of the bound fraction recovery half time. The slow binding kinetics of Utr230 implied by our FRAP data suggest that Utr230 can only bind to highly stable actin structures.
Discussion
Our results argue that there is no ‘perfect’ probe for studying the actin cytoskeleton in live cells. Even when expressed in the same cell, different actin probes display different patterns of localization as well as different dynamics (). Moreover, none of the live-cell actin probes we tested exactly reproduces the localization pattern of phalloidin. Great care must therefore be taken when interpreting images and time-lapse movies of live-cell actin probes. We suggest three basic rules for selecting and using live-cell actin probes: (1) a probe should efficiently incorporate into the cytoskeletal structure under study; (2) expression of the probe should not alter cell morphology or behavior; and (3) conclusions should be based on experiments performed with multiple, different probes. Although biases in the localization of actin probes have clear potential to introduce experimental artifacts, they also present an exciting opportunity. Once we understand the mechanisms by which various probes are excluded from certain populations of filaments, we can then use these probes to study biochemical differences between actin networks in vivo. In other words, biochemically defined actin probes could become powerful tools, providing mechanistic insight into the formation of actin networks in live cells.
Table 3. Binding preferences of in vivo actin reporters
Several factors could account for differences in live-cell actin probe localization, including: (1) local barriers to probe diffusion; (2) incompatibility or competition with endogenous actin-associated proteins; and (3) differential rates of actin filament turnover. The fact that only eGFP-actin incorporates well into filopodia, for example, likely reflects poor penetration of filament-binding probes into the filopodial compartment. Weak incorporation of eGFP-actin into stress fibers and lamellar filament bundles, on the other hand, probably reflects biophysical constraints imposed by actin-binding proteins associated with these structures. Previous work revealed, for example, that fusion of fluorescent proteins to actin monomers makes them poor substrates for formin-family nucleation and elongation factors,Citation5 so filaments created by formin-family proteins generally exclude eGFP-actin. Finally, the absence of utrophin-based probes from Arp2/3-generated lamellipodial actin networks likely reflects either occlusion of the Utr261 binding site by other filament side-binding proteins or possibly the unique geometry of these networks.
We observed the clearest biases between probes in their ability to bind dynamic lamellipodial actin networks. Judged by their ability to recapitulate the steep rise in phalloidin staining near advancing or ruffling membranes (), the probes we tested recognize lamellipodial actin networks in the following order: F-tractin>eGFP-actin>Lifeact>Utr261>>Utr230.
In addition to the fidelity with which they reveal the architecture of the actin cytoskeleton, we must also pay attention to the effects of live-cell actin probes on cell morphology and cytoskeletal function. In our hands the F-tractin-based probe most accurately reproduced actin structures visualized by phalloidin in a wide range of cells, but expression of this probe in Xenopus XTC cells perturbed their overall morphology and altered the organization of the actin cytoskeleton. On the other hand, a recent study that employed multiple fluorescent probes to label actin networks in various stages of Drosophila oogenesis found that expression of Utr261 or Lifeact in nurse cells can perturb the cytoskeleton and produce sterility. This study also found that fluorescent F-tractin fusions labeled multiple actin networks in nurse cells without perturbing cell morphology or function.Citation9
With the possible exception of the truncated utrophin construct, Utr230, none of the filament-binding probes is suitable for photo-bleaching or photo-activation studies of actin dynamics. Interestingly, while the Utr230-based probe does not label dynamic lamellipodial or lamellar networks, it dissociates much more slowly from stable actin structures than its longer cousin, Utr261. The fluorescence recovery kinetics of Utr230 bound to stress fibers are similar to those we previously reported for Utr230-based probes bound to filamentous actin in somatic nuclei,Citation8 suggesting that this rate reflects the slow dissociation of Utr230 from actin. Although in most cells it binds to stress fibers, in Drosophila S2 cells Utr230 associates with Golgi membranes. More work will be required to determine whether this probe actually reveals an endogenous Golgi-associated actin network, but previous studies provide evidence for filamentous actin associated with Golgi membranes (reviewed in ref. 17).Citation12 These Golgi-associated filaments were not recognized by phalloidin but reacted with actin-specific antibodies, raising the more general question of what fraction of actin filaments in a cell are detectable by phalloidin. Stress-induced cofilin-actin rods, for example, do not bind phalloidin and, in vitro, cofilin disrupts the phalloidin binding site on actin filaments by increasing their helical twist.Citation18,19
Materials & Methods
Molecular biology
Utrophin 230 and Utrophin 261 were cloned from full-length human cDNA (Open Biosystems). Human actin was cloned from a full-length human recombinant construct. Drosophila actin was cloned from Drosophila cDNA.10 F-tractin and Lifeact sequences were generated by gene synthesis (GenScript) and annealed primers, respectively. We used pEGFP-C1 (Clontech) as the host vector for EGFP fusions in B16-F10, U2-OS and XTC cells, with N-terminal EGFP fusions inserted into the unique AgeI and NheI sites. For S2 cell expression, all EGFP fusions were subcloned into the pMT copper inducible protomer vector using Gateway cloning technology (Invitrogen).
Cell lines and culturing
Human U2-OS and mouse B16-F10 cells were grown in Dulbecco's Modified Eagle Medium supplemented with 10% FBS and penicillin-streptomycin (UCSF Cell Culture facility) at 37°C with 5% CO2. Xenopus XTC cells were grown in 66% Liebovitz L-15 media diluted in sterile-filtered water and supplemented with 10% FBS and penicillin-streptomycin (UCSF Cell Culture facility) at 25°C. Transfections for the these cell lines were performed using Lipofectamine LTX (Invitrogen) using the standard manufacturer's protocol. Stable lines of U2-OS and B16-F10 cells were selected in media supplemented with 0.5 mg/mL G418 (Roche). Stable XTC cells could not be generated and transient transfections were instead performed. Drosophila S2 cells were grown in Schneider's medium (Invitrogen) supplemented with 10% FBS and penicillin-streptomycin (UCSF Cell Culture facility) at 25C. Transfections were performed using Effectene transfection reagent (Qiagen) using the standard manufacturer's protocol. Stable S2 cell lines were selected in complete media supplemented with 450 ug/mL Hygromycin (Life Technologies).
Imaging and Analysis
Each cell line was plated on clean glass coated with the corresponding preferred substrate. Coverslips for B16-F10 cells were coated with 0.01% poly-L-lysine (Sigma) in PBS for 15 minutes at room temperature (RT), washed in PBS and coated with 25 ug/mL purified mouse laminin in PBS for 2 hours at 37°C. B16-F10 cells were allowed to settle onto coverslips overnight before fixation. Coverslips for U2-OS cells were coated with 10 ug/mL fibronectin, incubated in PBS overnight at 4°C. U2-OS cells were allowed to settle onto coverslips overnight before fixation. XTC coverslips were coated with 0.01% poly-L-lysine for 15 minutes at RT. XTC cells were pelleted and resuspended in serum-free medium before plating to enhance spreading, and were fixed 60 minutes after plating. Expression of fusion proteins in S2 cells was induced with 100 uM CuSO4 overnight. Coverslips for S2 cells spreading on ConA were coated with 1:5 dilution of ConA in ddH2O and allowed to dry overnight. S2 cells were allowed to adhere and spread on ConA for at least two hours before fixation. Coverslips for S2 cells adhering to poly-D-lysine (PDL, Sigma) were coated with 1 mg/mL PDL in 10 mM Tris Buffer, pH 8 for 2 hours at RT, washed twice with ddH2O and allowed to dry at RT. S2 cells were plated and allowed to adhere to PDL coated surfaces for at least one hour before fixation.
All cells were fixed for 15 minutes at RT in 4% paraformaldehyde (Electron Microscopy Sciences) in PBS (B16-F10, U2-OS, XTC, and S2 cells on PDL) or in 1X HL3 bufferCitation20 (S2 cells on ConA). Coverslips were stained with 0.7 U/ml Alexa Fluor 568-phalloidin (Invitrogen) in 0.1% Triton-X 100 (Sigma) in PBS for 15 minutes at RT, then washed in PBS and mounted onto slides with Dako mounting media (Agilent). Phalloidin-stained cells were imaged using a DeltaVision RT system (Applied Precision) with a Photometrics CoolSnapHQ camera using a 100x 1.40NA UPlanSApo objective (Olympus). All images were collected as 0.25 micron z-stacks, with image stacks collected for >20 cells for each cell line/actin reporter combination.
Comparative image analysis was performed using ImageJ (National Institutes of Health). For subtraction-generated images, each image was converted to RGB and scaled to an intensity of 0–100, following background subtraction and removal of outlier pixels above or below 1.5 standard deviations from the mean. Images from the EGFP channel were subtracted from the corresponding image in the phalloidin channel, and a grayscale difference image was generated. All pixels in the difference image with values < = 0 are given an intensity value of 0, so that pixels in which the normalized phalloidin signal is lower than the normalized EGFP signal are not displayed.
For ratiometric images, each image was converted to RGB and pixel values were divided by the average pixel intensity, following background subtraction and removal of outlier pixels above or below 1.5 standard deviations from the mean. Images from the EGFP channel were divided from the corresponding image in the phalloidin channel, and a grayscale ratiometric image was generated. All pixels in the difference image with values < = 1 are given an intensity value of 0 (e.g. intensities form 0-1 indicate pixels where the normalized phalloidin signal is greater than the normalized EGFP signal).
For linescan analysis, plot profiles of unprocessed images were calculated by ImageJ. These plot profiles were normalized and figures were generated using the R software package.
FRAP Imaging and Analysis
Cell plating for live cell imaging was performed on substrate-coated glass as described above, in 35 cm diameter glass-bottom dishes (MatTek). U2-OS and B160-F10 cells were imaged at 37°C. FRAP experiments were performed on a DeltaVision RT system, also described above, with a 488nm laser. Fifteen to twenty movies were collected for each cell line/actin reporter combination, with post-bleaching intervals as indicated in the Results. Images were corrected for photobleaching and intensities in the bleached ROI were measured in ImageJ (National Institutes of Health) for each movie. Average recovery curves and curve normalizations were performed in R and curves were fit to single, double and triple exponential curves using CurveExpertPro with the following equations, respectively: y = k-A1*e(-x/t1); y = k-A1*e(-x/t1) A2*e(-x/t2); y = k-A1*e(-x/t1)-A2*e(-x/t2)-A3*e(-x/t3). An initial value of 1 as an estimate for k, based on the predicted plateau of the normalized recovery curves, was provided for improved parameter fitting. Plots of raw data and calculation of recovery equations, fast and slow fractions and their corresponding t1/2 values were generated in the R software package.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental_Figures.zip
Download Zip (4.8 MB)Supplemental_Files.zip
Download Zip (11.1 MB)Acknowledgements
We are grateful to the laboratory of Elizabeth Blackburn for use of their DeltaVision microscope and to Beth Cimini for training and technical assistance in performing FRAP experiments. We thank Scott Hansen for stocks and protocols for the culture of X. laevis XTC and mouse B16-F10 cells, Elena Ingerman and Eric Griffis for S2 cells lines, expression constructs, and protocol assistance, and Lillian Fritz-Laylin and members of the Ron Vale laboratory for thoughtful discussions on the use of actin reporter constructs.
Funding
The bulk of this work was supported by grants from the National Institutes of Health to RDM (GM061010 and GM079556) and funding from the Howard Hughes Medical Institute. Additional support was provided by a National Science Foundation Predoctoral Fellowship (BJB and LMG) and a National Institutes of Health Ruth L. Kirschstein Predoctoral Fellowship (BJB).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- De La Cruz, EM, Pollard, TD. Transient kinetic analysis of rhodamine phalloidin binding to actin filaments. Biochemistry 1994; 33:14387-14392
- Riedl, J., Crevenna, AH, Kessenbrock, K., Yu, JH, Neukirchen, D., Bista, M., Bradke, F., Jenne, D., Holak, TA, Werb, Z., Sixt, M., Wedlich-Soldner, R. Lifeact: a versatile marker to visualize F-actin. Nat. Meth. 2008; 5:606-607
- Burkel, BM, Von Dassow, G. and Bement, WM Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil. Cytoskeleton 2007; 64:822-832
- Schell, MJ, Erneux, C., Irvine, RF Inositol 1,4,5-trisphosphate 3-kinase A associates with F-actin and dendritic spines via its N terminus. J. Biol. Chem. 2001; 276(40):37537-46
- Chen, Q., Nag, S., TD. Formins filter modified actin subunits during processive elongation. J Struct Biol. 2012; 177(1):32-9
- Sanders, TA, Llagostera, E., Barna, M. Specialized filopodia direct long-range transport of SHH during vertebrate tissue patterning. Nature 2013; 497(7451)
- Munsie, LN, Caron, N., Desmond, CR, Truant, R. Lifeact cannot visualize some forms of stress-induced twisted F-actin. Nat. Methods 2009; 6(317)
- Belin, BJ, Cimini, BA, Blackburn, EH, Mullins, RD. Visualization of actin filaments and monomers in somatic cell nuclei. Mol. Biol. Cell 2013; 24(7):982-94
- Spracklen, AJ, Fagan, TN, Lovander, KE, Tootle, TL. The pros and cons of common actin labeling tools for visualizing actin dynamics during Drosophila oogenesis. Dev. Biol. 2014; 393(2):209-26
- Iwasa, JH, Mullins, RD. Spatial and temporal relationships between actin-filament nucleation, capping and disassembly. Curr. Biol. 2007; 17(5):395-406
- Ponti, A., Machacek, M., Gupton, SL, Waterman-Storer, CL, Danuser, G. Two distinct actin networks drive the protrusion of migrating cells. Science 2004; 305(5691):1782-6
- Goins, LM, Mullins, RD. A novel tropomyosin isoform functions at the mitotic spindle and Golgi in Drosophila. Mol Biol Cell. 2015; 26(13):2491-504.
- Kondylis, V., van Nispen tot Pannerden, HE, Herpers, B., Friggi-Grelin, F., Rabouille, C. The Golgi comprises a paired stack that is separated at G2 by Modulation of the Actin Cytoskeleton through ABi and Scar/WAVE. Dev. Cell 2007; 12:901-915
- Percival, JM, Hughes, JA, Brown, DL, Schevzov, G., Heimann, K., Vrhovski, B., Bryce, N., Stow, JL, Gunning, PW. Targeting of a tropomyosin isoform to short microfilaments associated with the Golgi complex. Mol. Biol. Cell 2004; 15:268-280
- Hotulainen, P., Lappalainen, P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J. Cell Biol. 2008; 17(3):383-394
- Hotulainen, P., Paunola, E., Vartiainen, MK, Lappalainen, P. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol. Biol. Cell 2005; 16(2):649-664
- Gurel, PS., Hatch, AL, Higgs, HN. Connecting the cytoskeleton to the endoplasmic reticulum and Golgi. Curr. Biol. 2014; 24(14):R660-R672
- Minamide, LS, Striegl, A.M., Boyle, JA, Meberg, PJ, Bamburg, JR. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat. Cell Biol. 2000; 2:628-636
- McGough, A., Pope, B., Chiu, W., Weeds, A. Cofilin changes the twist of F-actin: implications for actin filament dynamics and cellular function. J. Cell Biol. 1997; 138(4):771-781
- Rogers, SL, Wiedemann, U., Stuurman, N., Vale, RD. Molecular requirements for actin-based lamella formation in Drosophila S2 cells. J. Cell Biol. 2003; 162(6): 1079