
ABSTRACT
Epithelial-to-mesenchymal transition (EMT) is physiological in embryogenesis and wound healing but also associated with the formation of cancer stem cells (CSCs). Many EMT signaling pathways are implicated in CSC formation, but the precise underlying mechanisms of CSC formation remain elusive. We have previously demonstrated that PKC is critical for EMT induction and CSC formation in inducible breast EMT/CSC models. Here, we used formaldehyde-assisted isolation of regulatory elements-sequencing (FAIRE-seq) to investigate DNA accessibility changes after PKC activation and determine how they influence EMT and CSC formation. During EMT, DNA accessibility principally increased in regions distant from transcription start sites, low in CpG content, and enriched with chromatin enhancer marks. ChIP-sequencing revealed that a subset of these regions changed from poised to active enhancers upon stimulation, with some even more acteylated in CSCs. While regions with increased accessibility were enriched for FOX, AP-1, TEAD, and TFAP2 motifs, those containing FOX and AP-1 motif were associated with increased expression of CSC-associated genes, while those with TFAP2 were associated with genes with increased expression in non-CSCs. Silencing of 2 members of the FOX family, FOXN2 and FOXQ1, repressed CSCs and the mesenchymal phenotype and inhibited the CSC gene signature. These novel, PKC-induced DNA accessibility regions help explain how the epigenomic plasticity of cells undergoing EMT leads to CSC gene activation.
Introduction
Cancer stem cells (CSCs) are a distinct population of tumor cells with high proliferative and survival potential. CSCs have the capacity to self-renew and generate heterogeneous cancer cells, have high tumourigenic potential, and tend to be more chemoresistant.Citation1,2 CSCs exist in different cancers and, accordingly, are implicated in driving tumor initiation, dissemination, and resistance to therapy.Citation3
Breast cancers have been robustly categorised based on their gene expression profiles into different molecular subtypes related to breast epithelial cell differentiation.Citation4,5The CSC gene expression signature is associated with the aggressive claudin-low subtype derived from mammary stem cells (MaSCs), which are progenitors of both the luminal and myoepithelial cells that line the normal breast duct.Citation4 The basal-like subtype is likely to be derived from luminal progenitors, but it is unclear if the HER2+ and luminal subtypes arise from luminal progenitors or by ‘de-differentiation’ of existing luminal cells.Citation5 While epigenomic changes (such as H3K27me3 deposition) are known to occur during mammary epithelial development,Citation6 the evidence suggests that these cells maintain plasticity and can obtain the properties and expression profiles of the more de-differentiated state.Citation7
Numerous studies have demonstrated that epithelial-to-mesenchymal transition (EMT) is critical to CSC development.Citation8-10 EMT is a differentiation process by which epithelial cells undergo cytoskeletal reorganisation, loss of cell-cell junctions, and loss of apical–basal polarity, culminating in the acquisition of a mesenchymal phenotype that endows cells with increased motility and invasiveness.Citation9,11 EMT is a normal physiological process in embryonic developmentCitation12 and wound healing.Citation13 Hepatocyte growth factor (HGF), epidermal growth factor (EGF),Citation14 platelet-derived growth factor (PDGF),Citation15 insulin-like growth factor (IGF),Citation16 and transforming growth factor-b (TGF-β)Citation17 activate the Wnt, Notch, and Ras-ERK signaling pathways to induce EMT. The coordinated action of these pathways activates a set of EMT-inducing transcription factors (TFs) including Snail, Slug, Twist, and Zeb that collaborate to initialise and maintain the mesenchymal phenotype.Citation18 The recruitment of a variety of cells including lymphocytes, fibroblasts, and granulocytes to tumor-associated stroma creates an inflammatory microenvironment that delivers pro-EMT signals to the tumor.Citation19 NF-κB is a master regulator of inflammatory signaling that has specifically been shown to be critical for inducing and maintaining EMT in breast cancer.Citation20 Furthermore, we have previously reported that the signaling kinase protein kinase C-theta (PKC-θ) promotes this inflammatory pathway during EMT and CSC formation by acting as a critical chromatin-anchored switch for inducible genes via cross-talk with the TGF-β pathway.Citation21
One challenge in understanding transcriptional regulation is that enhancer regulatory elements can occur far from transcription start sites (TSSs). When used with sequencing, the formaldehyde-assisted isolation of regulatory elements (FAIRE-seq) technique facilitates the discovery of regulatory elements by unbiased mapping of open chromatin regions. This is achieved by exploiting the difference in densities of chromatin-bound and exposed DNA regions to isolate and characterize the latter, which are more likely to be involved in gene regulation. Isolation and sequencing of open regions provides genome-wide insights into the sequence-specific regulatory factors that may associate with these regions. FAIRE-seq has been used to identify regulatory regions controlling cellular differentiation,Citation22 pluripotency,Citation23 and carcinogenesis.Citation24
Here we sought to identify novel DNA accessibility changes that expose or conceal regulatory elements during EMT and determine whether these regions are likely to influence CSC gene expression. Since CSCs drive tumourigenesis and drug resistance, the identification of unique CSC properties would be useful so that they can be exploited for therapy. Although some of the genes and signaling pathways that contribute to EMT and CSC formation are characterized, the epigenetic events that contribute to these processes are less well understood.
We applied FAIRE-seq and expression array profiling to a well-characterized in vitro inducible model of EMT/CSCs, referred to as the MCF-IM model.Citation21 In this MCF-IM model, phorbol myristyl acetate (PMA), a key protein kinase C activator, induces EMT in the luminal human MCF-7 breast cancer cell line and generates a characteristic CD44high/CD24low CSC subpopulation.Citation21 We show that thousands of regions exhibit altered DNA accessibility during PMA-induced EMT, a large number of which occur at regions distant from TSSs and coincident with enhancer marks. Furthermore, novel accessibility changes are present near PMA-induced genes, a proportion of which are in the vicinity of genes exhibiting significantly altered expression in the CSC subpopulation. These data highlight how transcriptional plasticity develops via alterations in DNA accessibility in cells undergoing EMT. In this way, cells that have undergone EMT rapidly alter their transcriptional program according to newly emerged regulatory elements to trigger CSC gene expression.
Results
EMT induces DNA accessibility away from the TSS in regions with enhancer chromatin marks
The MCF-IM model () is useful for investigating early epigenomic changes in EMT and CSC formation. As detailed in Zafar et al.,Citation21 PMA alters epithelial MCF-7 morphology and gene expression to produce 2 mesenchymal subpopulations. The larger (90–95%) subpopulation maintains CD24 expression and does not have CSC properties (referred to here as NCSCs), while a smaller CD44high subpopulation (referred to here as CSCs) forms spherical colonies in suspension (mammospheres). The transcriptomes of non-stimulated (NS) MCF-7 cells, NCSCs, and CSCs and PKC-θ's influence on them have been analyzed previously.Citation21,25
Figure 1. Comparison of regions that change in DNA accessibility upon stimulation to those that do not. (A) The MCF-7 model of EMT and CSC formation. Non-stimulated (NS) cells are treated with PMA (ST) to produce mesenchymal cells with non-CSC (NCSC) and CSC properties. (B) Accessible regions were grouped according to accessible in NS and ST cells, only accessible in NS or ST (only), more accessible in NS or ST (more), or equal accessibility. Regions for each group were annotated by distance to the nearest transcription start site (TSS) (B), genomic region (C), summarised chromatin environment in different cell types and (D) summarised CTCF/chromatin in MCF-7 and mammary epithelial cells (HMEC) (E). For summarised chromatin (D, E), each region is colored by their state. DNA accessibility at specific regions (F). Dots show biological replicates, dashes show averages. FAIRE-PCR normalized to PPIA and scaled to average of NS. purple: ST, orange: MDA-MB-231, * p < 0.05 compared to NS.
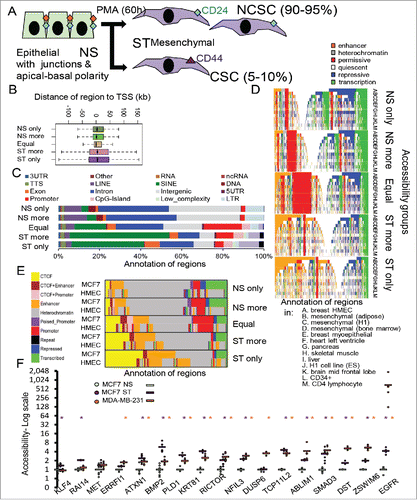
FAIRE-seq was used to examine NS and PMA-stimulated (ST) epigenomes. Using SICER, 126,124 (replicate 1) and 140,857 (replicate 2) accessible regions were identified in NS cells and 73,797 (replicate 1) and 72,341 (replicate 2) regions were identified in ST cells. Accessible regions ranged from 0.1 to 190.3 kb in size. The vast majority (>98%) of regions were less than 2.5 kb (Supplementary Fig. 1A).
Some regions greater than 2.5 kb were more accessible in ST (Supplementary Fig. 1B) while others were more accessible in NS (Supplementary Fig. 1C). They occurred frequently in areas of high copy number or with heterochromatin marks and their highly variable size precluded systematic analysis in the present study.
To investigate DNA accessibility at smaller regions, all enriched regions less than 2.5 kb in size were refined to 0.3 kb and the reads counted and normalized for all 4 samples (see Methods). The NS and ST replicates had Pearson correlation values of 0.96 and 0.94, respectively (Supplementary Fig. 1D). The regions were divided into 5 groups: (i) ‘NS only’, (ii) ‘NS more’, (iii) ‘equal’, (iv) ‘ST more’, and (v) ‘ST only’. Regions with greater (>1.5-fold) accessibility in NS (than ST) for both replicates were classified as either NS only (if they were only detected in NS samples) or NS more (Supplementary Fig. 1E-G, Supplementary Fig. 2). ST only and ST more groups were similarly defined, except that their accessibility was higher in ST. Peaks defined as ‘equal’ were less than 1.5-fold higher in NS or ST in both replicates.
Compared to the other groups, the more stable ‘equal’ accessibility regions had, on average, higher accessibility levels and were closer to TSSs (), with a small skew to regions downstream of TSSs. Regions with altered (especially increased) accessibility were located further from a TSS (), less were annotated as promoters/5′UTRs, and few ST only/more regions were in CpG islands (). While a large proportion of regions with higher accessibility in NS occurred in SINE repeats, regions with increased accessibility occurred in intergenic and intronic regions, with some in LINE or LTR repeats ().
Many regions of ‘equal’ accessibility had permissive chromatin (as summarised by the RoadMap project,Citation26 primarily defined by H3K4me3) marks across a range of cell types. While peaks with increased accessibility in ST had enhancer element chromatin marks (primarily defined by H3K4me1) in breast and mesenchymal cells, these tended to be more cell-type specific (). Chromatin marks together with CTCF have also been profiled in MCF-7 and human mammary epithelial cells (HMECs), and we used this data to confirm the high proportion of enhancer regions in the ST only and ST more groups ().Citation27 Furthermore, we discovered that a greater proportion of regions with increased accessibility showed evidence of CTCF binding.Citation27 CTCF is a major regulator of higher order chromatin structure with a particularly important role in the looping involved in long range interactions and its occupancy has been associated with nucleosome positioning in MCF-7 cells.Citation28
It has previously been shown that DNA methylation may negatively regulate DNA accessibility in MCF-7 cells.Citation27 We utilised this Citation27 data to determine if the regions that had increased accessibility upon stimulation had higher DNA methylation levels in NS MCF-7 cells than the group with unchanged accessibility. However, neither group with increased accessibility showed evidence of high levels of DNA methylation in NS MCF-7 cells (Supplementary Fig. 3A), and it is therefore unlikely that decreases in DNA methylation are a major cause of increased accessibility in these regions.
We next used FAIRE-PCR to confirm accessibility increases on stimulation for 2 ‘ST more’ and 15 ‘ST only’ regions within 100 kb of genes with higher expression in CSCs than NCSCs ( and Supplementary Fig. 2). Regions were identified by the name of the nearest gene. Fourteen of the 17 regions predicted to increase in accessibility had significantly higher values in the mesenchymal state for KLF4, RAI14, ATXN1, BMP2, PLD1, KRT81, RICTOR, NFIL3, TCP11L2, ABLIM1, SMAD3, DST, ZSWIM6, and EGFR, (p = 0.04, 0.0005, 0.0009, 0.0004, 0.01, 0.003, 0.006, 0.002, 0.005, 0.001, 0.005, 0.04, 0.02, and 0.007, respectively) in MCF-IMs. The accessibility of our candidate regions was also compared in MCF-7 and MDA-MB-231 cells. The MDA-MB-231 cell line is an ER-negative breast cancer cell line with CSC properties (˜95% CD44high CD24low CSCs) and a transcriptome enriched for the ‘stromal’ signature.Citation4 There was significantly greater accessibility of ERRFI1, ATXN1, BMP2, PLD1, KRT81, RICTOR, NFIL3, DUSP6, TCP11L2, DUSP6, ABLIM1, SMAD3, DST, ZSWIM6, and EGFR in MDA-MB-231 cells compared to NS MCF-7 cells (p = 0.02, 0.0008, 0.003, 0.0003, 0.0006, 0.003, 0.00005, 0.00002, 0.01, 0.001, 0.000006, 0.00005, and 0.03, respectively; ). For EGFR, DUSP6, ZSWIM6, DST, ABLIM1 and to a lesser degree ERRFI1, MDA-MB-231 accessibility was greater than ST MCF-7 accessibility, suggesting that the CSC subpopulation in ST MCF-7s had increased accessibility than NCSCs.
A subset of regions with increased accessibility is associated with poised to active enhancers in the mesenchymal state
The chromatin marks H3K4me1 and H3K27ac have been used to characterize enhancers as “poised” (with H3K4me1 but not H3K27ac) and “active” (with H3K27ac).Citation29 Active enhancers have more accessible chromatin. Citation29 Since a large subset of the regions with increased accessibility occurred in regions already marked as enhancers in NS MCF-7 cells, we sought to determine if these enhancers changed state, if their state differed in the bulk ST (with ˜7–10% CSC) population and the ‘CSC like’ MDA-MB-231 cell line (˜95% CD44high CD24low CSCs), or if any of the regions not marked as enhancers were ‘latent’ enhancersCitation30 that only become detectable upon stimulation. Thus, we profiled H3K4me1 and H3K27ac in NS, ST MCF-7, and MDA-MB-231 cells.
Examining the regions with equal accessibility in NS and ST cells revealed that those with higher accessibility had H3K27ac and diffuse H3K4me1 on histones flanking the accessible region (). For regions with equal accessibility, the levels of H3K27ac were similar in NS and ST cells. In contrast, a large proportion of the ST more and ST only regions showed increased H3K27ac in the ST and/or MDA-MB-231 cells.
Figure 2. Identification of the regions with increased accessibility that have changes in enhancer histone marks and are within certain distances of genes with differential expression. (A) Enhancer histone marks around the regions with different accessibility ordered by strength of accessibility. Enhancer marks are shown for non-stimulated and PMA-stimulated (ST) MCF-7 cells and NS MDA-MB-231 (MDA) cells. (B) For the ST only accessibility regions, marks were clustered into different patterns and the chromatin state of the regions in MCF-7 cells annotated. The average ChIP and FAIRE-seq profiles are shown for each subset of the resulting clusters with the number of regions in each cluster indicated. (C) Genes nearest to the regions were profiled for enrichment of Gene Ontology, Reactome, and KEGG pathways. The significance of the enrichment of different gene groups is indicated. (D) Number of regions with accessibility only in stimulated MCF-7 cells within different distances of genes with differential expression in NS and ST cells or non-cancer stem cells (NCSCs) and CSCs. Error bars show SD of 100 sets of random genes with the sets the same size as the gene set of interest. * z-score > 2. Marks are shown +/− 1 kb around the region, reads binned by 0.1 kb and adjusted by total mapped reads (A,B).
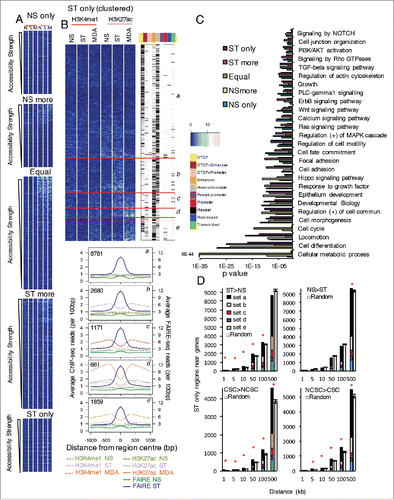
The ST only regions could be subdivided into those with low or unchanged H3K27ac (set a), increased H3K27ac in ST but not MDA-MB-231 (set b), increased H3K27ac in ST and MDA-MB-231 (set c), increased H3K27ac that was higher in MDA-MB-231 (set d), and increased H3K27ac in MDA-MB-231 (set e) (). We speculated that the CSC subpopulation in the ST sample would have higher levels of H3K27ac for set e genes that were not evident by bulk sample ChIP-seq. No large increases in H3K4me1 levels were seen that could be considered ‘latent’ enhancer activation. Furthermore, the verified regions that had higher accessibility in MDA-MB-231 than MCF-7 ST cells such as EGFR, DUSP6, ZSWIM6, ABLIM1, DST, and ERRFI1 were in set d or e. In contrast SMAD3, TCP11L2, NFIL3, RICTOR, PLD1, ATXN1, and KRT81 (which had similar accessibility in ST and MDA-MB-231 cells) were in set c, and BMP2 and KLF4 were in set b. Thus, the H3K27ac profiling results provided a good indicator of accessibility differences between the MCF-7 ST bulk population and the CSC-enriched MDA-MB-231 population for non-CTCF regions.
Interestingly, the majority of all genes in the genome had multiple regions of accessibility. There was usually at least one region of equal accessibility within 50 kb of the TSS and, while most had generally less than 8, some genes had up to 32 (Supplementary Fig. 3B). In addition to regions of equal accessibility, genes often had nearby regions with both increased and decreased accessibility (Supplementary Fig. 3B). As suggested by the large proportion of ‘equal’ regions at promoters, genes often had unchanged accessibility at their TSS and several regions of accessibility up- and downstream that varied in response to stimulation (Supplementary Fig. 2). Examination of the distances between regions from the same and different groups revealed that while many regions with increased accessibility were within 10 kb of a region with decreased accessibility, ST only regions tended to be nearer equal or ST more regions than NS-associated regions (Supplementary Fig. 3C).
Identification of regions with increased accessibility associated with genes with higher expression in CSCs and NCSCs
We next examined whether the genes closest to the accessible regions were enriched for any biological processes or Reactome/KEGG pathways. Regions with increased (and decreased/NS only) accessibility were close to genes involved in cell differentiation, migration, development, cell adhesion, and growth and signaling pathways such as calcium signaling, Notch, TGF, Wnt, MAPK, and PI3K (). Genes with ‘equal’ accessibility were also enriched for these pathways. However, genes close to regions of ‘equal’ accessibility were more highly enriched for cell cycle and metabolic processes.
To further explore the relationship between accessibility and expression and to identify which regions were associated with expression changes, we examined regions within different cut-off distances from a gene more (log2 0.5 difference) or less in stimulated cells and more or less in CSCs compared to NCSCs. Significance was determined by comparing the numbers to those expected by chance (see Methods).
More regions accessible only in ST cells were within 1–100 kb of a gene and induced upon stimulation (z-scores 2.1 – 7.6) than they were close to a random gene set of the same size (measured in 100 such random gene sets) (). This was also seen for the other accessibility groups for at least some distances (Supplementary Fig. 4). This was not the case for genes with decreased expression (NS>ST). Many genes induced upon stimulation are induced to a similar extent in NCSCs and CSCs; however, some are induced differentially.Citation21 Several different patterns of differential induction were observed (set i-x, Supplementary Fig. 5), but overall the genes could be grouped into those with higher expression in CSCs than NCSCs and those with lower expression in CSCs than NCSCs. More regions accessible only in ST cells were close to a gene that had higher expression in CSCs than NCSCs (z-scores 2.8–6.6) or in NCSCs than CSCs (z-scores 3 to 5.4) (). Interestingly, the regions near to these genes were not just those with enhancer chromatin marks but also those without (set a) (). The other accessibility groups (except for the ‘ST more’) showed no relationship with genes with higher expression in CSCs than NCSCs.
The genes with higher expression in CSCs than NCSCs occurring near regions with accessibility only in ST cells and that had increased H3K27ac were especially interesting (Supplementary Fig. 5A), as were genes with higher expression in NCSCs than CSCs that occurred near regions with accessibility only in ST and that had increased H3K27ac only in the ST population and not MDA-MB-231s (Supplementary Fig. 5B).
Enrichment of FOX, AP-1, CTCF, TEAD, and TFAP motifs in regions with increased accessibility
We next examined the regions in the 5 accessibility groups for over-represented sequence motifs to determine if they could be bound by different TFs using HOMER, which compares regions to those with similar GC content.Citation31 AP-1 (including Jun and Fos), TFAP2 (also known as AP2), Forkhead (FOX), TEAD, and CTCF-binding motifs were over-represented in regions with accessibility only in ST cells (, Supplementary Table 1).
Figure 3. Identification of the DNA binding motifs in regions with increased accessibility and how the motifs occur in the regions with respect to expression of nearby genes. (A) The proportion of regions with differential or equal (Eq.) accessibility containing motifs for AP1 (FOSL1), FOX (FOXA2), CTCF, TFAP (AP-2G), and TEAD (TEAD4) compared to background regions. (B) Expression in NS, NCSCs, and CSCs of the factors that can bind the motifs. (C) The transcription factor motifs enriched in regions near to genes with differential expression and with accessibility only in ST MCF-7 cells. Expression was compared between (i) NS and ST and (ii) NCSCs and CSCs. CLOVER enrichment scores are shown in brackets after the motif colored by the comparison they are significant in. All motifs with a score had p-value< 0.05 for enrichment when the regions within 50 kb of a gene in the gene set were compared to all regions with ‘ST only’ accessibility (C). * q-value (Benjamini) = 0 and p < 10−15 (A).
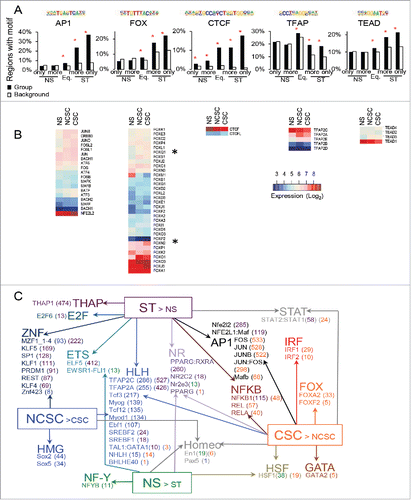
We used our previously publishedCitation21 expression profiles of NS, CSC, and NCSC populations to determine which motif-binding factors were expressed and how expression altered with stimulation (). Several proteins can bind the AP-1 and many are expressed in NS cells, with some (including FOSL1/2) showing induction with PMA. TFAP2C and TFAP2A were the most highly expressed TFAP2-binding proteins, while TEAD1 was the highest expressed TEAD. Of the FOX family members, FOXN2 was the most induced in CSCs compared to NCSCs, while FOXO1, FOXO3, FOXL2, FOXQ1, FOXS1, FOXJ1, and FOXN2 were all higher in CSCs compared to NS cells ().
Having identified motifs associated with increased accessibility, we next wanted to determine if those ST only regions that were near genes with differential expression contained different motifs to those that were not. CLOVER was used (with JASPAR motifs) to investigate how the occurrence of motifs in exposed regions related to expression of nearby (within 50kb) genes. The significance of motif enrichment in a subset of regions was tested against their occurrence in the whole ST only population. In contrast to examining occurrences relative to genomic background, this approach helps to determine which TFs may be important in controlling expression in regions that have become accessible. With the aim of identifying TFs that play a role in EMT and CSC development, our region subsets included those near genes with differential expression in: a) NS and ST cells (); b) NCSCs and CSCs (); and c) stromal, MaSC, and ‘basal’ breast epithelial cells (Supplementary Fig. 6).
Examination of differential motif enrichment in the ST-only regions near genes with increased expression in ST compared to NS cells and CSCs compared to NCSCs supported a role for FOX members in regulating CSC gene expression (), as well as suggesting a role for NF-κB, IRF, and STAT members. Interestingly, the Nfe2l2 AP-1 motif variant was more associated with genes with increased expression in the whole ST cell population, while the FOS/JUN AP-1 motif was more significantly associated with CSC > NCSC genes. In contrast, the TFAP2C/A motifs (part of the HLH family), which were also associated with upregulated genes, associated with genes showing higher expression in NCSCs. Interestingly, annotation of TFAP motif occurrence in the regions with H3K27ac only in ST and not MDA-MB-231s and that were near genes with higher expression in NCSCs supported a role for TFAP in regulating gene expression in NCSCs upon PMA stimulation (Supplementary Fig. 5B).
Prat et al.Citation4 defined ‘stromal’, ‘MaSc’, ‘basal/progenitor luminal’ (Basal/pL), and ‘mature luminal’ (mL) breast gene signatures, with the stromal (claudin-low) signature being associated with CSCs. When the expression of these signature genes was examined in our model (Supplementary Fig. 6A), overall the MaSC and basal genes had increased expression upon stimulation while the stromal genes had the strongest shift in expression to the CSC profile. Examining the enriched motifs in the regions with increased accessibility within 50 kb of a stromal, MaSc, or basal signature gene revealed that TFAP2A/C was enriched in MaSC cells and AP-1, NF-κB, TEAD, motifs were associated with all groups. Several FOX motifs were more significantly enriched in the regions near stromal genes (Supplementary Fig. 6B).
FOXN2 and FOXQ1 play a role in regulating the CSC phenotype
There was enrichment of the FOX motif in regions with increased accessibility and in: (a) the vicinity of genes that increased expression in CSCs, and (b) ‘stromal’ signature genes. FOXQ1 and FOXN2 expression increase upon stimulation and increased to a greater extent in the CSC population. Several members of the FOX family (including FOXQ1)Citation32 have been found to play a role in EMT and the acquisition of CSC properties.
While MCF-7 stimulation induces adherent mesenchymal cells, of which a subpopulation has CSC properties (), it also induces a suspended (‘floating’) cell population with a higher proportion of cells with CSC properties. To confirm the microarray results (), we examined FOXN2 and FOXQ1 expression in the MCF-7 model, measuring the stimulated adherent and floating cells separately (). We confirmed an increase in expression of both genes upon stimulation and both showed a greater increase in the floating cells. We next examined the level and localization of the proteins (). Nuclear protein levels were higher in both populations of stimulated cells and in MDA-MB-231 cells compared to NS MCF-7 cells and, in most cases, the nuclear to cytoplasmic ratio was higher.
Figure 4. Expression and localization of FOXN2 and FOXQ1 and their role in regulating the CSC phenotype. (A) Expression of FOXN2 and FOXQ1 in non-stimulated (NS), PMA-stimulated (ST), adherent (AD), and floating (FLT) cells. (B) Levels of FOXN2 and FOXQ1 in the nucleus as measured by total nuclear fluorescence intensity (TNFI) and the nuclear/cytoplasmic ratio (Fn/c) in the MCF-7 model and MDA-MB-231 (MDA) cells. (C) The effect of FOXN2 and FOXQ1 siRNA on the CD44Hi and CH24lo proportion of ST-AD and ST-FLT MCF-7 cells compared to control ST (Ctrl). (D) The effect of FOXN2 and FOXQ1 siRNA on the levels of laminin V and cytoplasmic fibronectin in MCF-7 cells. Expression normalized to PPIA, average of n = 3 +/− SEM (A). For protein localization, DAPI was used to identify the nucleus, image scale bar is 10 μm, and averages of 20 cells are shown +/− SEM * for p ≤ 0.05, ** for p ≤ 0.01, *** for p ≤ 0.001, and **** for p ≤ 0.0001. (B, D). FACS n = 3, error bars = SEM, red stars show significance against control siRNA (C).
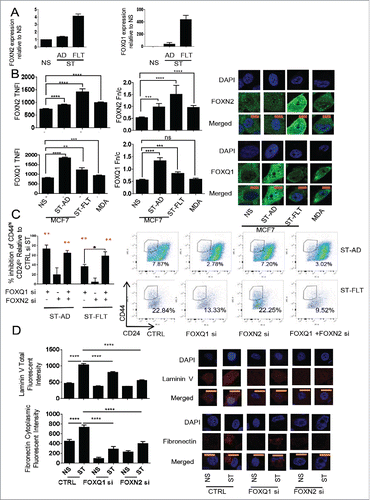
siRNA was next used to examine the roles of FOXN2 and FOXQ1 in the CD44HiCD24Lo () phenotype and on levels of the mesenchymal markers laminin V and fibronectin (). Confirmation of reduced FoxN2 and FoxQ1 protein levels in the presence of the siRNA was confirmed by immunohistochemistry (Supplementary Fig. 7). FOXQ1 siRNA significantly inhibited the induction of the CD44HiCD24LoCSC population (). Interestingly, the combination of FOXQ1 and FOXN2 siRNA inhibited the CSC population of the floating cells significantly more than FOXQ1 siRNA alone. Both FOXN2 and FOXQ1 siRNA inhibited the increase in laminin V and fibronectin proteins upon stimulation ().
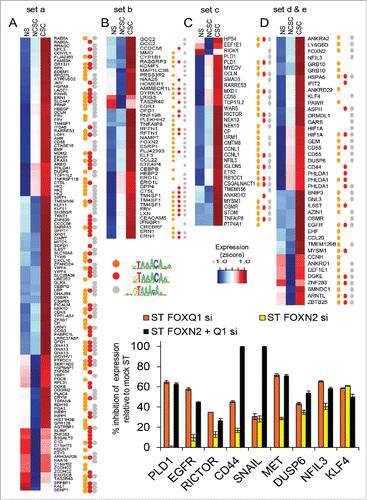
Many members of the FOX family, including FoxQ1 and FoxN2,Citation33 can bind the primary forkhead motif FkhP (gTAAAcA), which is similar to the FOX motif detected as enriched by HOMER. However, family members can have different efficiencies for versions of FkhP. Furthermore, a secondary forkhead motif (FkhS) and an additional motif FHL (GACGC) exist that are primarily bound by FOXN1–4.Citation33 Many interesting CSC-associated genes had regions containing the HOMER FOX motif (Supplementary Fig. 5A). However, since this motif is not optimised for Foxq1 or Foxn2 binding, we scanned the ST only regions for different FOX motif version using the Nakagawa et al.Citation33 and Jolma et al.Citation34 FOX position weight matrices. We confirmed that the FkhP motif was over-represented, and annotated CSC-associated genes with ST only accessibility with 3 GTAAACA variants, including one thought to have greater affinity for FOXN2 (red dot, , Supplementary Table 2). We profiled expression of 8 of these putative targets (KLF4, DUSP6, CD44, RICTOR, NFIL3, MET, EGFR and PLD1) and the mesenchymal-associated gene SNAIL and determined how FOXQ1, FOXN2, and combination siRNAs affected their induction with PMA (). All were at least partially dependent on FOXQ1 and all except PLD1 showed some dependence on FOXN2. The expression of CD44 and SNAIL in stimulated cells was completely inhibited by the combination of FOXQ1 and FOXN2 siRNAs.
Figure 5. Putative FOX targets in cancer stem cell (CSC) gene expression. (A-D) Expression of genes with increased expression in CSCs than NCSCs that have regions with increased accessibility upon stimulation and that contain FOX binding motifs. Genes are grouped A-D by how H3K27ac differs in ST and MDA-MB-231s (see ). Regions could contain any of 3 variants of the core primary FOX motif. Microarray expression values are scaled (z-score). (E) The level of inhibition of expression due to FOXN2 and FOXQ1 siRNA for putative targets in MCF-7 cells stimulated (ST) with PMA. PCR expression normalized to PPIA and expressed as percent inhibition of the control siRNA ST value, n = 3 PCR replicates, error bars SEM (E).
Discussion
Here we present novel changes in DNA accessibility that are induced by PKC activation and are highly likely to influence the expression of distant genes crucial for EMT and CSC induction. While the accessibility of many regions remained stable, especially those with high accessibility at promoter regions, wider epigenomic changes were also observed. Increases in the accessibility of regions about one to several nucleosomes in size occurred in intergenic and intronic regions, many of which were pre-decorated with chromatin enhancer marks. Several accessibility regions were detected near/within genes and many, including SMAD3, RICTOR, ATXN1, and KLF4, had highly stable accessibility at their TSS and changes at putative enhancer regions.
Promoter accessibility remains largely constant during differentiation and between different cell lineages.Citation35,36 In contrast, enhancer accessibility changes and functional activation are largely cell-type specific. Differences in chromatin and DNA accessibility have been associated with the expression of genes 750 kb away in MDA-MB-231 and HMEC cells.Citation37 Cell-type specific epigenetic modifications that activate enhancers allow modulation of promoter activity and activation of global cell type-specific transcriptional programs.
Clusters of active enhancers have recently been termed ‘super-’ (or stretch) enhancers.Citation29 Super-enhancers are often bookmarked by CTCF, which prevents genes outside these regions from being affected.Citation29 We found that many genes had multiple regions of accessibility that commonly occurred within 10 kb of each other (Supplementary Fig. 2). We observed large areas of higher H3K27ac near genes such as ZSWIM6 (Supplementary Fig. 2) that could be classified as super-enhancers using the definition of >3 kb of active histone marks.Citation29 However, we also found that genes could be near accessible regions that did not always change in the same way or to the same extent. We propose that the graduated expression of genes such as SMAD3 and KLF4 with a NS < NCSC < CSC expression pattern (Supplementary Fig. 5 set ii), could be a result of multiple regulatory elements being ‘activated’ to different extents in NCSCs and CSCs. SMAD3 contains several regions with increased accessibility, with at least one set c region that increased in H3K27ac in ST and MDA-MB-231 cells and at least one set d region that increased more in MDA-MB-231 cells ().
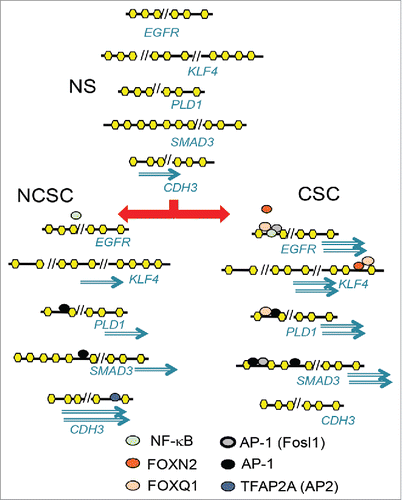
Figure 6. Putative models of how the factors binding the motifs may act differently in NS, NCSCs, and CSCs to give rise to genes with higher expression in CSCs than NCSCs or NCSCs than CSCs. Upon stimulation of MCF-7 cells (NS), transcription factors such as AP-1 increase chromatin accessibility at regulatory regions away from the transcription start site. The differential activation of AP-1 and FOX members and TFAP2A in cancer stem cells (CSC) and non-CSC (NCSC) regulates differential gene expression. Genes can have more than one regulatory region that become accessible to different extents in NCSC and CSC, giving rise to different levels of expression.
A number of genes in EMT and CSC-associated pathways such as PI3K showed altered accessibility and, although gene expression was not automatically increased, opening of the chromatin increased the likelihood of transcription by exposing motifs for other TFs. How these TFs are affected by intrinsic and extrinsic signals influences expression. PKC activates many TFs including NF-κB and STAT, and regions containing these motifs were linked to increased expression with stimulation. The expression outcome ultimately depends on many factors such as the combination of active TFs, the strength of the enhancer-promoter interaction, and other post-transcriptional regulation. In the majority of cells in our model the combined effects on the transcriptome result in a NCSC phenotype. However, in a proportion of the cells, this increased epigenomic plasticity allows for expression of genes that promote CSC development. It is likely that a initial small stochastic increase in certain genes such as those expressing TFs or receptors leads to a positive-feedback amplification process, with receptors increasing activation of signaling pathways and their target TFs and leading to further increases in receptor expression.
The addition of H3K27ac profiling in the ST and CSC-like MDA-MB-231 cell line helped us to distinguish between regions likely to be different in the NCSC and CSC population. However, an important finding of this paper is that stimulation affects many regions in both NCSCs and CSCs, and while some genes are expressed equally in the 2, others show different expression levels due to the transcription factors present that can bind the newly exposed regions. The combination of motif analysis of the newly accessible regions and which are associated with NCSC or CSC expression suggests that AP-1 plays a major role in creating accessibility upon stimulation. TFAP family members can then promote a NCSC gene expression pattern while a subset of FOX family members such as Foxq1 and Foxn2 divert the stimulated cells to the CSC fate ().
Regions becoming accessible on PKC activation were most enriched for motifs binding AP-1. Members of the AP-1 family have previously been found to have a role in EMT and in nucleosome remodelling in response to extracellular signals.Citation38-41 In addition to the association of AP-1 with increased accessibility, expression of genes near a large subset of these regions increased after stimulation in both NCSCs and CSCs. However, AP-1 is a dimer and can differentially bind DNA and regulate genes based on the different combinations of Fos, Jun, and Maf/Nfe variants. Interestingly, FOS and JUN motifs were differentially enriched to the NFE2l2/MAF variants of the AP-1 motif. Fra-1 (encoded by FOSL1), a member of the Fos family of basic leucine zipper domain proteins, dimerises with c-Jun, JunB, or JunD to form the prototypical AP-1 TF complex.Citation42 PKCα activation supports the mesenchymal state by preferentially inducing and utilizing Fra-1 (over c-Fos) in the AP-1 transcriptional complex during EMT, and PKCα-induced FOSL1 is a key event in breast CSC formation.Citation39,40 It is probable that some differential accessibility and expression in CSCs compared to NCSCs is related to different levels of the various AP-1 subunits in the 2 populations ().
TEAD family members have also have previously been implicated in nucleosome remodelling. They are important endpoints of the hippo signaling pathway and are associated with both transcriptional activation and repression.Citation43 TEADs and their co-activators are directly implicated in aggressive breast cancer phenotypes.Citation44,45 The findings presented here suggest it also plays an important role in increasing accessibility upon stimulation.
Many ST only regions were close to genes underexpressed in CSCs compared to NCSCs; these genes could be subdivided based on their expression and many, such as CDH3, were induced by stimulation. The H3K27ac patterns suggest that for some regions (such as CDH3) this increase occurs predominantly in NCSCs compared to CSCs (Supplementary Fig. 5B). The TFAP2 motif was enriched in increased accessibility regions and in regions within 50 kb of a gene, with higher expression in NCSCs than CSCs or the MaSC signature. TFAP2A and TFAP2C are the predominant TFs to bind this motif, and TFAP2A was expressed at higher levels in NCSCs () and has been shown to play a role in TGF-β signaling.Citation46 The role of TFAP2s in cellular differentiation and cancer is complex, and SMADs can also bind TFAP2 motifs. Citation46,47 Our results suggest that the activity of TFs binding to TFAP sites (perhaps via SMAD) is increased upon stimulation but in CSCs (compared to NCSCs) this activity is at least partially inhibited ().
Stimulation of MCF-7 cells opened up DNA near genes that increased expression to a greater extent (set i and set ii, Supplementary Fig. 5A) or only (set iii) in CSCs compared to NCSCs. While the H3K27ac results suggests that many of these regions did have increased accessibility in the CSCs compared to NCSCs, others did not. There was FOX motif enrichment in regions with increased accessibility and in (a) the vicinity of genes that increased expression in CSCs, and (b) ‘stromal’ signature genes. This suggests that FOX family members can act at these newly accessible regions to active gene expression in CSC progenitors, possibly by regulating accessibility as well as expression of the nearby gene. In support of this, the FOX motif was enriched in MDA-MB-231-specific regions in a FAIRE-seq comparison of HMECs.Citation37
The FOX family of transcriptional regulators is defined by the presence of highly conserved ‘Forkhead’ or ‘winged-helix’ DNA-binding domains.Citation33,48 Several FOX proteins can act as pioneer factors and contain a domain that is similar to linker histones, allowing them to open DNA.Citation49,50 FOX proteins are involved in proliferation, apoptosis, differentiation, and metabolism.Citation51 Since FOX proteins share 2 main DNA-binding motifs,Citation33 it is difficult to implicate a specific FOX protein or combination of FOX factors in CSC development in our breast cancer model based on the consensus motif alone. Several FOX family members including FOXQ1, FOXC2 and FOXA1 have been positively or negatively implicated in EMT and/or CSC development. Citation48 It is likely that several family members act to regulate accessibility and/or expression upon stimulation and progression along the CSC pathway and that there is some level of redundancy. However, the results presented here show that FOXQ1 and FOXN2 have non-redundant roles in optimal expression of several CSC-associated genes and the protein levels of fibronectin and laminin V.
Dissection of the gene regulatory network of sea urchin EMT has proposed specific roles for FOXN2 in motility, apical constriction, and polarity.Citation52 FOXQ1 depletion in MDA-MB-231 cells increases epithelial morphology and reduces invasiveness,Citation32 and it also plays a role in other metastasis models.Citation48 It has been suggested that this role may be mediated through its repression of E-cadherin via direct binding to an upstream E-box.Citation48 However, the identification of FOX motifs in regulatory regions of CSC-associated genes raises the possibility that an important role of FOXQ1 and FOXN2 in CSC formation is mediated via their positive regulation of the expression of these genes. The most distinguishing feature of the FOX motif is that it is enriched in the newly accessible regions closer to genes with higher expression in CSCs than NCSCs. These regions had varying H3K27ac states in NS, ST, and MDA-MB-231s. For example, PLD1 has a FOX motif-containing set c region, while EGFR has a FOX motif-containing set e region (). Thus, in addition to any role they may play in increasing DNA accessibility, these results support a role for Fox members in converting DNA enhancer accessibility to gene expression.
The use of FAIRE-seq to identify regulatory elements distant from the TSS, emerging technologies to associate distant elements with their gene targets, evolving computational techniques to predict TF binding sites,Citation34 and high-throughput sequencing of TF binding in different cell types will allow the mapping of complex transcriptional networks underlying biological processes such as EMT and CSC formation. This combination of approaches will be invaluable for discovering the potential pathological consequences of human sequence variation.
Materials and methods
Cells were cultured at 37°C in low glucose Dulbecco's modified Eagle medium (DMEM) (Life Technologies) supplemented with 10% foetal calf serum, 2mM L-glutamine, and 0.1% antibiotics. MCF-7 cells were either untreated or stimulated with PMA (1.29ng/ml, Sigma-Aldrich) for 60 h. For siRNA experiments, MCF-7 cells were treated with Lipofectamine 2000 (Life Technologies) and either 20 nM mock (sc-36869, Santa Cruz Biotechnology), FOXN2 (sc-62341), or FOXQ1 (sc-60660) siRNA for 48 h before stimulation with PMA. Transfection efficiency was assessed with microscopy to detect FOXN2 or FOXQ1 staining.
FAIRE and ChIP-seq
FAIRE and ChIP were performed as outlined previously.Citation53 Briefly, 5 × 106 cells were cross-linked with formaldehyde (1%, 10 min) then quenched with glycine (125mM). Fixed cells were lysed in SDS lysis buffer and chromatin-bound DNA sheared by sonication with a Misonix Sonicator S-4000. For FAIRE, nucleosome-free DNA was extracted using phenol-chloroform. ChIP samples were incubated overnight at 4°C with Protein A Magnetic Beads (Millipore) and 5 μg of either H3K4me1 (ab8895; Abcam) or H3K27ac (ab4729; Abcam) antibodies. For FAIRE and ChIP, protein-DNA cross-links were reversed by incubating overnight at 65°C and purified with the Zymo-Spin™ I kit (Zymo Research).
Libraries were prepared from FAIRE DNA (20ng) or ChIP DNA (10ng) using the NEBNext® Ultra DNA Library Prep Kit or ChIP-Seq Library for Illumina (New England Biolabs), respectively. After Ampure XP Bead clean-up, libraries were single-end 50bp sequenced on a HiSeq2000 (at the Ramaciotti Center for Genomics, NSW).
FAIRE for specific regions was assessed with SYBR Green real-time PCR using the primers listed in Supplementary Table 3. The PPIA TSS region was used for normalization across treatments. A t-test presuming unequal variance was used to test for significance.
Confocal laser scanning microscopy
Cells were permeabilised with Triton X-100 (1%, 20 min) and probed with an antibody to FOXQ1 (sc-47597), FOXN2 (sc-67490), fibronectin (MAB 1926, Millipore) or laminin V (MAB 1949, Millipore) followed by visualization with a secondary antibody conjugated to Alexa-Fluor 488 (A11001; Life Technologies) or Alexa-Flour 568 (A11055; Life Technologies) and co-staining with DAPI on a Nikon C1 plus confocal system. Expression was quantified using Fiji-ImageJ. The Mann–Whitney non-parametric test was used to determine significance (Prism).
Flow cytometry (FACS)
FACs was performed as previously describedCitation21 with cells stained with anti-CD44-APC (559942), anti-CD24-PE (555428), and Hoechst 33258 (BD Biosciences). The one-sided t-test was used to determine significance against control siRNA.
RNA isolation and real time PCR
Total RNA was extracted from cells using TRI Reagent® (Sigma-Aldrich) as previously described.Citation54 The RNA (1 μg) was DNase I treated (Roche Life Science) and reverse-transcribed (Maxima First Strand cDNA Synthesis kit, Thermo Fisher Scientific). Real-time PCR was performed as previously described using TaqMan probe sets.Citation54
Bioinformatics
Reads were cleaned with cutadaptCitation55 and mapped to Hg19 with Bowtie2Citation56 to produce 58,339,489 – 73,355,539 reads per FAIRE sample and 34,222,132–45,348,533 reads per ChIP sample. Duplicate reads were removed with Picard, and only uniquely mapping reads were used for further analysis. Enriched FAIRE regions were determined by comparison with a total input sample using SICER (FDR 0.01, 100bp).Citation57 Enriched regions were also called using MACS2, producing largely similar results. However, while MACS2 called more refined peaks it did not detect many smaller peaks that were subsequently confirmed by FAIRE-PCR. Reads were extended to 200bp. FAIRE-seq reads were visualised in UCSC using bedGraph files created in HTSeq.Citation58 All UCSC images have the same y-axis scale with maximums at 117, 126, 122, 147, 185, 19, 17, and 22 for the NS replicate 1, NS replicate 2, ST replicate 1, ST replicate 2, total input, NS (H3K27ac), ST (H3K27ac), and MDA-MB-231 (H3K27ac), respectively, which scaled for library size (FAIRE and H3K27ac scaled independently).
Enriched regions <2.5 kb (306,828 regions) were refined to 0.3 kb by determining the highest point within the region for the sample they were called in and using this as the center of a 0.3 kb region (R). DNA accessibility was quantified for all 4 treatments using these peaks by counting how many read midpoints occurred within the peak. By trialling different approaches to normalize the counts (analysis not shown), loess normalization (utilizing LPE in R) was selected as the optimal method (Supplementary Fig. 1E and F). This approach reduced the accessibility in the NS samples, and the resulting values were more consistent with the FAIRE-PCR results obtained previously Citation21 and those shown here (). To maximise the chances of identifying changes occurring in the small CSC sub-population in the ST group, the peak detection criteria were intentionally sensitive, despite the increased likelihood of detecting false positives. Using the replicate treatments as a guide, we estimated that using a 1.5-fold cutoff with one replicate gives a ∼0.5 false discovery rate (FDR). However, requiring the regions to be >1.5-fold in 2 replicates decreases the FDR to 0.16–0.25. Note that only 2.4% of the ST only regions had an average fold change <2, while 10.0% of the ST more regions had an average fold change <2. Regions whose center was within 240bp of a region with a higher maximum read count were removed. Correlation of read levels in the regions was calculated in R using Pearson coefficients. The vioplot package was used to create violin plots.
To determine changes in H3K27ac, tags were counted +/− 1 kb around each FAIRE region, normalized to total library size (HOMERCitation31), and a 1.5-fold cut-off and 20 tag minimum was used to call greater H3K27ac in ST and MDA-MB-231 samples compared to NS.
The region centers were annotated using their summarised chromatin state from different cell types in Roadmap Citation26 with the ‘TSS’ state renamed ‘permissive’. CTCF and DNA methylation data were from GSE57498.Citation27 Results were compiled and visualised in R using ggplot2 and reshape2 libraries. HOMERCitation31 (annotatePeaks.pl; findMofifsGenome.pl) was used to determine average profiles, motif enrichment (p-value cut-off <10−15 for known motifs), and peak annotation (including GO and genome enrichment). Average profile values were adjusted by total mapped reads. CLOVERCitation59 (p < 0.05) and the non-redundant 2014 version of transcription factor binding motifs from JASPAR Citation60 and Fox motifs from Nakagawa et al. Citation33 and Jolma et al. Citation34 were also used to determine enriched motifs.
Gene expression data were from Zafar et al. Citation25 and a log2 0.5 difference was considered differential expression. To assess the significance of the number of regions within different distances of a gene set, the number of regions within the same distances was measured for 100 random microarray probe sets of the same size as the gene set of interest.
FAIRE-seq and ChIP-seq data are deposited in GEO under accession GSE71023.
Abbreviations
| AD | = | adherent |
| AP | = | Activator Protein |
| bHLH | = | basic helix–loop-helix |
| CSC | = | cancer stem cells |
| EMT | = | epithelial-to-mesenchymal transition |
| FAIRE-seq | = | formaldehyde-assisted isolation of regulatory elements-sequencing |
| FLT | = | floating |
| FOX | = | forkhead |
| HMEC | = | human mammary epithelial cells |
| MaSC | = | mammary stem cell |
| MCF-IM | = | MCF-7 inducible model |
| mL | = | mature luminal |
| NS | = | non-stimulated |
| NCSC | = | non-cancer stem cells |
| PMA | = | phorbol myristyl acetate |
| pL | = | progenitor luminal |
| PKC | = | protein kinase C |
| ST | = | PMA-stimulated |
| TF | = | transcription factor |
| TSS | = | transcription start site |
| ZNF | = | zinc finger |
| TFAP2 | = | Transcription Factor AP-2 |
| TEAD | = | TEA domain. |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
2014NUCLEUS0038R-file004.xlsx
Download MS Excel (51.3 KB)2014NUCLEUS0038R-file003.xlsx
Download MS Excel (76.1 KB)2014NUCLEUS0038R-file002.xlsx
Download MS Excel (32.1 KB)2014NUCLEUS0038R-f07-z-4c.pptx
Download MS Power Point (15.3 MB)Funding
This work was funded by an Australian, National Health and Medical Research Council (NHMRC) grant APP1068065.
References
- Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget 2015; 6:10697-711; PMID:25986923; http://dx.doi.org/10.18632/oncotarget.4037
- Ajani JA, Song S, Hochster HS, Steinberg IB. Cancer stem cells: the promise and the potential. Semin Oncol 2015; 42 Suppl 1:S3-17; PMID:25839664; http://dx.doi.org/10.1053/j.seminoncol.2015.01.001
- Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol 2007; 18:460-6; PMID:18023337; http://dx.doi.org/10.1016/j.copbio.2007.10.007
- Prat A, Karginova O, Parker JS, Fan C, He X, Bixby L, Harrell JC, Roman E, Adamo B, Troester M, et al. Characterization of cell lines derived from breast cancers and normal mammary tissues for the study of the intrinsic molecular subtypes. Breast Cancer Res Treat 2013; 142:237-55; PMID:24162158; http://dx.doi.org/10.1007/s10549-013-2743-3
- Visvader JE, Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev 2014; 28:1143-58; PMID:24888586; http://dx.doi.org/10.1101/gad.242511.114
- Maruyama R, Choudhury S, Kowalczyk A, Bessarabova M, Beresford-Smith B, Conway T, Kaspi A, Wu Z, Nikolskaya T, Merino VF, et al. Epigenetic regulation of cell type-specific expression patterns in the human mammary epithelium. PLoS Genet 2011; 7:e1001369; PMID:21533021; http://dx.doi.org/10.1371/journal.pgen.1001369
- Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A 2011; 108:7950-5; PMID:21498687; http://dx.doi.org/10.1073/pnas.1102454108
- Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Ann Rev Cell Dev Biol 2011; 27:347-76; PMID:21740232; http://dx.doi.org/10.1146/annurev-cellbio-092910-154036
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139:871-90; PMID:19945376; http://dx.doi.org/10.1016/j.cell.2009.11.007
- Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PloS One 2008; 3:e2888; PMID:18682804; http://dx.doi.org/10.1371/journal.pone.0002888
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15:178-96; PMID:24556840; http://dx.doi.org/10.1038/nrm3758
- Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 2005; 24:7443-54; PMID:16288291; http://dx.doi.org/10.1038/sj.onc.1209091
- Weber CE, Li NY, Wai PY, Kuo PC. Epithelial-mesenchymal transition, TGF-beta, and osteopontin in wound healing and tissue remodeling after injury. J Burn Care Res: Off Pub Am Burn Assoc 2012; 33:311-8; PMID:22561306; http://dx.doi.org/10.1097/BCR.0b013e318240541e
- Davis FM, Azimi I, Faville RA, Peters AA, Jalink K, Putney JW Jr., Goodhill GJ, Thompson EW, Roberts-Thomson SJ, Monteith GR. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene 2014; 33:2307-16; PMID:23686305; http://dx.doi.org/10.1038/onc.2013.187
- Jechlinger M, Sommer A, Moriggl R, Seither P, Kraut N, Capodiecci P, Donovan M, Cordon-Cardo C, Beug H, Grunert S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J Clin Investigat 2006; 116:1561-70; PMID:16741576; http://dx.doi.org/10.1172/JCI24652
- Malaguarnera R, Belfiore A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol 2014; 5:10; PMID:24550888; http://dx.doi.org/10.3389/fendo.2014.00010
- Katsuno Y, Lamouille S, Derynck R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol 2013; 25:76-84; PMID:23197193; http://dx.doi.org/10.1097/CCO.0b013e32835b6371
- Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Investigat 2009; 119:1417-9; PMID:19487817; http://dx.doi.org/10.1172/JCI39675
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science (New York, NY) 2011; 331:1559-64; PMID:21436443; http://dx.doi.org/10.1126/science.1203543
- Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, Chao CH, Yamaguchi H, Yang NK, Ding Q, et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res 2012; 72:1290-300; PMID:22253230; http://dx.doi.org/10.1158/0008-5472.CAN-11-3123
- Zafar A, Wu F, Hardy K, Li J, Tu WJ, McCuaig R, Harris J, Khanna KK, Attema J, Gregory PA, et al. Chromatinized protein kinase C-theta directly regulates inducible genes in epithelial to mesenchymal transition and breast cancer stem cells. Mol Cell Biol 2014; 34:2961-80; PMID:24891615; http://dx.doi.org/10.1128/MCB.01693-13
- Waki H, Nakamura M, Yamauchi T, Wakabayashi K, Yu J, Hirose-Yotsuya L, Take K, Sun W, Iwabu M, Okada-Iwabu M, et al. Global mapping of cell type-specific open chromatin by FAIRE-seq reveals the regulatory role of the NFI family in adipocyte differentiation. PLoS Genet 2011; 7:e1002311; PMID:22028663; http://dx.doi.org/10.1371/journal.pgen.1002311
- Murtha M, Strino F, Tokcaer-Keskin Z, Sumru Bayin N, Shalabi D, Xi X, Kluger Y, Dailey L. Comparative FAIRE-seq analysis reveals distinguishing features of the chromatin structure of ground state- and primed-pluripotent cells. Stem Cells (Dayton, Ohio) 2015; 33:378-91; PMID:25335464; http://dx.doi.org/10.1002/stem.1871
- Davie K, Jacobs J, Atkins M, Potier D, Christiaens V, Halder G, Aerts S. Discovery of transcription factors and regulatory regions driving in vivo tumor development by ATAC-seq and FAIRE-seq open chromatin profiling. PLoS Genet 2015; 11:e1004994; PMID:25679813; http://dx.doi.org/10.1371/journal.pgen.1004994
- Zafar A, Hardy K, Wu F, Li J, Rao S. The role of protein kinase-C theta in control of epithelial to mesenchymal transition and cancer stem cell formation. Genomics Data 2015; 3:28-32; PMID:26484144; http://dx.doi.org/10.1016/j.gdata.2014.11.002
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Systematic analysis of chromatin state dynamics in nine human cell types. Nature 2011; 473:43-9; PMID:21441907; http://dx.doi.org/10.1038/nature09906
- Taberlay PC, Statham AL, Kelly TK, Clark SJ, Jones PA. Reconfiguration of nucleosome-depleted regions at distal regulatory elements accompanies DNA methylation of enhancers and insulators in cancer. Genome Res 2014; 24:1421-32; PMID:24916973; http://dx.doi.org/10.1101/gr.163485.113
- Marshall AD, Bailey CG, Rasko JE. CTCF and BORIS in genome regulation and cancer. Curr Opin Genet Dev 2014; 24:8-15; PMID:24657531; http://dx.doi.org/10.1016/j.gde.2013.10.011
- Witte S, O'Shea JJ, Vahedi G. Super-enhancers: Asset management in immune cell genomes. Trends Immunol 2015; 36:519-26; PMID:26277449; http://dx.doi.org/10.1016/j.it.2015.07.005
- Romanoski CE, Link VM, Heinz S, Glass CK. Exploiting genomics and natural genetic variation to decode macrophage enhancers. Trends Immunol 2015; 36:507-18; PMID:26298065; http://dx.doi.org/10.1016/j.it.2015.07.006
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010; 38:576-89; PMID:20513432; http://dx.doi.org/10.1016/j.molcel.2010.05.004
- Qiao Y, Jiang X, Lee ST, Karuturi RK, Hooi SC, Yu Q. FOXQ1 regulates epithelial-mesenchymal transition in human cancers. Cancer Res 2011; 71:3076-86; PMID:21346143; http://dx.doi.org/10.1158/0008-5472.CAN-10-2787
- Nakagawa S, Gisselbrecht SS, Rogers JM, Hartl DL, Bulyk ML. DNA-binding specificity changes in the evolution of forkhead transcription factors. Proc Natl Acad Sci U S A 2013; 110:12349-54; PMID:23836653; http://dx.doi.org/10.1073/pnas.1310430110
- Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G, et al. DNA-binding specificities of human transcription factors. Cell 2013; 152:327-39; PMID:23332764; http://dx.doi.org/10.1016/j.cell.2012.12.009
- Poletti V, Delli Carri A, Malagoli Tagliazucchi G, Faedo A, Petiti L, Mazza EM, Peano C, De Bellis G, Bicciato S, Miccio A, et al. Genome-wide definition of promoter and enhancer usage during neural induction of human embryonic stem cells. PloS One 2015; 10:e0126590; PMID:25978676; http://dx.doi.org/10.1371/journal.pone.0126590
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108-12; PMID:19295514; http://dx.doi.org/10.1038/nature07829
- Rhie SK, Hazelett DJ, Coetzee SG, Yan C, Noushmehr H, Coetzee GA. Nucleosome positioning and histone modifications define relationships between regulatory elements and nearby gene expression in breast epithelial cells. BMC Genomics 2014; 15:331; PMID:24885402; http://dx.doi.org/10.1186/1471-2164-15-331
- Diesch J, Sanij E, Gilan O, Love C, Tran H, Fleming NI, Ellul J, Amalia M, Haviv I, Pearson RB, et al. Widespread FRA1-dependent control of mesenchymal transdifferentiation programs in colorectal cancer cells. PloS One 2014; 9:e88950; PMID:24658684; http://dx.doi.org/10.1371/journal.pone.0088950
- Desmet CJ, Gallenne T, Prieur A, Reyal F, Visser NL, Wittner BS, Smit MA, Geiger TR, Laoukili J, Iskit S, et al. Identification of a pharmacologically tractable Fra-1/ADORA2B axis promoting breast cancer metastasis. Proc Natl Acad Sci U S A 2013; 110:5139-44; PMID:23483055; http://dx.doi.org/10.1073/pnas.1222085110
- Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, Wu ZJ, Krall JA, Bierie B, Guo W, et al. Protein kinase C alpha is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 2013; 24:347-64; PMID:24029232; http://dx.doi.org/10.1016/j.ccr.2013.08.005
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell 2011; 43:145-55; PMID:21726817; http://dx.doi.org/10.1016/j.molcel.2011.06.016
- Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Sci 2004; 117:5965-73; PMID:15564374; http://dx.doi.org/10.1242/jcs.01589
- Kim M, Kim T, Johnson RL, Lim DS. Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep 2015; 11:270-82; PMID:25843714; http://dx.doi.org/10.1016/j.celrep.2015.03.015
- Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011; 147:759-72; PMID:22078877; http://dx.doi.org/10.1016/j.cell.2011.09.048
- Hiemer SE, Szymaniak AD, Varelas X. The transcriptional regulators TAZ and YAP direct transforming growth factor beta-induced tumorigenic phenotypes in breast cancer cells. J Biol Chem 2014; 289:13461-74; PMID:24648515; http://dx.doi.org/10.1074/jbc.M113.529115
- Koinuma D, Tsutsumi S, Kamimura N, Taniguchi H, Miyazawa K, Sunamura M, Imamura T, Miyazono K, Aburatani H. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor beta signaling. Mol Cell Biol 2009; 29:172-86; PMID:18955504; http://dx.doi.org/10.1128/MCB.01038-08
- Cyr AR, Kulak MV, Park JM, Bogachek MV, Spanheimer PM, Woodfield GW, White-Baer LS, O'Malley YQ, Sugg SL, Olivier AK, et al. TFAP2C governs the luminal epithelial phenotype in mammary development and carcinogenesis. 2015; 34:436-44; PMID:24469049
- Feuerborn A, Kuffer S, Grone HJ. Forkhead factors regulate epithelial plasticity: impact on cancer progression. Cell Cycle 2011; 10:2454-60; PMID:21685726; http://dx.doi.org/10.4161/cc.10.15.16306
- Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev 2011; 25:2227-41; PMID:22056668; http://dx.doi.org/10.1101/gad.176826.111
- Vernimmen D, Bickmore WA. The hierarchy of transcriptional activation: from enhancer to promoter. Trends Genet: TIG 2015; 31:696-708; PMID:26599498; http://dx.doi.org/10.1016/j.tig.2015.10.004
- Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer 2007; 7:847-59; PMID:17943136; http://dx.doi.org/10.1038/nrc2223
- Saunders LR, McClay DR. Sub-circuits of a gene regulatory network control a developmental epithelial-mesenchymal transition. Development (Cambridge, England) 2014; 141:1503-13; PMID:24598159; http://dx.doi.org/10.1242/dev.101436
- Simon JM, Giresi PG, Davis IJ, Lieb JD. A detailed protocol for formaldehyde-assisted isolation of regulatory elements (FAIRE). Current protocols in molecular biology / edited by Frederick M Ausubel [et al] 2013; Chapter 21:Unit21 6.
- Sutcliffe EL, Parish IA, He YQ, Juelich T, Tierney ML, Rangasamy D, Milburn PJ, Parish CR, Tremethick DJ, Rao S. Dynamic histone variant exchange accompanies gene induction in T cells. Mol Cell Biol 2009; 29:1972-86; PMID:19158270; http://dx.doi.org/10.1128/MCB.01590-08
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 2011; 17
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9:357-9; PMID:22388286; http://dx.doi.org/10.1038/nmeth.1923
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 2009; 25:1952-8; PMID:19505939; http://dx.doi.org/10.1093/bioinformatics/btp340
- Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 2015; 31:166-9; PMID:25260700; http://dx.doi.org/10.1093/bioinformatics/btu638
- Frith MC, Fu Y, Yu L, Chen JF, Hansen U, Weng Z. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res 2004; 32:1372-81; PMID:14988425; http://dx.doi.org/10.1093/nar/gkh299
- Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res 2004; 32:D91-4; PMID:14681366; http://dx.doi.org/10.1093/nar/gkh012