
ABSTRACT
Phosphatase and tensin homolog (PTEN) is an important tumor-suppressor gene which constitutes an important PI3K/Akt pathway by regulating the signaling of multiple biological processes, including apoptosis, metabolism, cell proliferation, and cell growth has been gaining increasing attention. However, the role of PTEN in regulating apoptosis of canine mammary tumors cells still needs further investigation. In this experiment, the effect of PTEN on proliferation and apoptosis in canine mammary tumors (CMT) cells was analyzed. As a result, gene and protein expression levels of apoptosis-related genes were detected. Eukaryotic expression vector pcDNA3.1+-PTEN were successfully constructed and stably transferred into canine CMT cells after geneticin (G418) selection. After pcDNA3.1+-PTEN transfection, compared with control group, the cells proliferation was inhibited and the cell apoptosis was increased in CMT cells. The expression of p-Akt was decreased and the apoptosis-related genes, such as caspase-3, caspase-9, and Bax, were increased. These data serve to demonstrate the function of PTEN on apoptosis and gene regulatory in PI3K/Akt pathway in CMT cells. Collectively, our data link the tumor-suppressor activities of PTEN to the machinery controlling cell cycle through the modulation of signaling molecules whose signal target is the functional inactivation of the apoptosis gene product.
KEYWORDS:
Introduction
Canine mammary tumors (CMTs) are the most common types of canine cancer forms (Rutteman et al. Citation2001; Jemal et al. Citation2011), approximately 50% of which are malignant (Sleeckx et al. Citation2011) and are the most frequent cause of death in dogs. There are similarities in important clinical and pathophysiologic characteristics between canine and human mammary cancers. Therefore, comparative molecular analysis in human provides an opportunity to gain deeper insight into pathway implications in mammary tumorigenesis in both species and to reveal additional regions which are not yet identified in human breast cancer. It may also be an important step in the work towards developing a new cancer treatment.
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is an important tumor-suppressor and is a key negative regulator of PI3K/AKT pathway regulating the signaling of multiple biological processes such as apoptosis, metabolism, cell proliferation, and cell growth (Cantley & Neel Citation1999; Vazquez & Sellers Citation2000). Deletions and mutations of PTEN are widely reported in several human malignancies (Worby & Dixon Citation2014) and in canine osteosarcoma (Levine et al. Citation2002), melanoma (Koenig et al. Citation2002), hemangiosarcoma (Dickerson et al. Citation2005), and mammary tumors (Ressel et al. Citation2009), suggesting that PTEN is associated with tumor development and growth in veterinary oncology.
Furthermore, PTEN is also found in the nucleus where it may contribute to tumor genesis through a mechanism that is dependent of PIP3 dephosphorylation (Carnero & Paramio Citation2014). PIP3 could activate phosphoinositol-dependent kinase (PDK-1), induce several important protein kinases which could stimulate cellular proliferation and enhance cell survival (Schwartzbauer & Robbins Citation2001; Yuan & Whang Citation2002). The PDK-1 substrate Akt blocks apoptosis by inactivating apoptosis-associated proteins such as BAD, FKHR, caspase-3, and caspase-9 (Datta et al. Citation1997; Cardone et al. Citation1998; Brunet et al. Citation1999). Furthermore, previous studies have demonstrated that activation of AKT plays a pivotal role in mammary cancer, by stimulating cell cycle progress, survival, metabolism, and migration through phosphorylation of many physiological substrates (Blanco-Aparicio et al. Citation2007). Hyperactivated AKT has been also shown to promote cell proliferation, possibly through down-regulation of the cyclin-dependent kinase inhibitor p27 as well as up-regulation and stabilization of cyclin D1 (Blume-Jensen & Hunter Citation2001).
We hypothesized that PTEN modulates the PI3K/Akt signaling pathway within CMTs cell by altering the expression of locally produced AKT, mammalian target of rapamycin (mTOR), and apoptosis genes. Therefore, in this study, our aim was to identify the influence of PTEN over-expression in regulation of neoplastic growth and survival in canine mammary tumor cells by introducing PTEN cDNA into canine mammary tumors cells lines (CHMm and CHMp), which provides a theoretical basis for tumor gene therapy.
Materials and methods
Canine breast tumor cell lines, CHMp and CHMm, which were obtained from breast tumor tissue of a female canine (Uyama et al. Citation2006), were provided by the Veterinary Surgeon Laboratory (Tokyo University, Tokyo, Japan). The cells were cultured in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal bovine serum (FBS) (Gibco, Langley, OK), 1% glutamine (Gibco), and 1% penicillin–streptomycin (Gibco) at 37°C under 5% CO2.
RNA isolation and cDNA synthesis
According to the manufacturer’s instructions, total RNA was isolated from cultured cells using Trizol Reagent (Invitrogen, Carlsbad, CA) and the concentration of the extracted total RNA was quantified with a spectrophotometer. RNA integrity was then evaluated through the observation of 18S and 28S ribosomal bands after electrophoresis on 1% agarose gel with the presence of ethidium bromide. cDNA was synthesized from 1 µg of RNA using arbitrary primers and the Prime Script® RT reagent Kit (TaKaRa, Dalian, China) at 37°C for 15 min, 85°C for 5 s.
PTEN gene amplification
The target PCR product size of PTEN gene was 1212 bp, and primers with particular restriction enzyme sites were as follows. Forward primer 5′-CGGAATTCGCCACCATGGGGACAGCCATCATCAAGGAGATCG-3′(EcoRI) and reverse primer 5′-CGGATATCTCAGACTTTTGTGATTTGTGTGTGC-3′ (EcoRV) (GenBank accession number NM_001003192.1). PCR was performed under the following condition: after denaturation at 95°C for 5 min, DNA amplification was performed for 30 cycles at 94°C for 45 s, 55°C for 1 min, and 72°C for 100 s, with a final extension at 72°C for 5 min, and then was kept at 4°C. The PCR product was then sequenced to identify its correctness.
Construction of eukaryotic expression vector of PTEN
To obtain the final recombinant vector, cloned PTEN cDNA was inserted into the EcoRI and EcoRV site of pcDNA3.1+vector. The products were double enzyme digested and sequenced to identify their correctness.
Stable transfection
According to the manufacturer’s instructions, in order to obtain a stable in recombinant clones, CHMp and CHMm were plated into six-well plates at a density of 2 × 105 cells per well and transfected the following day with 3 µg of the recombinant vector or pcDNA3.1+vector, using Lipofectamine 2000 (Invitrogen). Twenty fours after transfection, the medium was replaced, with additional 10% FBS (Gibco) and geneticin (G418) (Gibco) (500 μg/mL-CHMm, 600 μg/mL-CHMp), and the cells were cultured for the next 3 weeks.
Quantitative real-time PCR
According to the manufacturer’s instructions, real-time PCR was performed in a total volume of 20 μL using 96-well micro well plates using SYBR® Premix Ex TaqTM (TaKaRa) on an ABI Prism 7500 (Applied Biosystems, Foster City, CA, USA). The relative expression levels were calculated with the 2−ΔΔCT method (Livak & Schmittgen Citation2001).
The primers were designed by Integrated DNA Technologies Inc. (Integrated DNA Technologies, Coralville, IA; ) and synthesized by Invitrogen (Shanghai, China). All qRT-PCR amplifications were performed in duplicate and repeated three times.
Table 1. Analysis of primer sequences for real-time PCR.
Western blot analysis
Cells were briefly washed twice with ice-cold PBS before scraping on ice with lysis buffer containing 10 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.5% Nonidet P40 containing protease inhibitors (2 mM PMSF, 10 mg/mL leupeptin, and 10 mg/mL aprotinin), and phosphatase inhibitors (100 mM sodium fluoride, 10 mM sodium pyrophosphate, and 2 mM sodium orthovanadate). Cellular debris was removed by centrifugation (12,000 g for 15 min at 4°C). Total cell lysate was quantified by the BCA protein assay (Beyotime, China) and denatured at 100°C for 5 min in a protein sample buffer containing 1% sodium dodecyl sulfate and 1% dithiothreitol. Thirty micrograms of total protein extracts were loaded in each lane and subjected to 10% or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were blocked with 5% skim milk in Tris-buffered saline-Tween for 2 h at room temperature (RT) and incubated with primary antibodies mouse anti-PTEN (1:300 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) in blocking solution for 10–12 h at 4°C. Primary antibodies were detected with horseradish-peroxidase-conjugated goat IgG raised against the corresponding species in blocking solution (2 h, RT) goat anti-rabbit (1:2000 dilution; ZSGB-BIO, China) and goat anti-mouse (1:2000 dilution; ZSGB-BIO, China). Antibody binding was detected with an automatic chemiluminescence imaging analysis system Tanon 5200 (Tanon, China) using the ECL kit (PPLYGEN, China). Bolts were analyzed by densitometry and quantified with Tanon Gis software (Tanon, China).
Cell proliferation assays
According to the manufacturer’s instructions, cell proliferation was assessed by CCK-8 assay as described previously (Cui et al. Citation2013). Stable transfected cells and control cells were plated in 96-well plates with a density of 3000 cells per well. After 48 h cells seeding, cell proliferation was examined using the Cell Counting Kit-8 (DOJINDO, Japan).
Determination of apoptosis by flow cytometry
As described by the manufacturer’s instructions, apoptosis was quantified with Annexin V-FITC apoptosis detection kit (Beyotime Institute of Biotechnology). After 48 h cells seeding, the cells were collected and washed with PBS, gently resuspended in Annexin V binding buffer and incubated with Annexin V-FITC/PI. Afterwards, Flow cytometry was performed using Cell Quest software (BD Biosciences, San Jose, CA, USA).
Statistical analysis
Data were expressed as the mean ± SEM. Each experiment was performed in triplicate. Statistical analyses were performed with SPSS version 11.0 (SPSS, Chicago, IL, USA). All of the significances of difference were analyzed by unpaired Student’s t-test and ANOVA. The p-value of <.05 was considered to be statistically significant.
Results
Cloning of PTEN gene and identification of pcDNA3.1+-PTEN
PTEN gene was amplified by PCR, and the predicted fragment was obtained ((a)). Sequencing results confirmed that PTEN gene sequence was identical with that of PTEN in Genebank. PcDNA3.1+-PTEN vector was double enzyme digested by EcoRI/EcoRV (TaKaRa) ((b)), and then, PTEN gene was cloned into pcDNA3.1+ vector.
Figure 1. (a) PCR detection. M, Marker; 1, PCR product. (b) pcDNA3.1+-PTEN vector digested by EcoRI/EcoRV. M, Marker; 1, pcDNA3.1+-PTEN vector digested by EcoRI/EcoRV. (c) The expression of PTEN gene after stable transfection was detected by RT-PCR. (d) Detection of PTEN protein by Western blot.
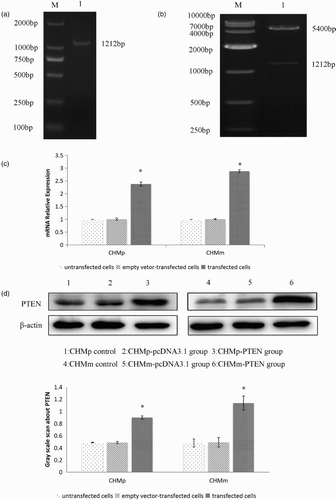
The PTEN expression vector pcDNA3.1+-PTEN and the control vector pcDNA3.1+ were transfected into CHMm and CHMP cells. The cells were selected with the G418, resistant clones were obtained after 3 weeks as CHMm/PTEN, CHMm/DNA3.1+, CHMp/PTEN, and CHMp/DNA3.1 + . When compared with control cells (p < .05, (c)), the expression of PTEN gene in CHMm and CHMp cells rose significantly to 2.38 and 2.88 times, respectively. As shown in , PTEN protein expression was detected by Western blot. Transfection group was significantly higher (45.72% and 57.61%, respectively) than empty vector group and the control group in both CHMp and CHMm cells ((d)).
Evaluation of pcDNA3.1+-PTEN proliferation in CHMp and CHMm cells
To assess the effect of pcDNA3.1+-PTEN expression on proliferation, CHMp and CHMm were maintained in complete culture for 48 h. CCK-8 assay showed that PTEN transfected CHMp and CHMm cells can inhibit the cell proliferation. Compared with the empty vector group and the control group, cells proliferation was significantly reduced (p < .05) ((a)).
Figure 2. (a) Detection of cell proliferation ability after transfection in both CHMp and CHMm cells. (b) Detection of cell apoptosis ability after transfection in both CHMp and CHMm cells. (c) Detection of Bcl-2, Bax, caspase-3, and caspase-9 gene expression by RT-PCR.
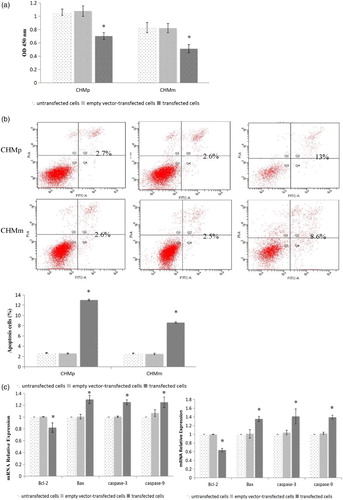
Effects of pcDNA3.1+-PTEN apoptosis in CHMp and CHMm cells
To determine the effect of pcDNA3.1+-PTEN expression on apoptosis in CHMp and CHMm, the cells were maintained in complete culture for 48 h. Annexin V-PI assay showed that PTEN transfected CHMp and CHMm cells increase the cell apoptosis. Compared with the empty vector group and the control group, cells proliferation was significantly reduced (p < .05) ((b)).
RT-PCR results in (c) showed that caspase-3, caspase-9, and Bax mRNA were expressed in each cell group. Compared with the empty vector group and the control group, caspase-3, caspase-9, and Bax mRNA expression level of transfected PTEN groups were significantly increased in CHMp and CHMm cells (p < .05).
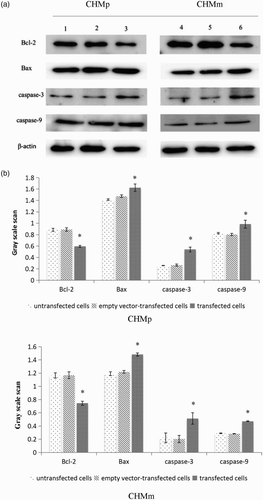
Western blot in results showed that caspase-3, caspase-9, and Bax protein were expressed in each cell group. Compared with the empty vector group and the control group, caspase-3, caspase-9, and Bax protein expression level of transfected PTEN groups were both significantly increased in CHMp and CHMm cells (p < .05).
Figure 3. Detection of Bcl-2, Bax, caspase-3, and caspase-9 protein expression by Western blot in CHMp and CHMm cells.
Effects of pcDNA3.1+-PTEN in PI3K/Akt signal pathway
RT-PCR results showed that Akt mRNA was expressed in each cell group. Compared with the empty vector group and the control group, Akt mRNA expression level was not significantly changed in CHMp and CHMm cells (p > .05) ((a)).
Figure 4. (a) Detection of Akt, mTOR, P27kip, and CyclinD1 gene expression by RT-PCR. (b–d) Detection of Akt, pAKT, mTOR, P27, and CyclinD1 protein expression by Western blot in CHMp and CHMm cells.
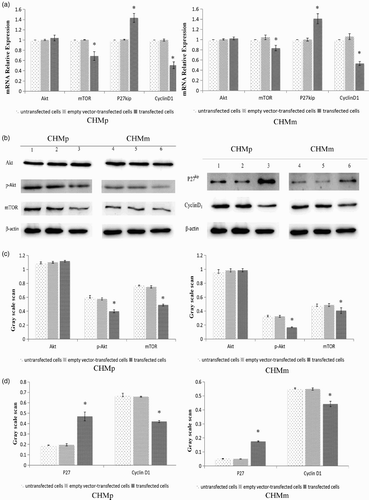
Western blot results showed that Akt and p-Akt protein were both expressed in each of the cell groups. Compared with the empty vector groups and the control groups, there was no significant change in the total Akt protein expression level in CHMp and CHMm cells (p > .05). However, compared with the empty vector groups and the control groups, p-Akt protein expression levels of transfected PTEN groups were significantly decreased (p < .05) ((b–d)).
Discussion
Although there has been a proposed role of PTEN in cancer, the molecular mechanism by which signal pathway can modulate cells proliferation or apoptosis in canine mammary tumor is still unknown. Moreover, we showed that over-expression of PTEN in CHMp and CHMm cells, which promotes cell apoptosis and downstream effects involving the PI3K/Akt signaling pathway, and the expression of caspase-3, caspase-9, and Bax. The identification of PTEN as an important tumor suppressor in cell proliferation and apoptosis provides insight into the contribution of altered PTEN expression in contributing to tumor therapy and animal welfare.
PTEN, a tumor-suppressor gene, has been shown that deletion is the most common event observed in breast tumor (Kechagioglou et al. Citation2014; Worby & Dixon Citation2014). Moreover, it is well known that loss of PTEN plays an important role in tumor cell proliferation, metastasis, cell cycle arrest, regulation of cell adhesion and differentiation (Borge et al. Citation2015). It has been hypothesized that this is due to lack of control of the signaling pathways that mediate cellular processes such as apoptosis and migration (Huang & Kontos Citation2002; Koul et al. Citation2002; Pene et al. Citation2002). Additionally, in this study, the over-expression PTEN genes in CHMp and CHMm cells were successfully reconstructed, thus providing a model to study the PI3K/Akt signal pathway transduction modulated by PTEN expression in canine.
The involvement of over-expression PTEN-induced cell proliferation was further evaluated. In fact, it has been shown that PTEN over-expression inhibits cell growth in a variety of normal and transformed, human and murine cells (Paramio et al. Citation1999). This study is in line with previous reports, compared to the control group, that over-expression PTEN cells led to a decrease in cell proliferation. We have therefore studied the expression of p27 and cyclinD1 genes, which was in accordance with Weng et al. (Citation2001), which showed that there was an increase in the abundance of p27, a decrease in the protein levels of cyclin D1 and the inhibition of Akt phosphorylation. This might reflect that PTEN may have a broader role other than only on the cell’s physical aspect. Thus, for these analysis purposes, this strongly suggests that tumors of different gene expression along specific pathway (Borge et al. Citation2015) and that different mechanisms of genomic instability give rise to the different breast tumors progress and may contribute to their characteristic biological and clinical behavior (Bergamaschi et al. Citation2006). However, functional evaluation of the over-expression PTEN in this present study would greatly add to our understanding of their importance in CMT development and should be addressed in future studies.
In this study, our observations indicated that PTEN could induce apoptosis. Based on the flow of cytometric analysis of CMTs, the proportion of apoptosis cells was greater than the control cells in this study. In addition, previous studies have shown that there has been PI3K/Akt pathway activation due to apoptosis proteins deactivation including caspase-3, caspase-9, and Bax (Manning & Cantley Citation2007). Thus, to further evaluate the contribution of PTEN on the biological effects of apoptosis, we assessed the impact of over-expression of PTEN in PI3K/Akt signaling pathway by RT-PCR and Western blot analysis. We found out that there was a significant decrease in apoptosis-related protein, such as Bcl-2 and Bax, inhibitor of apoptosis protein caspase-3 and caspase-9. An important example of such changes is supported by Li et al. (Citation2013), PTEN exhibits strong tumor-suppressive activities, activating intrinsic apoptotic pathway in human small-cell lung cancer. In view of our experimental finding, we speculate that the apoptosis genes may serve to modulate the effect of PTEN in PI3K/Akt signal pathway and thereby may contribute to suppressing the progression of the tumor.
PI3K/Akt signal pathway is a critical event in a wide variety of cellular processes, including proliferation, migration, and survival (Huang & Kontos Citation2002; Liu et al. Citation2008). In a very interesting study, AKT was one of the proteins recruited to the membrane by PIP3 where it was activated by other PIP3-activated protein, PDK1 (Mora et al. Citation2004). Similarly, Paramio showed that PI3K and Akt or PDK1 could be active by PTEN which could then induce growth arrest in human retinoblastoma. Furthermore, Akt and PTEN are associated with breast cancer malignancy and a poor prognosis (Stal et al. Citation2003; Tsutsui et al. Citation2005). Akt activation inhibition leads to the consequent modulation of multiple downstream effector pathways (Blanco-Aparicio et al. Citation2007). However, we observed that, although Akt genes has no significant changes after PTEN over-expression, p-Akt was obviously decreased, which is in line with the previous study which showed that PTEN acts as a key negative regulator in the PI3K/AKT pathway (Cantley & Neel Citation1999; Yuan & Cantley Citation2008). It has also been reported that loss of functional PTEN could lead to increased activity of AKT and the mTOR kinase pathways, which can promote both cell survival and proliferation (Tamura et al. Citation1998; Sarbassov et al. Citation2005). On the contrary, in this study, over-expression of PTEN canine tumor cells, mTOR, and the cell proliferation were decreased. Furthermore, it has been shown that there are potential regulatory mechanisms between the PTEN/PI3K/AKT pathway and the cell cycle target gene-cyclinD1with a clear observation of a decrease in mRNA and protein level leading to up-regulation of p27 in our study, which is in accordance with Weng et al. (Citation2001). Therefore, PTEN appears to inhibit cell cycle progression through the cooperation of its protein phosphatase activity in PI3K/Akt signal pathway.
In summary, it is shown here that PTEN over-expression could induce canine mammary tumor cells apoptosis and down-regulated PI3K/Akt pathway. Therefore, based on target predication, further work is warranted to evaluate the role of tumor signal transduction. Knowledge of specific processes such as tumor cell proliferation, invasion, and migration that are regulated by cell signal pathway, and the identification of critical targets individual genes, such as PTEN, could provide novel insight into the mechanisms of tumorigenesis and metastasis in canine mammary tumor.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bergamaschi A, Kim YH, Wang P, Sørlie T, Hernandez-Boussard T, Lonning PE, Tibshirani R, Børresen-Dale AL, Pollack JR. 2006. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer. 45:1033–1040. doi: 10.1002/gcc.20366
- Blanco-Aparicio C, Renner O, Leal JF, Carnero A. 2007. PTEN, more than the AKT pathway. Carcinogenesis. 28:1379–1386. doi: 10.1093/carcin/bgm052
- Blume-Jensen P, Hunter T. 2001. Oncogenic kinase signalling. Nature. 411:355–365. doi: 10.1038/35077225
- Borge KS, Nord S, Van Loo P, Lingjærde OC, Gunnes G, Alnæs GI, Solvang HK, Lüders T, Kristensen VN, Børresen-Dale AL. 2015. Canine mammary tumours are affected by frequent copy number aberrations, including amplification of MYC and loss of PTEN. PloS ONE. 10:e0126371. doi: 10.1371/journal.pone.0126371
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 96:857–868. doi: 10.1016/S0092-8674(00)80595-4
- Cantley LC, Neel BG. 1999. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 96:4240–4245. doi: 10.1073/pnas.96.8.4240
- Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. 1998. Regulation of cell death protease caspase-9 by phosphorylation. Science. 282:1318–1321. doi: 10.1126/science.282.5392.1318
- Carnero A, Paramio JM. 2014. The PTEN/PI3K/AKT pathway in vivo, cancer mouse models. Front Oncol. 4:252. doi: 10.3389/fonc.2014.00252
- Cui Z, Ren S, Lu J, Wang F, Xu W, Sun Y, Wei M, Chen J, Gao X, Xu C. 2013. The prostate cancer-up-regulated long noncoding RNA PlncRNA-1 modulates apoptosis and proliferation through reciprocal regulation of androgen receptor. Urol Oncol Semin Ori Invest. 31:1117–1123. doi: 10.1016/j.urolonc.2011.11.030
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. 1997. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 91:231–241. doi: 10.1016/S0092-8674(00)80405-5
- Dickerson E, Thomas R, Fosmire S, Lamerato-Kozicki A, Bianco S, Wojcieszyn J, Breen M, Helfand S, Modiano J. 2005. Mutations of phosphatase and tensin homolog deleted from chromosome 10 in canine hemangiosarcoma. Vet Pathol Online. 42:618–632. doi: 10.1354/vp.42-5-618
- Huang J, Kontos CD. 2002. Inhibition of vascular smooth muscle cell proliferation, migration, and survival by the tumor suppressor protein PTEN. Arterioscler Thromb Vasc Biol. 22:745–751. doi: 10.1161/01.ATV.0000016358.05294.8D
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. 2011. Global cancer statistics. CA Cancer J Clin. 61:69–90. doi: 10.3322/caac.20107
- Kechagioglou P, Papi RM, Provatopoulou X, Kalogera E, Papadimitriou E, Grigoropoulos P, Nonni A, Zografos G, Kyriakidis DA, Gounaris A. 2014. Tumor suppressor PTEN in breast cancer: heterozygosity, mutations and protein expression. Anticancer Res. 34:1387–1400.
- Koenig A, Bianco S, Fosmire S, Wojcieszyn J, Modiano J. 2002. Expression and significance of p53, rb, p21/waf-1, p16/ink-4a, and PTEN tumor suppressors in canine melanoma. Vet Pathol Online. 39:458–472. doi: 10.1354/vp.39-4-458
- Koul D, Shen R, Garyali A, Ke L, Liu TJ, Yung WA. 2002. MMAC/PTEN tumor suppressor gene regulates vascular endothelial growth factor-mediated angiogenesis in prostate cancer. Int J Oncol. 21:469–475.
- Levine R, Forest T, Smith C. 2002. Tumor suppressor PTEN is mutated in canine osteosarcoma cell lines and tumors. Vet Pathol Online. 39:372–378. doi: 10.1354/vp.39-3-372
- Li D, Zhang Y, Xie Y, Xiang J, Zhu Y, Yang J. 2013. Enhanced tumor suppression by adenoviral PTEN gene therapy combined with cisplatin chemotherapy in small-cell lung cancer. Cancer Gene Ther. 20:251–259. doi: 10.1038/cgt.2013.14
- Liu Z, Hou P, Ji M, Guan H, Studeman K, Jensen K, Vasko V, El-Naggar AK, Xing M. 2008. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J Clin Endocrinol Metab. 93:3106–3116. doi: 10.1210/jc.2008-0273
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods. 25:402–408. doi: 10.1006/meth.2001.1262
- Manning BD, Cantley LC. 2007. AKT/PKB signaling: navigating downstream. Cell. 129:1261–1274. doi: 10.1016/j.cell.2007.06.009
- Mora A, Komander D, van Aalten DM, Alessi DR. 2004. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 15:161–170. doi: 10.1016/j.semcdb.2003.12.022
- Paramio JM, Navarro M, Segrelles C, Gomez-Casero E, Jorcano JL. 1999. PTEN tumour suppressor is linked to the cell cycle control through the retinoblastoma protein. Oncogene. 18:7462–7468. doi: 10.1038/sj.onc.1203151
- Pene F, Claessens YE, Muller O, Viguie F, Mayeux P, Dreyfus F, Lacombe C, Bouscary D. 2002. Role of the phosphatidylinositol 3-kinase/Akt and mTOR/P70S6-kinase pathways in the proliferation and apoptosis in multiple myeloma. Oncogene. 21:6587–6597. doi: 10.1038/sj.onc.1205923
- Ressel L, Millanta F, Caleri E, Innocenti V, Poli A. 2009. Reduced PTEN protein expression and its prognostic implications in canine and feline mammary tumors. Vet Pathol Online. 46:860–868. doi: 10.1354/vp.08-VP-0273-P-FL
- Rutteman G, Withrow S, MacEwen E. 2001. Tumors of the mammary gland. Small Anim Clin Oncol. 3:455–477.
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 307:1098–1101. doi: 10.1126/science.1106148
- Schwartzbauer G, Robbins J. 2001. The tumor suppressor gene PTEN can regulate cardiac hypertrophy and survival. J Biol Chem. 276:35786–35793. doi: 10.1074/jbc.M102479200
- Sleeckx N, De Rooster H, Veldhuis Kroeze E, Van Ginneken C, Van Brantegem L. 2011. Canine mammary tumours, an overview. Reprod Domest Anim. 46:1112–1131. doi: 10.1111/j.1439-0531.2011.01816.x
- Stal O, Pérez-Tenorio G, Akerberg L, Olsson B, Nordenskjold B, Skoog L, Rutqvist LE. 2003. Akt kinases in breast cancer and the results of adjuvant therapy. Breast Cancer Res. 5:37–44. doi: 10.1186/bcr569
- Tamura M, Gu J, Matsumoto K, Aota SI, Parsons R, Yamada KM. 1998. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 280:1614–1617. doi: 10.1126/science.280.5369.1614
- Tsutsui S, Inoue H, Yasuda K, Suzuki K, Higashi H, Era S, Mori M. 2005. Reduced expression of PTEN protein and its prognostic implications in invasive ductal carcinoma of the breast. Oncology. 68:398–404. doi: 10.1159/000086981
- Uyama R, Nakagawa T, Hong SH, Mochizuki M, Nishimura R, Sasaki N. 2006. Establishment of four pairs of canine mammary tumour cell lines derived from primary and metastatic origin and their E-cadherin expression. Vet Comp Oncol. 4:104–113. doi: 10.1111/j.1476-5810.2006.00098.x
- Vazquez F, Sellers WR. 2000. The PTEN tumor suppressor protein: an antagonist of phosphoinositide 3-kinase signaling. BBA-Rev Cancer. 1470:21–35.
- Weng LP, Brown JL, Eng F. 2001. PTEN coordinates G1 arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum Mol Genet. 10:599–604. doi: 10.1093/hmg/10.6.599
- Worby CA, Dixon JE. 2014. Pten. Annu Rev Biochem. 83:641–669. doi: 10.1146/annurev-biochem-082411-113907
- Yuan T, Cantley L. 2008. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 27:5497–5510. doi: 10.1038/onc.2008.245
- Yuan XJ, Whang YE. 2002. PTEN sensitizes prostate cancer cells to death receptor-mediated and drug-induced apoptosis through a FADD-dependent pathway. Oncogene. 21:319–327. doi: 10.1038/sj.onc.1205054