
ABSTRACT
To explore the function of VIG-1 in Caenorhabditis elegans, we analyzed the phenotypes of two vig-1 deletion mutants: vig-1(tm3383) and vig-1(ok2536). Both vig-1 mutants exhibited phenotypes associated with genome instability, such as a high incidence of males (Him) and increased embryonic lethality. These phenotypes became more evident in succeeding generations, implying that the germline of vig-1 accumulates DNA damage over generations. To examine whether vig-1 causes a defect in the DNA damage response, we treated worms with UV or camptothecin, a specific topoisomerase I inhibitor. We observed that the embryonic survival of the vig-1 mutants was reduced compared with that of the wild-type worms. Our results thus suggest that VIG-1 is required for maintaining genome stability in response to endogenous and exogenous genotoxic stresses.
1. Introduction
Genome integrity is critical for the survival and reproduction of living organisms. The genome is constantly damaged by diverse chemical and physical agents from endogenous and exogenous sources. Thus, cells have evolved intricate mechanisms, collectively termed the DNA damage response (DDR), to overcome the genotoxic stresses that disturb genome integrity. The DDR detects DNA damage and evokes appropriate cellular processes (Ciccia and Elledge Citation2010). The failure of the DDR to preserve genome integrity often results in diseases, including cancers and neurodegenerative disorders (Jackson and Bartek Citation2009; O'Driscoll Citation2012).
The nematode Caenorhabditis elegans (C. elegans) is a good model for studying the DDR mechanisms (Stergiou and Hengartner Citation2004; Rose Citation2014). Genes responsible for the DDR can be readily identified because the corresponding mutants frequently show increased embryonic lethality and/or a high incidence of males (Him) as phenotypes. As most of the genes involved in the DDR are conserved between worms and mammals, studies of C. elegans could provide valuable information for understanding the human DDR and for treating relevant human diseases.
C. elegans germline is a lineage of immortal cells that are specialized to produce gametes (Smelick and Ahmed Citation2005). Defects in the DDR system often make germ cells mortal in a generation-dependent fashion, and the animals with these defects eventually become sterile. For example, animals carrying a loss-of-function mutation in the mrt-2 gene, which encodes a checkpoint protein for the DDR, reproduce normally in early generations, but progressively lose their fertility in later generations (Ahmed and Hodgkin Citation2000).
Caudy et al. (Citation2003) reported that VIG-1 is a putative component of the RNA-induced silencing complex (RISC) in C. elegans and that depletion of VIG-1 by RNAi disrupts the let-7 miRNA function. Recently, it has been reported that a vig-1 deletion leads to the reduction of several miRNA species, including let-7 (Wang et al. Citation2017). However, the biological function of VIG-1 is still poorly understood. In this study, we provide evidence that VIG-1 is involved in the maintenance of genome stability.
2. Materials and methods
2.1. C. elegans strains and maintenance
The strains used in this study were Bristol N2 (wild type), FX03383 vig-1(tm3383), and RB1933 vig-1(ok2536). FX03383 vig-1(tm3383) was obtained from Shohei Mitani, and RB1933 vig-1(ok2536) was obtained from the C. elegans Genetics Center. Worms were cultivated using standard methods (Brenner Citation1974). All strains were maintained at 20°C unless otherwise stated. Before initiating experiments, the vig-1 mutants were outcrossed to wild-type worms for six times.
2.2. Phenotypic analyses
Worms were cultured at 20°C for various generations, and single L4 hermaphrodites of each generation were placed on NGM plates seeded with E. coli OP50. They were allowed to lay eggs at 20 or 25°C, and further experiments were carried out at the constant temperature of 20 or 25°C. Worms were transferred to fresh plates every 24 h until the worms no longer laid eggs.
2.2.1. Eggs laid per worm
The number of eggs laid by each worm was counted while transferring the worms to fresh NGM plates seeded with OP50.
2.2.2. Embryonic lethality
The number of unhatched eggs was counted 24 h after the eggs were laid. The embryonic lethality (%) was determined by counting the number of unhatched eggs per 100 eggs laid.
2.2.3. Male ratio
The males were counted when the hatched progeny reached the adult stage. The adult male can be distinguished from the hermaphrodite by its small size and unique tail shape. The male ratio (%) was determined by counting the number of males per 100 adult worms.
2.3. Hoechst staining
To observe the chromosomes at the diakinesis oocytes, adult worms were placed in a drop of M9 buffer on a slide, fixed with 95% ethanol for 3-5 min, treated with 76 µM Hoechst 33342 (Sigma), and immediately mounted in Prolong Gold antifade reagent (Molecular Probes). Microscopy images were captured using a fluorescence microscope (LSM 710, Carl Zeiss). For quantification of chromosomes, two diakinesis oocytes, which are close to the anterior spermatheca, were analyzed.
2.4. UV sensitivity assay
UV sensitivity assays were performed as described by Craig et al. (Citation2012) with minor modifications. Synchronized L4 hermaphrodites were irradiated with various doses of UV at an emission peak of 254 nm (GS gene linker UV chamber, Bio-Rad). The UV-irradiated worms were transferred to fresh NGM plates seeded with OP50 and grown at 20°C. The eggs laid during 20-40 h following UV exposure were collected. The number of hatched worms was counted 24 h after the eggs were laid. The embryonic survival (%) was determined by counting the number of hatched animals per 100 eggs laid. The embryonic survival of the UV-irradiated worms was normalized to that of the nonirradiated control worms.
2.5. Camptothecin (CPT) sensitivity assay
CPT sensitivity assay was performed as described by Saito et al. (Citation2012) with minor modifications. Synchronized L4 hermaphrodites were grown on NGM plates seeded with OP50 for 19 h at 20°C. The worms were then treated with various concentrations of CPT (Sigma) in M9 buffer containing OP50 with gentle shaking in the dark for 19 h. After the CPT-treated worms were allowed to recover for 3 h on NGM plates seeded with OP50, eggs were collected for 4 h. The number of hatched worms was counted 24 h after the eggs were laid. The embryonic survival (%) was determined by counting the number of hatched animals per 100 eggs laid. The embryonic survival of the CPT-treated worms was normalized to that of the nontreated control worms.
3. Results
3.1. vig-1 mutants show a progressive loss of fertility
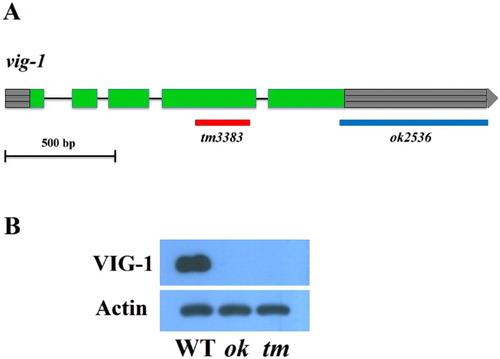
To explore the biological function of VIG-1, we analyzed the phenotypes of two vig-1 deletion mutants: vig-1(tm3383) and vig-1(ok2536). vig-1(tm3383) has a 250-bp deletion within the protein coding sequence, whereas vig-1(ok2536) has a 676-bp deletion, which corresponds to the protein coding sequence of the last 14 amino acids and entire predicted 3′ untranslated region (UTR) ((A)). We tested whether these two vig-1 mutants produce the VIG-1 protein by performing western blot experiments using an anti-VIG-1 monoclonal antibody. The antibody detected a protein band of approximately 50 kDa in the wild-type sample, but no band was found in the vig-1 mutants ((B)), showing that the mutants are defective in producing the VIG-1 protein.
Figure 1. Lack of the VIG-1 protein in two vig-1 deletion mutants. (A) Deleted DNA regions in the two vig-1 mutants are shown. Green boxes represent exons, lines represent introns, and gray boxes with horizontal lines at both ends represent 5′ and 3′ UTRs. (B) Immunoblot analysis of VIG-1 expression. Protein lysates were prepared from mixed-stage populations of the indicated strains, and immunoblotted with a monoclonal antibody against VIG-1. Actin was used as a loading control. WT, wild type; ok, vig-1(ok2536); tm, vig-1(tm3383).
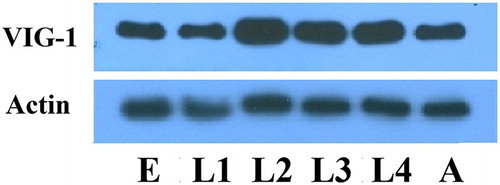
We next determined the temporal expression pattern of VIG-1 in the wild-type animals with the anti-VIG-1 monoclonal antibody. VIG-1 expression was observed at each developmental stage examined (), implying that VIG-1 plays a role in all the developmental stages.
Figure 2. Temporal pattern of VIG-1 expression. Protein lysates were prepared from wild-type worms at each developmental stage, and immunoblotted with an anti-VIG-1 antibody. Actin was used as a loading control. E, egg stage; L1-L4, larval stages; A, adult stage.
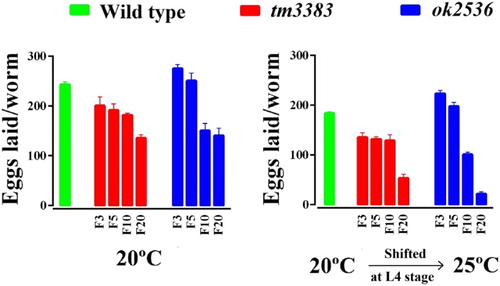
When we analyzed the phenotypes of homozygous vig-1 mutants (vig-1/vig-1) directly derived from a heterozygote (vig-1/+), very mild to no difference from the wild-type animals was noted at 20°C. However, the vig-1 mutants of succeeding generations laid a smaller number of eggs than the wild-type animals at 20°C (, left panel). In generation F3, vig-1(tm3383) laid 201 ± 17.4 eggs, but only 135 ± 7.0 eggs were laid at F20. In the case of vig-1(ok2536), 276 ± 7.4 and 141 ± 14.7 eggs were laid at F3 and F20, respectively. By comparison, wild-type animals laid ∼250 eggs in all the generations tested.
Figure 3. The total number of eggs laid by the vig-1 mutants decreases over generations. Worms were cultured at 20°C for various generations, and single L4 hermaphrodites at the indicated generation were transferred to individual plates and allowed to lay eggs at either 20°C (left panel) or 25°C (right panel). The total number of eggs laid by the worms was determined as described in the Materials and methods. Error bars indicate SEM (n = 10).
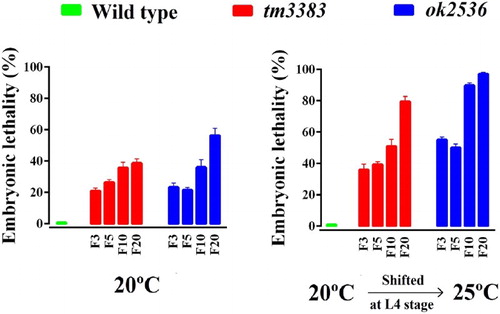
We also observed that the vig-1 mutants displayed higher embryonic lethality than the wild-type animals at 20°C (, left panel). In generation F3, vig-1 deletion caused ∼20% embryonic lethality, which gradually increased in subsequent generations. As a consequence, the vig-1 mutants produced smaller brood sizes (total number of progeny/worm) than the wild-type animals at 20°C. For example, in generation F3, brood sizes of vig-1(tm3383) and vig-1(ok2536) were 158 ± 12.1 and 211 ± 7.0, respectively. In generation F10, brood sizes decreased to 117 ± 7.0 for vig-1(tm3383) and 93 ± 9.1 for vig-1(ok2536). In generation F20, brood sizes were further reduced to 83 ± 6.5 for vig-1(tm3383) and 65 ± 12.5 for vig-1(ok2536). The brood size of wild-type animals was ∼240, regardless of the generation number. This progressive decrease in brood size evoked by vig-1 deletion suggests that the germline of vig-1 mutants gradually accumulates genetic alterations over generations.
Figure 4. The embryonic lethality of the vig-1 mutants increases over generations. Worms were cultured at 20°C for various generations, and single L4 hermaphrodites at the indicated generation were transferred to individual plates and allowed to lay eggs at either 20°C (left panel) or 25°C (right panel). The embryonic lethality (%) was determined as described in the Materials and methods. Error bars indicate SEM (n = 10).
We also found that the hatched progeny of the vig-1 mutants showed diverse types of morphological abnormalities, including a growth arrest, protruding vulva, and aberrant male tail structure (Supplementary Figure 1). Again, these abnormalities were less frequent in earlier generations, but were clearly visible in later generations.
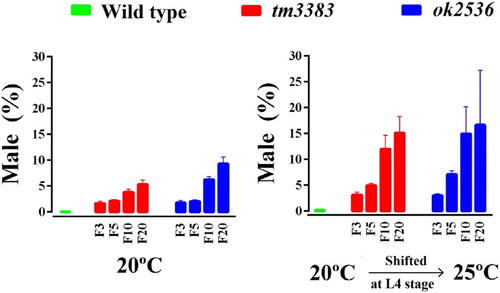
As many mutations affecting genome stability are temperature-sensitive (Ahmed and Hodgkin Citation2000; Lee et al. Citation2003; Lee et al. Citation2004), we examined whether the vig-1 mutants are sensitive to high temperature. When the vig-1 mutants were shifted to 25°C during the L4 stage, the phenotypes of the vig-1 mutants were enhanced. The number of eggs laid per worm was greatly reduced in the vig-1 mutants, especially at F20, whereas that in the wild-type animals was moderately reduced, regardless of generation (, right panel). Temperature-shift of the vig-1 mutants to 25°C resulted in a further ∼2-fold increase in embryonic lethality compared with the values at 20°C in all generations tested (, right panel). Furthermore, if the hatched progeny of the vig-1 mutants were allowed to grow at 25°C, they often exhibited developmental abnormalities, such as a growth arrest. Although some progeny could survive and reach adulthood, they usually failed to lay eggs. This high-temperature sterility was seen even in early generations. The biochemical reason for the sensitivity of the vig-1 mutants to high temperature is currently unknown.
3.2. vig-1 mutants are defective in meiotic chromosome segregation
C. elegans populations are normally XX hermaphrodites (5 pairs of autosomes and 2X chromosomes), but XO males arise due to X-chromosome loss during meiosis with a frequency of ∼0.2% (Hodgkin et al. Citation1979). We found that both vig-1 mutants had a high incidence of the males (Him) phenotype at 20°C (, left panel). In generation F3, vig-1 deletion caused ∼2% of the worms to be males. When the vig-1 mutant worms reached F20, the male ratio increased to 5-9%, whereas wild-type worms showed less than 0.1% males. We also observed that temperature-shift of the vig-1 mutants to 25°C markedly increased the male ratio (, right panel). These data indicate that X-chromosome nondisjunction is promoted due to the lack of VIG-1. Conceivably, the VIG-1 protein is necessary for proper meiotic chromosome segregation.
Figure 5. The male ratio of vig-1 mutants increases over generations. Worms were cultured at 20°C for various generations, and single L4 hermaphrodites at the indicated generation were allowed to lay eggs at either 20°C (left panel) or 25°C (right panel). The male (%) was determined as described in the Materials and methods. Error bars indicate SEM (n = 10).
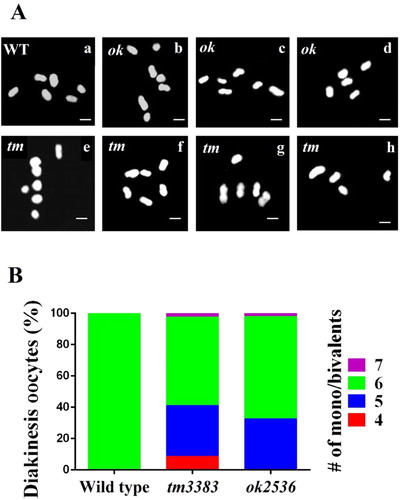
As a high incidence of the males (Him) phenotype was evident in the vig-1 mutants, we asked if vig-1 induces chromosomal alterations in the diakinesis oocytes. During diakinesis, which is the last stage of meiotic prophase I, C. elegans chromosomes are highly condensed and easily identified by staining DNA (Hillers et al. Citation2017). When the wild-type hermaphrodites were treated with Hoechst dye, typical 6 bivalents were observed; however, various abnormalities in chromosome number and size were detected in the vig-1 mutants ((A)). Some diakinesis oocytes of the vig-1 mutants had more than six Hoechst-stained bodies, and many other diakinesis oocytes possessed less than six bodies. In generation F3, the frequencies of the diakinesis oocytes with aberrant chromosome structure were 43.5% (20 out of 46) for vig-1(tm3383) and 34.6% (18 out of 52) for vig-1(ok2536) ((B)). Overall, our results strongly suggest that VIG-1 serves an important function in meiotic processes.
Figure 6. vig-1 deletion causes defects in chromosomal segregation during meiosis. (A) vig-1 deletion induces chromosomal aberrations in the diakinesis oocytes. F3 generation adult hermaphrodites were stained with Hoechst dye, and the chromosomes in the diakinesis oocytes were observed using a fluorescence microscope. Whereas all the wild-type animals (WT) examined had normal 6 chromosomal bivalents (a), the vig-1 mutants exhibited 4-7 chromosomal bodies (b-h). The vig-1 mutants with 7 mono/bivalents (b and e), 6 mono/bivalents (c and f), 5 mono/bivalents (d and g), and 4 mono/bivalents (h) are shown. ok, vig-1(ok2536); tm, vig-1(tm3383). Scale bar, 2 µm. (B) Chromosomal abnormalities seen in F3 generation vig-1 mutants are quantified.
3.3. vig-1 mutants are sensitive to exogenous DNA damage
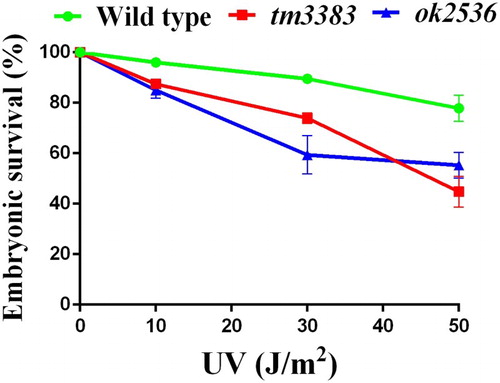
Like other living organisms, C. elegans possesses elaborate DDR mechanisms to protect the genome from genotoxic stresses (Stergiou and Hengartner Citation2004). In fact, C. elegans mutants with a defect in the DDR exhibit decreased embryonic survival in response to DNA-damaging agents, such as UV radiation (Park et al. Citation2004) and camptothecin (CPT) treatment (Hyun et al. Citation2016). To investigate whether the vig-1 mutants are defective in eliciting the DDR, we subjected L4-stage vig-1 mutant worms to UV radiation. Although the embryonic survivals of both the wild-type worms and vig-1 mutants were reduced in a dose-dependent manner, the vig-1 mutants were more sensitive to UV exposure than the wild-type worms (). In response to 50 J/m2 UV, vig-1(tm3383), vig-1(ok2583), and wild-type worms showed 45 ± 6.5%, 55 ± 5.1%, and 78 ± 5.2% embryonic survival, respectively. This hypersensitivity of the vig-1 mutants implies that VIG-1 participates in the DDR process against UV.
Figure 7. The vig-1 mutants are sensitive to UV. F3 generation L4 hermaphrodites were irradiated with various doses of UV, and the embryonic survival (%) was determined as described in the Materials and methods. More than 100 embryos were used for each data point. Similar results were obtained from three separate experiments, and error bars indicate SEM.
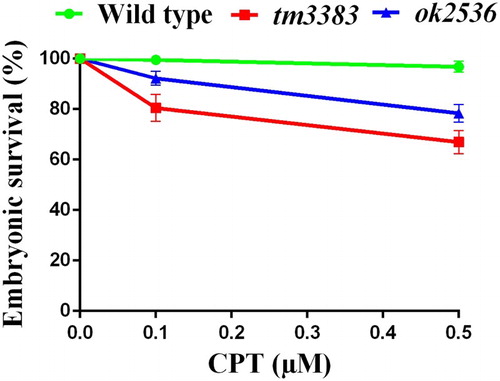
Double strand breaks (DSBs) are generated by DNA damage signals from either physiological (endogenous) or environmental (exogenous) sources, and cells activate the homologous recombination (HR) or non-homologous end joining (NHEJ) pathway to repair DSBs (Ciccia and Elledge Citation2010; Rastogi et al. Citation2010). To test the possibility that VIG-1 plays a role in DSB repair, we treated the worms with CPT, a specific topoisomerase I inhibitor, which makes DSBs at replication forks (Liu et al. Citation2000; Sakasai and Iwabuchi Citation2015). We observed that the embryonic survival of the vig-1 mutants was substantially diminished compared with that of the wild-type worms (). Upon the exposure to 0.5 µM CPT, vig-1(tm3383) and vig-1(ok2583) showed 67 ± 4.6% and 78 ± 3.5% embryonic survival, respectively. By comparison, the embryonic survival of wild-type worms was 97 ± 2.1% with 0.5 µM CPT treatment. These results support the notion that VIG-1 is involved in the CPT-induced DSB repair.
Figure 8. The vig-1 mutants are sensitive to CPT. F3 generation young adults were treated with 0.1 or 0.5 μM CPT for 19 h, and the embryonic survival (%) was determined as described in the Materials and methods. More than 100 embryos were used for each data point. Similar results were obtained from four separate experiments, and error bars indicate SEM.
4. Discussion
Several lines of evidence described in this study indicate that VIG-1 is a regulator of genome stability in C. elegans. First, vig-1 mutants exhibited a progressive loss of fertility. Second, vig-1 mutants revealed aberrant chromosome segregation during meiosis. Third, vig-1 mutants were more sensitive to DNA-damaging agents (UV and CPT) than wild-type worms.
The high degree of embryonic lethality seen in vig-1 mutants is likely due to meiotic chromosome missegregation because embryos with a loss of autosome usually fail to hatch. It is also possible that a defective DDR system for endogenous DNA damage contributes to embryonic lethality. For instance, nonviable embryos can be produced if SPO-11-induced DSBs, which trigger meiotic recombination, are not properly repaired (Alpi et al. Citation2003; Penkner et al. Citation2007; Lemmens et al. Citation2013).
UV induces a wide variety of DNA lesions, such as cyclobutane pyrimidine dimers, 6-4 photoproducts, and DNA strand breaks (Rastogi et al. Citation2010). UV-induced DNA alterations can be repaired via diverse processes, including the nucleotide excision repair (NER), HR, and NHEJ pathways. The hypersensitivity of vig-1 mutants to UV raises the possibility that VIG-1 serves as a component of the molecular machinery that participates in the repair of DNA damage. CPT, a specific inhibitor of topoisomerase I, generates DSBs during S phase when DNA is being replicated (Hsiang et al. Citation1989; Ryan et al. Citation1991; Sakasai and Iwabuchi Citation2015). The hypersensitivity of vig-1 mutants to CPT implies that VIG-1 might play a role in DSB repair.
VIG-1 contains the RGG/RG motif, which is found in many RNA-binding proteins and shown to affect various aspects of RNA metabolism (Thandapani et al. Citation2013). In addition to its function in RNA binding, the RGG/RG motif provides the site for protein interaction. It thus can be postulated that VIG-1 modulates genome stability by binding to RNA molecules and/or proteins. Of note, several proteins with the RGG/RG motif (e.g. MRE11 and 53BP1) have roles in the maintenance of genome stability (Thandapani et al. Citation2013).
Sequence comparison indicates that VIG-1 is homologous to human SERPINE1 mRNA binding protein 1 (SERBP1, also called PAI-RBP1). Recently, Ahn et al. (Citation2015) reported that SERBP1 influences the DDR by promoting the translation of CtIP, a key regulator of the HR pathway (You and Bailis Citation2010). Deletion analysis revealed that a RGG-rich region of SERBP1 is necessary for CtIP mRNA binding (Ahn et al. Citation2015). It will be of interest to see if VIG-1 controls the level of the C. elegans homolog of CtIP, COM-1, which blocks the NHEJ pathway and facilitates the HR-mediated DSB repair during meiosis (Lemmens et al. Citation2013).
VIG-1 is considered to be a component of the RISC of C. elegans (Caudy et al. Citation2003). Knockdown of the vig-1 gene interferes with the let-7 miRNA function, suggesting that VIG-1 modulates the miRNA pathway. The role of VIG-1 in miRNA function is also supported by the observation that GLD-1, a genetic interactor of VIG-1, is required for the functions of let-7 and mir-35 miRNA families (Akay et al. Citation2013). Interestingly, accumulating evidence indicates that miRNAs play essential roles in the DDR (Landau and Slack Citation2011). For example, C. elegans miR-34 expression is upregulated in response to radiation, and the mir-34 mutant exhibits altered sensitivity to radiation (Kato et al. Citation2009). It remains to be determined whether VIG-1 participates in the DDR by modulating the miRNA function.
In conclusion, our data show that VIG-1 is involved in the maintenance of genome stability by protecting DNA from endogenous and exogenous genotoxic stresses. Elucidating the molecular mechanism by which VIG-1 performs its function(s) will help us to better understand how the DDR system works in metazoans.
SUPPORTING_INFORMATION.docx
Download MS Word (808.7 KB)Acknowledgments
We thank Boram Choi and Ziyu Liu for their contributions in the early phase of this work. We are grateful to Shohei Mitani and the C. elegans Genetics Center for providing vig-1(tm3383) and vig-1(ok2536), respectively. The anti-VIG-1 monoclonal antibody was generated by Eun Sung Jeon and Chom-Kyu Chong.
Disclosure statement
The authors have no conflicting interests.
Additional information
Funding
Related Research Data
References
- Ahmed S, Hodgkin J. 2000. MRT-2 checkpoint protein is required for germline immortality and telomere replication in C. elegans. Nature. 403:159–164. doi: 10.1038/35003120
- Ahn JW, Kim S, Na W, Baek SJ, Kim JH, Min K, Yeom J, Kwak H, Jeong S, Lee C et al. 2015. Serbp1 affects homologous recombination-mediated DNA repair by regulation of CtIP translation during S phase. Nucleic Acids Res. 43:6321–6333. doi: 10.1093/nar/gkv592
- Akay A, Craig A, Lehrbach N, Larance M, Pourkarimi E, Wright JE, Lamond A, Miska E, Gartner A. 2013. RNA-binding protein GLD-1/quaking genetically interacts with the mir-35 and the let-7 miRNA pathways in Caenorhabditis elegans. Open Biol. 3:130151. doi: 10.1098/rsob.130151
- Alpi A, Pasierbek P, Gartner A, Loidl J. 2003. Genetic and cytological characterization of the recombination protein RAD-51 in Caenorhabditis elegans. Chromosoma. 112:6–16. doi: 10.1007/s00412-003-0237-5
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics. 77:71–94.
- Caudy AA, Ketting RF, Hammond SM, Denli AM, Bathoorn AM, Tops BB, Silva JM, Myers MM, Hannon GJ, Plasterk RH. 2003. A micrococcal nuclease homologue in RNAi effector complexes. Nature. 425:411–414. doi: 10.1038/nature01956
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell. 40:179–204. doi: 10.1016/j.molcel.2010.09.019
- Craig AL, Moser SC, Bailly AP, Gartner A. 2012. Methods for studying the DNA damage response in the Caenorhabdatis elegans germ line. Methods Cell Biol. 107:321–352. doi: 10.1016/B978-0-12-394620-1.00011-4
- Hillers KJ, Jantsch V, Martinez-Perez E, Yanowitz JL. 2017. Meiosis. WormBook. 1-43.
- Hodgkin J, Horvitz HR, Brenner S. 1979. Nondisjunction mutants of the nematode Caenorhabditis elegans. Genetics. 91:67–94.
- Hsiang YH, Lihou MG, Liu LF. 1989. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 49:5077–5082.
- Hyun M, Choi S, Stevnsner T, Ahn B. 2016. The Caenorhabditis elegans Werner syndrome protein participates in DNA damage checkpoint and DNA repair in response to CPT-induced double-strand breaks. Cell Signal. 28:214–223. doi: 10.1016/j.cellsig.2015.12.006
- Jackson SP, Bartek J. 2009. The DNA-damage response in human biology and disease. Nature. 461:1071–1078. doi: 10.1038/nature08467
- Kato M, Paranjape T, Muller RU, Nallur S, Gillespie E, Keane K, Esquela-Kerscher A, Weidhaas JB, Slack FJ. 2009. The mir-34 microRNA is required for the DNA damage response in vivo in C. elegans and in vitro in human breast cancer cells. Oncogene. 28:2419–2424. doi: 10.1038/onc.2009.106
- Landau DA, Slack FJ. 2011. Micrornas in mutagenesis, genomic instability, and DNA repair. Semin Oncol. 38:743–751. doi: 10.1053/j.seminoncol.2011.08.003
- Lee MH, Han SM, Han JW, Kim YM, Ahnn J, Koo HS. 2003. Caenorhabditis elegans dna-2 is involved in DNA repair and is essential for germ-line development. FEBS Lett. 555:250–256. doi: 10.1016/S0014-5793(03)01243-2
- Lee SJ, Yook JS, Han SM, Koo HS. 2004. A Werner syndrome protein homolog affects C. elegans development, growth rate, life span and sensitivity to DNA damage by acting at a DNA damage checkpoint. Development. 131:2565–2575. doi: 10.1242/dev.01136
- Lemmens BB, Johnson NM, Tijsterman M. 2013. COM-1 promotes homologous recombination during Caenorhabditis elegans meiosis by antagonizing Ku-mediated non-homologous end joining. PLoS Genet. 9:e1003276. doi: 10.1371/journal.pgen.1003276
- Liu LF, Desai SD, Li TK, Mao Y, Sun M, Sim SP. 2000. Mechanism of action of camptothecin. Ann N Y Acad Sci. 922:1–10. doi: 10.1111/j.1749-6632.2000.tb07020.x
- O'Driscoll M. 2012. Diseases associated with defective responses to DNA damage. Cold Spring Harb Perspect Biol. 4:a012773. doi: 10.1101/cshperspect.a012773
- Park HK, Suh D, Hyun M, Koo HS, Ahn B. 2004. A DNA repair gene of Caenorhabditis elegans: a homolog of human XPF. DNA Repair (Amst). 3:1375–1383. doi: 10.1016/j.dnarep.2004.04.008
- Penkner A, Portik-Dobos Z, Tang L, Schnabel R, Novatchkova M, Jantsch V, Loidl J. 2007. A conserved function for a Caenorhabditis elegans Com1/Sae2/CtIP protein homolog in meiotic recombination. EMBO J. 26:5071–5082. doi: 10.1038/sj.emboj.7601916
- Rastogi RP, Richa, Kumar A, Tyagi MB, Sinha RP. 2010. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids. 2010:592980. doi: 10.4061/2010/592980
- Rose A. 2014. Replication and repair. WormBook.1-16.
- Ryan AJ, Squires S, Strutt HL, Johnson RT. 1991. Camptothecin cytotoxicity in mammalian cells is associated with the induction of persistent double strand breaks in replicating DNA. Nucleic Acids Res. 19:3295–3300. doi: 10.1093/nar/19.12.3295
- Saito TT, Mohideen F, Meyer K, Harper JW, Colaiacovo MP. 2012. SLX-1 is required for maintaining genomic integrity and promoting meiotic noncrossovers in the Caenorhabditis elegans germline. PLoS Genet. 8:e1002888. doi: 10.1371/journal.pgen.1002888
- Sakasai R, Iwabuchi K. 2015. The distinctive cellular responses to DNA strand breaks caused by a DNA topoisomerase I poison in conjunction with DNA replication and RNA transcription. Genes Genet Syst. 90:187–194. doi: 10.1266/ggs.15-00023
- Smelick C, Ahmed S. 2005. Achieving immortality in the C. elegans germline. Ageing Res Rev. 4:67–82. doi: 10.1016/j.arr.2004.09.002
- Stergiou L, Hengartner MO. 2004. Death and more: DNA damage response pathways in the nematode C. elegans. Cell Death Differ. 11:21–28. doi: 10.1038/sj.cdd.4401340
- Thandapani P, O’Connor TR, Bailey TL, Richard S. 2013. Defining the RGG/RG motif. Mol Cell. 50:613–623. doi: 10.1016/j.molcel.2013.05.021
- Wang C, Gupta P, Fressigne L, Bosse GD, Wang X, Simard MJ, Hansen D. 2017. TEG-1 CD2BP2 controls miRNA levels by regulating miRISC stability in C. elegans and human cells. Nucleic Acids Res. 45:1488–1500.
- You Z, Bailis JM. 2010. Dna damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 20:402–409. doi: 10.1016/j.tcb.2010.04.002