
ABSTRACT
Autoimmune diseases are conditions in which the immune system mistakenly targets and damages healthy tissue in the body. In recent decades, the incidence of autoimmune diseases has increased, resulting in a significant disease burden. The current autoimmune therapies focus on targeting inflammation or inducing immunosuppression rather than addressing the underlying cause of the diseases. The activity of metabolic pathways is elevated in autoimmune diseases, and metabolic changes are increasingly recognized as important pathogenic processes underlying these. Therefore, metabolically targeted therapies may represent an important strategy for treating autoimmune diseases. This review provides a comprehensive overview of the evidence surrounding glucose metabolic reprogramming and its potential applications in drug discovery and development for autoimmune diseases, such as type 1 diabetes, multiple sclerosis, systemic lupus erythematosus, rheumatoid arthritis, and systemic sclerosis.
Introduction
The immune system is a crucial defense against infectious agents, including viruses, bacteria, and fungi. Although it is finely tuned not to attack our tissues without losing its response to foreign invaders, it may mistakenly attack healthy tissues, resulting in autoimmune diseases (Schwartz Citation2012; Wang et al. Citation2015). The frequency of autoimmune diseases has increased over recent decades (Miller Citation2023). The means of annual increases in the worldwide incidence and prevalence of autoimmune diseases are 19.1% and 12.5%, respectively (Lerner et al. Citation2015). Therefore, understanding the underlying mechanisms of autoimmune diseases and developing new therapeutic strategies have become increasingly important.
Genetic factors, environmental factors, and dysregulation of the immune system are believed to contribute to the development of autoimmunity (Rosenblum et al. Citation2015). However, the exact mechanisms underlying the development of autoimmune diseases remain unclear (Rosenblum et al. Citation2015). Therefore, the most current treatments for autoimmune diseases focus on replacement therapies, anti-inflammation, or immunosuppression rather than addressing the underlying issues that cause the initiation and progression of the autoimmune process (Streeter and Wraith Citation2021; Jung and Kim Citation2022). Moreover, long-term use of these anti-inflammatory or immunosuppressive medicines increases the likelihood of infections and cancer (Streeter and Wraith Citation2021). Therefore, understanding how abnormal immune responses are developed and sustained is necessary to overcome these limitations and effectively address the underlying cause of autoimmune diseases.
The metabolic shift is a well-established event in the development of tumorigenicity. However, metabolic reprogramming has become a key feature in numerous other disease pathologies, including autoimmune diseases (Pearce et al. Citation2013; Yin et al. Citation2016; Vander Heiden and DeBerardinis Citation2017). Cancer and immune cells have similarities in their need for sufficient metabolic flux and bioenergetics to support macromolecule synthesis, as well as cell growth and expansion (Andrejeva and Rathmell Citation2017). The activation, proliferation, and differentiation of immune cells can be further shaped and fine-tuned during metabolic reprogramming (Chou et al. Citation2022). Emerging evidence has revealed an important role for metabolites in regulating the phenotype and effector function of immune cells in response to energy demands in resting or stimulated states (Guijas et al. Citation2018; Kolan et al. Citation2020; Kolliniati et al. Citation2022). Moreover, by interrupting these metabolic pathways, it is possible to alter the fate of immune cells and modulate immunity (Patel et al. Citation2019).
In this review, we discuss the advances in the current understanding of the glucose metabolic signatures of various cells in autoimmune diseases and speculate on the prospect of targeting metabolism as a novel therapeutic strategy.
Glucose metabolism in autoimmunity
Glucose metabolism is an important metabolic pathway that provides energy to cells and is finely tuned to accommodate the cellular demands for energy and biosynthesis (Park et al. Citation2022) (). It comprises multiple enzymes that catalyze the conversion of glucose into metabolized products and energy in the form of ATP (Han et al. Citation2016). Pyruvate produced through glycolysis is mainly used in oxidative phosphorylation (OXPHOS) after the tricarboxylic acid (TCA) cycle to generate more ATP (Han et al. Citation2016). Glycolytic intermediates such as glucose-6-phosphate (G6P) and 3-phosphoglycerate (G3P) are indirectly involved in the pentose phosphate pathway (PPP) and amino acid synthesis, respectively (Ramos-Martinez Citation2017; Zhao et al. Citation2020).
Figure 1. Glucose metabolism and its inhibitors.
Note: After entering cells, glucose is phosphorylated by HK to form G6P. This step helps trap glucose inside the cells and initiate its metabolism. G6P serves as a substrate for the PPP, a metabolic pathway that runs parallel to glycolysis. PPP generates R5P, which is essential for nucleotide synthesis and produces abundant amounts of NADPH. NADPH is an important reducing agent used in various biosynthetic processes and helps protect cells from oxidative damage. G6P undergoes a series of oxidative decompositions known as glycolysis to generate pyruvate. Along the glycolytic pathway, intermediate metabolites are formed, providing raw materials for the biosynthesis of other molecules such as serine and glycine. PKM2 controls the final step of glycolysis, converting PEP to pyruvate. PKM2 is a specific isoform of pyruvate kinase, which is often observed in proliferating cells and plays a crucial role in regulating the balance between glycolysis and other metabolic pathways. Pyruvate generated by PKM2 can be utilized in multiple ways. One major pathway is OXPHOS, where pyruvate is converted to acetyl-CoA, which enters the TCA cycle. The TCA cycle further breaks down acetyl-CoA to produce ATP and reducing agents (NADH, FADH2) used in OXPHOS. Additionally, pyruvate can be converted to lactate through a pathway called anaerobic glycolysis. During glycolysis, glucose is metabolized to pyruvate, producing a small amount of ATP and NADH. The conversion of pyruvate to lactate is catalyzed by the enzyme LDH, and lactate is transported across the cell membrane by specific transporters known as monocarboxylate transporters (MCTs). Illustrations of human organs, obtained from Freepik at https://www.freepik.com, are presented to remind the main targets of autoimmune diseases. Abbreviations: GLUT: glucose transporter; HK: hexokinase; PFKFB3: 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; PFK1: phosphofructokinase 1; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; PGK1: phosphoglycerate kinase 1; PKM2: pyruvate kinase isoenzyme M2; LDH: lactate dehydrogenase; MCT: monocarboxylate transporter; OXPHOS: oxidative phosphorylation; 2-DG: 2-deoxy-D-glucose; 3-BrPA: 3-bromopyruvate; LND: lonidamine; 3-PO: 3-(3-pyridinyl)−1-(4-pyridinyl)−2-propen-1-one; DMF: dimethyl fumarate; MnP: manganese metalloporphyrin; T1D: type 1 diabetes; MS: multiple sclerosis; SLE: systemic lupus erythematosus; RA: rheumatoid arthritis; SSc: systemic sclerosis.
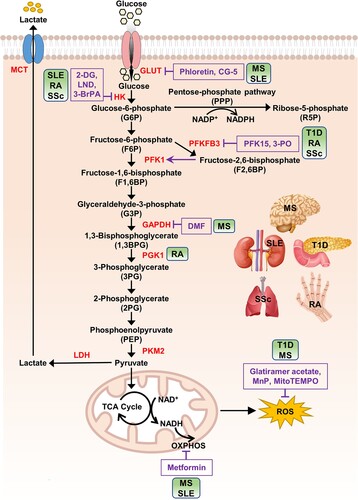
Glycolysis plays a crucial role in immune cells, particularly during immune activation and effector functions (Pearce and Pearce Citation2013). Immune cells, such as T cells, B cells, macrophages, and dendritic cells, undergo metabolic reprogramming to meet their energy demands and support their specialized functions (Pearce and Pearce Citation2013; Woo et al. Citation2021). Although glycolysis produces less ATP compared to that from the TCA cycle or OXPHOS, it serves a crucial role in providing energy to these cells. The glycolytic process enables innate immune cells to undergo functional changes, such as the production and secretion of cytokines (Rodríguez-Prados et al. Citation2010; O’Neill and Pearce Citation2016). Resting T cells obtain energy primarily through the oxidative phosphorylation of glucose, lipids, and amino acids (Chen et al. Citation2015). However, when T cells are activated, they switch to aerobic glycolysis as their primary source of energy to support the rapid changes in their phenotype (Chen et al. Citation2015). This change in metabolism provides the necessary energy and building blocks for the cells to grow and divide rapidly, and carry out their immune functions. Further, the shift to glycolysis in activated T cells plays an important role in regulating gene expression. For example, glucose uptake through the GLUT3 transporter plays a crucial role in Th17 cell function in autoimmune diseases (Shi et al. Citation2011; Gerriets et al. Citation2016), and it regulates the metabolic pathways that control the expression of genes involved in the inflammatory response of Th17 cells (Hochrein et al. Citation2022). Therefore, although metabolic reprogramming occurring in immune cells is necessary for cell differentiation, proliferation, and effector function, it may mediate the imbalance of immune homeostasis, contributing to the onset and progression of autoimmune diseases (Mohammadnezhad et al. Citation2022).
Besides generating energy, glycolytic intermediates play an important role in helping immune cells alter their characteristics in response to external signals (Ganeshan and Chawla Citation2014; Shyer et al. Citation2020). The PPP is a metabolic pathway that diverges from glycolysis at the first irreversible step and involves catalysis by glucose-6-phosphate dehydrogenase (G6PD) with G6P as its primary substrate (Ramos-Martinez Citation2017). The PPP is a major source of nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) and plays a pivotal role in maintaining the cellular redox state (Xiao et al. Citation2018). During oxidative stress, glycolytic enzymes such as phosphofructokinase 1 (PFK1) can become inactivated, increasing NADPH production through the diversion of G6P to the oxidative PPP (Patra and Hay Citation2014). This interaction between glycolysis and the PPP allows cells to adjust their metabolism for survival and proliferation (Patra and Hay Citation2014). NADPH is a crucial molecule that plays a key role in maintaining the balance of redox reactions within cells and defending against oxidative stress (Koju et al. Citation2022). Disruptions in the balance of NADPH levels within cells have been linked with the development of various diseases, including autoimmune diseases (Panday et al. Citation2015). According to a recent study, the PPP has been shown to regulate the removal of exhausted cells and immune tolerance (He et al. Citation2022). Further, enhancing PPP activity has been observed to reduce the removal of dead cells by macrophages and exacerbate the symptoms of systemic lupus erythematosus (SLE) in a mouse model (He et al. Citation2022).
Type 1 diabetes
Type 1 diabetes (T1D) is an organ-specific autoimmune disease caused by the selective destruction of pancreatic β-cells. T1D pathogenesis is mainly mediated by autoreactive T cells that infiltrate the islets, playing a key role in the process of β-cell destruction (Burrack et al. Citation2017). Emerging evidence suggests that metabolic perturbations of immune cells play an important role in initiating and exacerbating T1D (Sysi-Aho et al. Citation2011; Pflueger et al. Citation2011; Oresic et al. Citation2013; La Torre et al. Citation2013). Upon encountering islet β cell autoantigens, naïve T cells are activated and undergo robust metabolic reprogramming to utilize aerobic glycolysis, which is required to generate ATP quickly for supporting T cell activation, clonal expansion, and effector cytokine production (Pearce and Pearce Citation2013; Chang et al. Citation2013; Pearce et al. Citation2013).
Activation of T cells provided by proinflammatory cytokines and reactive oxygen species (ROS) is suggested to play a critical role in T1D pathogenesis (Tse et al. Citation2007; Padgett et al. Citation2013). ROS are required to drive and sustain T cell activation-induced metabolic reprogramming (Previte et al. Citation2017); ROS production and ATP synthesis are tightly regulated by maintaining mitochondrial membrane potential (Zorov et al. Citation2014). However, T cells from patients with T1D exhibit mitochondrial hyperpolarization, which is associated with increased ROS production and interferon (IFN)-γ secretion, indicating that mitochondrial dysfunction promotes the effector functions of autoreactive T cells due to an altered proinflammatory T cell effector response (Chen et al. Citation2017). Therefore, treatment of T cells with manganese metalloporphyrin (MnP), a ROS-scavenging and potent antioxidant, was observed to impede the metabolic transition from OXPHOS to aerobic glycolysis, indicating that ROS are required during the switch from OXPHOS to aerobic glycolysis during T cell activation (Previte et al. Citation2017). Further, PFK15, a small molecule inhibitor of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), inhibits the metabolic reprogramming of diabetogenic CD4+ T cells to glycolysis and delays T1D onset in an animal model by inducing T cell exhaustion (Martins et al. Citation2021). These findings indicate that metabolic regulation can be considered a new therapeutic strategy for T1D by controlling T cell metabolism and restoring immune tolerance.
Multiple sclerosis
Multiple sclerosis (MS) is an autoimmune disease characterized by chronic inflammation, demyelination, gliosis, and neurodegeneration of the central nervous system (Noyes and Weinstock-Guttman Citation2013). Although the precise cause of MS remains unclear, autoreactive CD4+ T cells are considered to recognize and destroy myelin in the central nervous system by releasing proinflammatory cytokines, including IFN-γ and IL-17, which play significant roles in MS pathogenesis (Kaskow and Baecher-Allan Citation2018; Segal Citation2019).
Several studies have demonstrated altered energy metabolism and mitochondrial injury in MS pathogenesis (Graumann et al. Citation2003; Mahad et al. Citation2008; Tavazzi et al. Citation2011; Braidy et al. Citation2013). Analysis of the gene expression profile in post-mortem MS brains revealed upregulation of genes reflecting higher energy metabolism (Graumann et al. Citation2003). Additionally, significantly elevated nitrite and nitrate levels were identified from the serum metabolic profile analyses of patients with MS, indicating that they suffered from severe purine and pyrimidine metabolic abnormalities, potentially because of altered mitochondrial function (Tavazzi et al. Citation2011). Higher levels of nicotinamide adenine dinucleotide hydrogen (NADH) were observed in the serum of patients with MS compared to those in healthy subjects; these are associated with disease progression considering the role of NADH in maintaining genomic integrity and mitochondrial energy production (Braidy et al. Citation2013). Defects in the mitochondrial respiratory chain complex IV, which are represented by the loss of cytochrome c oxidase-1 and 4, are observed in acute MS lesions (Mahad et al. Citation2008).
A recent study revealed the unknown mechanism of action of dimethyl fumarate (DMF), an immunomodulatory drug used to treat MS. DMF inactivates the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in peritoneal macrophages and CD4+ T cells in both mice and humans (Kornberg et al. Citation2018). Similarly, glatiramer acetate, a random copolymer of glutamic acid, lysine, alanine, and tyrosine, which is used for treating MS, enhances the mitochondrial activity of CD4+ T cells and improves their response to oxidative stress (De Riccardis et al. Citation2016). Metformin can function as an AMP-activated protein kinase (AMPK) activator or mitochondrial OXPHOS inhibitor, depending on its concentration, and is used for treating type 2 diabetes by suppressing liver glucose production and increasing insulin sensitivity (He Citation2020). A cohort study on metformin-treated patients with MS demonstrated a significant reduction in the number of brain lesions with elevated AMPK expression, decreased production of IFN-γ and IL-17, and an increased percentage of regulatory T cells (Tregs) (Negrotto et al. Citation2016).
In MS, the demyelination and remyelination processes are imbalanced, which may result in dysregulation of energy metabolism, followed by axon destabilization and damage (Nave Citation2010). Oligodendrocyte precursor cells (OPCs) can differentiate into myelinating oligodendrocytes (Kirby et al. Citation2019). Metformin restores the regenerative capacity of aged OPCs and promotes remyelination in the stem cells of elderly rats (Neumann et al. Citation2019). Additionally, a recent study showed that phloretin, an inhibitor of glucose transporter 1 (GLUT1), enhances remyelination by directly stimulating OPC maturation through peroxisome proliferator-activated receptor γ (PPARγ) activation (Dierckx et al. Citation2022). These results suggest that inhibition of glucose metabolism promotes remyelination, which could be a new therapeutic strategy for MS.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the inappropriate activation of autoreactive T and B cells, which leads to the production of autoantibodies and immune complexes that can cause chronic inflammation and multiple organ dysfunctions (Tsokos Citation2011). The clinical manifestations of SLE are diverse, including rash, arthritis, pleuritis, nephritis, and even neuropathy (Tsokos Citation2011; Hanly Citation2014).
CD4+ T cells from lupus-prone mice and patients with SLE exhibited enhanced glycolysis and mitochondrial metabolism, indicating that altered intrinsic metabolism reprogramming occurs in SLE (Wahl et al. Citation2010; Yin et al. Citation2015). During immune responses, T cells increase their glucose uptake and glycolysis through the action of phosphatidylinositol 3-kinase (PI3K) and Akt through CD28 costimulation (Frauwirth et al. Citation2002). Inhibition of glucose transporters by CG-5 ameliorated the SLE phenotypes in lupus-prone mice by suppressing Th1 and Th17 cell differentiation, inducing regulatory T cells, and reducing the expansion of germinal center B cells and the production of autoantibodies (Li et al. Citation2019). Further, the T cells of patients with SLE exhibited mitochondrial dysfunction, as evidenced by mitochondrial hyperpolarization, intracellular pH elevation, increased production of reactive oxygen intermediates (ROI), decreased glutathione levels, and ATP depletion (Gergely, Grossman, et al. Citation2002; Gergely, Niland, et al. Citation2002; Perl et al. Citation2004). Inhibition of mitochondrial ROS by MitoTEMPO reduces disease severity and type I IFN responses in lupus-prone mice (Lood et al. Citation2016). Further, treatment with a combination of glycolysis inhibitor 2-deoxy-D-glucose (2-DG) and mitochondrial OXPHOS inhibitor metformin restored T cell metabolism and reverted disease phenotypes in lupus-prone mice (Yin et al. Citation2015, Citation2016). These results suggest that inhibiting glycolysis or mitochondrial metabolism in T cells can be a promising therapeutic strategy for SLE.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease that affects joints and extra-articular tissues (Smolen et al. Citation2018). The main pathological features of RA include synovial hyperplasia, inflammatory cell infiltration, pannus formation, and erosion of cartilage and bone, ultimately leading to progressive joint destruction (Nygaard and Firestein Citation2020; Mahmoud et al. Citation2022). The accumulation of cells in the inflamed synovium induces hypoxic conditions, which modify the metabolic environment (Quiñonez-Flores et al. Citation2016). As a result, altered metabolites are observed in patients with RA, and the altered metabolic pathways can exacerbate synovial inflammation by activating immune cells and synovial fibroblasts (Young et al. Citation2013; Fearon et al. Citation2022).
Glucose metabolism is significantly increased in the inflamed joints of early-stage RA, which is associated with phenotypic changes in synovial cells (Chang and Wei Citation2011; Masoumi et al. Citation2020). Increased glycolysis shifts macrophages towards an inflammatory phenotype, producing IL-1β, IL-6, and TNF-α. Hexokinase 2 (HK2) and pyruvate kinase isoenzyme M2 (PKM2) are upregulated in RA macrophages, further promoting glycolytic flux (Xu et al. Citation2020; Hanlon et al. Citation2022). Further, the metabolic intermediates generated during glycolysis, such as lactate, can modulate immune responses and perpetuate chronic inflammation in RA joints (Biniecka et al. Citation2016; Fearon et al. Citation2022). Moreover, T cells from patients with RA display distinct metabolic alterations favoring PPP over glycolysis, which enables RA T cells to become invasive and pro-inflammatory (Weyand et al. Citation2017; Fearon et al. Citation2022).
Several studies have reported that glycolysis inhibitors suppress the aggressive phenotype of RA fibroblast-like synoviocytes (RA-FLS) and immune cells and alleviate arthritis in animal models. Inhibition of glycolysis using 2-DG, 3-bromopyruvate (3-BrPA), or 3-(3-pyridinyl)−1-(4-pyridinyl)−2-propen-1-one (3-PO) decreases the aggressive phenotypes of RA-FLS by reducing cytokine production, proliferation, and migration (Garcia-Carbonell et al. Citation2016; Biniecka et al. Citation2016; Abboud et al. Citation2018). In addition, 3-BrPA modulates Th17/Treg cell differentiation and suppresses dendritic cell activation (Okano et al. Citation2017). Accordingly, treatment with 2-DG or 3-BrPA decreased inflammatory arthritis in mouse models (Garcia-Carbonell et al. Citation2016; Okano et al. Citation2017; Abboud et al. Citation2018). Another glycolysis inhibitor, PFK15 reduces the activation and aggressive phenotypes of RA-FLS and attenuates the severity of arthritis in collagen-induced arthritis (CIA) mice (Zou et al. Citation2017). HK2 is specifically expressed in the RA synovial membrane and has been shown to regulate the aggressive function of RA-FLS (Bustamante et al. Citation2018). Lonidamine (LND), an HK2 inhibitor, induces apoptosis in RA-FLS, suppresses the production of inflammatory factors by RA-FLS, and attenuates arthritis phenotypes in the CIA model (Song et al. Citation2019). Similarly, PGK1 is increased in RA synovial tissues, whereas PGK1 knockdown decreases the proliferation and migration of RA-FLS, as well as reduces the production of IL-1β and IFN-γ in RA-FLS (Zhao et al. Citation2016). Therefore, targeting glycolysis can be a novel approach for treating RA by modulating the pathological activities of RA-FLS and immune cells.
Systemic sclerosis
Systemic sclerosis (SSc), also known as scleroderma, is an autoimmune disease characterized by vasculopathy and progressive fibrosis of various tissues and organs, including the skin, lungs, heart, and kidneys (Volkmann et al. Citation2023). SSc has the highest mortality among rheumatic diseases; interstitial lung disease (ILD) involving inflammation and fibrosis is one of the leading causes of death in patients with SSc (Elhai et al. Citation2017; Volkmann et al. Citation2023). Effective therapies to prevent fibrosis are currently limited.
Positron emission tomography/computed tomography (PET/CT) scans in patients with SSc have shown increased 18fluorodeoxyglucose (18F-FDG) uptake in the skin and soft tissue calcinosis (Oksuzoglu et al. Citation2015; Vadrucci et al. Citation2016). Further, glycolysis has been recently revealed as a key metabolic pathway in fibrosis (Henderson and O’Reilly Citation2021). SSc dermal fibroblasts have increased glycolysis and lactate levels, which leads to myofibroblast transition, the key process in SSc pathogenesis (Henderson et al. Citation2020; Andreucci et al. Citation2021). High glycolytic metabolism and the resulting acidic extracellular microenvironment contribute to the impairment of angiogenesis and the induction of endothelial cells to myofibroblast transdifferentiation, thereby driving the fibrotic process in SSc (Andreucci et al. Citation2021). Inhibition of glycolysis by 2-DG or 3-PO can attenuate transforming growth factor β1 (TGF-β1)-induced fibrosis in dermal fibroblasts (Henderson et al. Citation2020). Glycolytic reprogramming is an important driving force behind lung myofibroblast differentiation and pulmonary fibrosis (Kottmann et al. Citation2012; Xie et al. Citation2015). Additionally, microarray analysis of lung tissues from patients with SSc and pulmonary fibrosis demonstrated perturbation of bioenergetics, including glycolysis (Renaud et al. Citation2020). As a result, inhibition of glycolysis by 3-PO attenuated the profibrotic phenotypes of lung myofibroblasts in vitro and pulmonary fibrosis in vivo (Xie et al. Citation2015). According to a recent study, anlotinib, a multiple receptor tyrosine kinase inhibitor, reversed pulmonary fibrosis by inhibiting PFKFB3-driven glycolysis in myofibroblasts (Chen et al. Citation2021). These results suggest that glycolysis plays a key role in the pathogenesis of SSc by regulating fibrosis and can be a promising therapeutic target for SSc.
Conclusions
Glycolysis and OXPHOS are key metabolic processes involved in cellular energy metabolism. The balance between glycolysis and OXPHOS is tightly regulated and essential for maintaining cellular homeostasis. Under normal conditions, cells preferentially utilize OXPHOS to meet their energy demands. However, in certain situations, such as continuous stimulation or chronic inflammation, there can be a shift towards increased glycolysis, even in the presence of oxygen. This metabolic reprogramming, known as aerobic glycolysis, is a hallmark of many pathological conditions, including autoimmune diseases. Increased glycolysis provides immune cells with the necessary energy and metabolic intermediates to support proliferation, survival, and cytokine production. However, an imbalance favoring glycolysis over OXPHOS can lead to excessive activation of immune cells, the persistence of inflammation, and tissue damage that is observed in autoimmune diseases. Thus, understanding the factors that disrupt the balance between glycolysis and OXPHOS, which leads to excessive immune cell activation, is an active area of research in autoimmune diseases.
Several studies have shown that glycolysis is increased in autoimmune diseases such as T1D, MS, SLE, RA, and SSc, confirming that glycolysis plays a significant role in the pathogenesis of autoimmune diseases. Although autoimmune diseases affect various organs and tissues, they all share a common characteristic of metabolic disruption. Considering the importance of these specific metabolic changes in various autoimmune diseases, targeting glucose metabolism predictably shows beneficial effects for treating autoimmune diseases (). All these results highlight the significance of metabolic changes in autoimmune diseases and suggest promising therapeutic approaches for autoimmune diseases.
Table 1. Glucose metabolism inhibitors studied in autoimmune diseases.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Abboud G, Choi S, Kanda N, Zeumer-Spataro L, Roopenian DC, Morel L. 2018. Inhibition of glycolysis reduces disease severity in an autoimmune model of rheumatoid arthritis. Front Immunol. 9. doi:10.3389/fimmu.2018.01973.
- Andrejeva G, Rathmell JC. 2017. Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. 26(1):49–70. doi:10.1016/j.cmet.2017.06.004.
- Andreucci E, Margheri F, Peppicelli S, Bianchini F, Ruzzolini J, Laurenzana A, Fibbi G, Bruni C, Bellando-Randone S, Guiducci S, et al. 2021. Glycolysis-derived acidic microenvironment as a driver of endothelial dysfunction in systemic sclerosis. Rheumatology. 60(10):4508–4519. doi:10.1093/rheumatology/keab022.
- Biniecka M, Canavan M, McGarry T, Gao W, McCormick J, Cregan S, Gallagher L, Smith T, Phelan JJ, Ryan J, et al. 2016. Dysregulated bioenergetics: a key regulator of joint inflammation. Ann Rheum Dis. 75(12):2192–2200. doi:10.1136/annrheumdis-2015-208476.
- Braidy N, Lim CK, Grant R, Brew BJ, Guillemin GJ. 2013. Serum nicotinamide adenine dinucleotide levels through disease course in multiple sclerosis. Brain Res. 1537:267–272. doi:10.1016/j.brainres.2013.08.025.
- Burrack AL, Martinov T, Fife BT. 2017. T cell-mediated beta cell destruction: autoimmunity and alloimmunity in the context of type 1 diabetes. Front Endocrinol (Lausanne). 8:343. doi:10.3389/fendo.2017.00343.
- Bustamante MF, Oliveira PG, Garcia-Carbonell R, Croft AP, Smith JM, Serrano RL, Sanchez-Lopez E, Liu X, Kisseleva T, Hay N, et al. 2018. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann Rheum Dis. 77(11):1636–1643. doi:10.1136/annrheumdis-2018-213103.
- Chang C-H, Curtis JD, Maggi LB, Faubert B, Villarino AV, O’Sullivan D, Huang SC-C, van der Windt GJW, Blagih J, Qiu J, et al. 2013. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 153(6):1239–1251. doi:10.1016/j.cell.2013.05.016.
- Chang X, Wei C. 2011. Glycolysis and rheumatoid arthritis. Int J Rheum Dis. 14(3):217–222. doi:10.1111/j.1756-185X.2011.01598.x.
- Chen H, Yang T, Zhu L, Zhao Y. 2015. Cellular metabolism on T-cell development and function. Int Rev Immunol. 34(1):19–33. doi:10.3109/08830185.2014.902452.
- Chen J, Chernatynskaya AV, Li J-W, Kimbrell MR, Cassidy RJ, Perry DJ, Muir AB, Atkinson MA, Brusko TM, Mathews CE. 2017. T cells display mitochondria hyperpolarization in human type 1 diabetes. Sci Rep. 7(1):10835. doi:10.1038/s41598-017-11056-9.
- Chen W, Zhang J, Zhong W, Liu Y, Lu Y, Zeng Z, Huang H, Wan X, Meng X, Zou F, et al. 2021. Anlotinib inhibits PFKFB3-driven glycolysis in myofibroblasts to reverse pulmonary fibrosis. Front Pharmacol. 12. doi:10.3389/fphar.2021.744826.
- Chou W-C, Rampanelli E, Li X, Ting JP-Y. 2022. Impact of intracellular innate immune receptors on immunometabolism. Cell Mol Immunol. 19(3):337–351. doi:10.1038/s41423-021-00780-y.
- De Riccardis L, Ferramosca A, Danieli A, Trianni G, Zara V, De Robertis F, Maffia M. 2016. Metabolic response to glatiramer acetate therapy in multiple sclerosis patients. BBA Clin. 6:131–137. doi:10.1016/j.bbacli.2016.10.004.
- Dierckx T, Vanherle S, Haidar M, Grajchen E, Mingneau F, Gervois P, Wolfs E, Bylemans D, Voet A, Nguyen T, et al. 2022. Phloretin enhances remyelination by stimulating oligodendrocyte precursor cell differentiation. Proc Natl Acad Sci USA. 119(46):e2120393119. doi:10.1073/pnas.2120393119.
- Elhai M, Meune C, Boubaya M, Avouac J, Hachulla E, Balbir-Gurman A, Riemekasten G, Airò P, Joven B, Vettori S, et al. 2017. Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis. 76(11):1897–1905. doi:10.1136/annrheumdis-2017-211448.
- Fearon U, Hanlon MM, Floudas A, Veale DJ. 2022. Cellular metabolic adaptations in rheumatoid arthritis and their therapeutic implications. Nat Rev Rheumatol. 18(7):398–414. doi:10.1038/s41584-022-00771-x.
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB. 2002. The CD28 signaling pathway regulates glucose metabolism. Immunity. 16(6):769–777. doi:10.1016/S1074-7613(02)00323-0.
- Ganeshan K, Chawla A. 2014. Metabolic regulation of immune responses. Annu Rev Immunol. 32:609–634. doi:10.1146/annurev-immunol-032713-120236.
- Garcia-Carbonell R, Divakaruni AS, Lodi A, Vicente-Suarez I, Saha A, Cheroutre H, Boss GR, Tiziani S, Murphy AN, Guma M. 2016. Critical role of glucose metabolism in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol. 68(7):1614–1626. doi:10.1002/art.39608.
- Gergely P, Jr., Grossman C, Niland B, Puskas F, Neupane H, Allam F, Banki K, Phillips PE, Perl A. 2002. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 46(1):175–190. doi:10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H.
- Gergely P, Jr, Niland B, Gonchoroff N, Pullmann R, Jr, Phillips PE, Perl A. 2002. Persistent mitochondrial hyperpolarization, increased reactive oxygen intermediate production, and cytoplasmic alkalinization characterize altered IL-10 signaling in patients with systemic lupus erythematosus. J Immunol. 169(2):1092–1101. doi:10.4049/jimmunol.169.2.1092.
- Gerriets VA, Kishton RJ, MO J, Cohen S, Siska PJ, Nichols AG, Warmoes MO, de Cubas AA, MacIver NJ, Locasale JW, et al. 2016. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol. 17(12):1459–1466. doi:10.1038/ni.3577.
- Graumann U, Reynolds R, Steck AJ, Schaeren-Wiemers N. 2003. Molecular changes in normal appearing white matter in multiple sclerosis are characteristic of neuroprotective mechanisms against hypoxic insult. Brain Pathol. 13(4):554–573. doi:10.1111/j.1750-3639.2003.tb00485.x.
- Guijas C, Montenegro-Burke JR, Warth B, Spilker ME, Siuzdak G. 2018. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat Biotechnol. 36(4):316–320. doi:10.1038/nbt.4101.
- Han H-S, Kang G, Kim JS, Choi BH, Koo S-H. 2016. Regulation of glucose metabolism from a liver-centric perspective. Exp Mol Med. 48(3):e218–e218. doi:10.1038/emm.2015.122.
- Hanlon MM, McGarry T, Marzaioli V, Amaechi S, Song Q, Nagpal S, Veale DJ, Fearon U. 2022. Rheumatoid arthritis macrophages are primed for inflammation and display bioenergetic and functional alterations. Rheumatology. doi:10.1093/rheumatology/keac640.
- Hanly JG. 2014. Diagnosis and management of neuropsychiatric SLE. Nat Rev Rheumatol. 10(6):338–347. doi:10.1038/nrrheum.2014.15.
- He D, Mao Q, Jia J, Wang Z, Liu Y, Liu T, Luo B, Zhang Z. 2022. Pentose phosphate pathway regulates tolerogenic apoptotic cell clearance and immune tolerance. Front Immunol. 12. doi:10.3389/fimmu.2021.797091.
- He L. 2020. Metformin and systemic metabolism. Trends Pharmacol Sci. 41(11):868–881. doi:10.1016/j.tips.2020.09.001.
- Henderson J, Duffy L, Stratton R, Ford D, O’Reilly S. 2020. Metabolic reprogramming of glycolysis and glutamine metabolism are key events in myofibroblast transition in systemic sclerosis pathogenesis. J Cell Mol Med. 24(23):14026–14038. doi:10.1111/jcmm.16013.
- Henderson J, O’Reilly S. 2021. The emerging role of metabolism in fibrosis. Trends Endocrinol Metab. 32(8):639–653. doi:10.1016/j.tem.2021.05.003.
- Hochrein SM, Wu H, Eckstein M, Arrigoni L, Herman JS, Schumacher F, Gerecke C, Rosenfeldt M, Grün D, Kleuser B, et al. 2022. The glucose transporter GLUT3 controls T helper 17 cell responses through glycolytic-epigenetic reprogramming. Cell Metab. 34(4):516–532.e11. doi:10.1016/j.cmet.2022.02.015.
- Jung SM, Kim W-U. 2022. Targeted immunotherapy for autoimmune disease. Immune Netw. 22(1):e9. doi:10.4110/in.2022.22.e9.
- Kaskow BJ, Baecher-Allan C. 2018. Effector T cells in multiple sclerosis. Cold Spring Harb Perspect Med. 8(4):a029025. doi:10.1101/cshperspect.a029025.
- Kirby L, Jin J, Cardona JG, Smith MD, Martin KA, Wang J, Strasburger H, Herbst L, Alexis M, Karnell J, et al. 2019. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat Commun. 10(1):3887. doi:10.1038/s41467-019-11638-3.
- Koju N, Qin Z, Sheng R. 2022. Reduced nicotinamide adenine dinucleotide phosphate in redox balance and diseases: a friend or foe? Acta Pharmacol Sin. 43(8):1889–1904. doi:10.1038/s41401-021-00838-7.
- Kolan SS, Li G, Wik JA, Malachin G, Guo S, Kolan P, Skålhegg BS. 2020. Cellular metabolism dictates T cell effector function in health and disease. Scand J Immunol. 92(5):e12956. doi:10.1111/sji.12956.
- Kolliniati O, Ieronymaki E, Vergadi E, Tsatsanis C. 2022. Metabolic regulation of macrophage activation. J Innate Immun. 14(1):51–68. doi:10.1159/000516780.
- Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, Calabresi PA, Snyder SH. 2018. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science. 360(6387):449–453. doi:10.1126/science.aan4665.
- Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, Honnons S, Jones C, Isern NG, Hu JZ, et al. 2012. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am J Respir Crit Care Med. 186(8):740–751. doi:10.1164/rccm.201201-0084OC.
- La Torre D, Seppänen-Laakso T, Larsson HE, Hyötyläinen T, Ivarsson SA, Lernmark A, Oresic M, DiPiS Study Group. 2013. Decreased cord-blood phospholipids in young age-at-onset type 1 diabetes. Diabetes. 62(11):3951–3956. doi:10.2337/db13-0215.
- Lerner A, Jeremias P, Matthias T. 2015. The world incidence and prevalence of autoimmune diseases is increasing. Int J Celiac Dis. 3(4):151–155. doi:10.12691/ijcd-3-4-8.
- Li W, Qu G, Choi S, Cornaby C, Titov AA, Kanda N, Teng X, Wang H, Morel L. 2019. Targeting T cell activation and lupus autoimmune phenotypes by inhibiting glucose transporters. Front Immunol. 10. doi:10.3389/fimmu.2019.00833.
- Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ. 2016. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 22(2):146–153. doi:10.1038/nm.4027.
- Mahad D, Ziabreva I, Lassmann H, Turnbull D. 2008. Mitochondrial defects in acute multiple sclerosis lesions. Brain. 131(7):1722–1735. doi:10.1093/brain/awn105.
- Mahmoud DE, Kaabachi W, Sassi N, Tarhouni L, Rekik S, Jemmali S, Sehli H, Kallel-Sellami M, Cheour E, Laadhar L. 2022. The synovial fluid fibroblast-like synoviocyte: a long-neglected piece in the puzzle of rheumatoid arthritis pathogenesis. Front Immunol. 13. doi:10.3389/fimmu.2022.942417.
- Martins CP, New LA, Connor EC, Previte DM, Cargill KR, Tse IL, Sims- Lucas S, Piganelli JD. 2021. Glycolysis inhibition induces functional and metabolic exhaustion of CD4+ T cells in type 1 diabetes. Front Immunol. 12:669456. doi:10.3389/fimmu.2021.669456.
- Masoumi M, Mehrabzadeh M, Mahmoudzehi S, Mousavi MJ, Jamalzehi S, Sahebkar A, Karami J. 2020. Role of glucose metabolism in aggressive phenotype of fibroblast-like synoviocytes: latest evidence and therapeutic approaches in rheumatoid arthritis. Int Immunopharmacol. 89:107064. doi:10.1016/j.intimp.2020.107064.
- Miller FW. 2023. The increasing prevalence of autoimmunity and autoimmune diseases: an urgent call to action for improved understanding, diagnosis, treatment, and prevention. Curr Opin Immunol. 80:102266. doi:10.1016/j.coi.2022.102266.
- Mohammadnezhad L, Shekarkar Azgomi M, La Manna MP, Sireci G, Rizzo C, Badami GD, Tamburini B, Dieli F, Guggino G, Caccamo N. 2022. Metabolic reprogramming of innate immune cells as a possible source of new therapeutic approaches in autoimmunity. Cells. 11(10):1663. doi:10.3390/cells11101663.
- Nave K-A. 2010. Myelination and the trophic support of long axons. Nat Rev Neurosci. 11(4):275–283. doi:10.1038/nrn2797.
- Negrotto L, Farez MF, Correale J. 2016. Immunologic effects of metformin and pioglitazone treatment on metabolic syndrome and multiple sclerosis. JAMA Neurol. 73(5):520–528. doi:10.1001/jamaneurol.2015.4807.
- Neumann B, Baror R, Zhao C, Segel M, Dietmann S, Rawji KS, Foerster S, McClain CR, Chalut K, van Wijngaarden P, Franklin RJM. 2019. Metformin restores CNS remyelination capacity by rejuvenating aged stem cells. Cell Stem Cell. 25(4):473–485.e8. doi:10.1016/j.stem.2019.08.015.
- Noyes K, Weinstock-Guttman B. 2013. Impact of diagnosis and early treatment on the course of multiple sclerosis. PubMed. 19(17 Suppl):s321–s331. https://pubmed.ncbi.nlm.nih.gov/24494633.
- Nygaard G, Firestein GS. 2020. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol. 16(6):316–333. doi:10.1038/s41584-020-0413-5.
- Okano T, Saegusa J, Nishimura K, Takahashi S, Sendo S, Ueda Y, Morinobu A. 2017. 3-bromopyruvate ameliorate autoimmune arthritis by modulating Th17/Treg cell differentiation and suppressing dendritic cell activation. Sci Rep. 7(1):42412. doi:10.1038/srep42412.
- Oksuzoglu K, Ozen G, Inanir S, Direskeneli RH. 2015. Flip-flop phenomenon in systemic sclerosis on fluorodeoxyglucose positron emission tomography/computed tomography. Indian J Nucl Med. 30(4):350. doi:10.4103/0972-3919.164018.
- O’Neill LAJ, Pearce EJ. 2016. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 213(1):15–23. doi:10.1084/jem.20151570.
- Oresic M, Gopalacharyulu P, Mykkänen J, Lietzen N, Mäkinen M, Nygren H, Simell S, Simell V, Hyöty H, Veijola R, et al. 2013. Cord serum lipidome in prediction of islet autoimmunity and type 1 diabetes. Diabetes. 62(9):3268–3274. doi:10.2337/db13-0159.
- Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. 2013. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci. 1281(1):16–35. doi:10.1111/j.1749-6632.2012.06826.x.
- Panday A, Sahoo MK, Osorio D, Batra S. 2015. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 12(1):5–23. doi:10.1038/cmi.2014.89.
- Park D, Lim G, Yoon S-J, Yi H-S, Choi DW. 2022. The role of immunomodulatory metabolites in shaping the inflammatory response of macrophages. BMB Rep. 55(11):519–527. doi:10.5483/BMBRep.2022.55.11.128.
- Patel CH, Leone RD, Horton MR, Powell JD. 2019. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat Rev Drug Discov. 18(9):669–688. doi:10.1038/s41573-019-0032-5.
- Patra KC, Hay N. 2014. The pentose phosphate pathway and cancer. Trends Biochem Sci. 39(8):347–354. doi:10.1016/j.tibs.2014.06.005.
- Pearce EL, Pearce EJ. 2013. Metabolic pathways in immune cell activation and quiescence. Immunity. 38(4):633–643. doi:10.1016/j.immuni.2013.04.005.
- Pearce EL, Poffenberger MC, Chang C-H, Jones RG. 2013. Fueling immunity: insights into metabolism and lymphocyte function. Science. 342(6155):1242454. doi:10.1126/science.1242454.
- Perl A, Gergely P, Banki K. 2004. Mitochondrial dysfunction in T cells of patients with systemic lupus erythematosus. Int Rev Immunol. 23(3–4):293–313. doi:10.1080/08830180490452576.
- Pflueger M, Seppänen-Laakso T, Suortti T, Hyötyläinen T, Achenbach P, Bonifacio E, Orešič M, Ziegler A-G. 2011. Age- and islet autoimmunity-associated differences in amino acid and lipid metabolites in children at risk for type 1 diabetes. Diabetes. 60(11):2740–2747. doi:10.2337/db10-1652.
- Previte DM, O’Connor EC, Novak EA, Martins CP, Mollen KP, Piganelli JD. 2017. Reactive oxygen species are required for driving efficient and sustained aerobic glycolysis during CD4+ T cell activation. PLoS One. 12(4):e0175549. doi:10.1371/journal.pone.0175549.
- Quiñonez-Flores CM, González-Chávez SA, Pacheco-Tena C. 2016. Hypoxia and its implications in rheumatoid arthritis. J Biomed Sci. 23(1):62. doi:10.1186/s12929-016-0281-0.
- Ramos-Martinez JI. 2017. The regulation of the pentose phosphate pathway: remember Krebs. Arch Biochem Biophys. 614:50–52. doi:10.1016/j.abb.2016.12.012.
- Renaud L, Da Silveira WA, Takamura N, Hardiman G, Feghali-Bostwick C. 2020. Prominence of IL6, IGF, TLR, and bioenergetics pathway perturbation in lung tissues of scleroderma patients with pulmonary fibrosis. Front Immunol. 11. doi:10.3389/fimmu.2020.00383.
- Rodríguez-Prados J-C, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, Cascante M, Boscá L. 2010. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 185(1):605–614. doi:10.4049/jimmunol.0901698.
- Rosenblum MD, Remedios KA, Abbas AK. 2015. Mechanisms of human autoimmunity. J Clin Invest. 125(6):2228–2233. doi:10.1172/JCI78088.
- Schwartz RH. 2012. Historical overview of immunological tolerance. Cold Spring Harb Perspect Biol. 4(4):a006908. doi:10.1101/cshperspect.a006908.
- Segal BM. 2019. The diversity of encephalitogenic CD4+ T cells in multiple sclerosis and its animal models. J Clin Med. 8(1):120. doi:10.3390/jcm8010120.
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. 2011. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 208(7):1367–1376. doi:10.1084/jem.20110278.
- Shyer JA, Flavell RA, Bailis W. 2020. Metabolic signaling in T cells. Cell Res. 30(8):649–659. doi:10.1038/s41422-020-0379-5.
- Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, Kavanaugh A, McInnes IB, Solomon DH, Strand V, Yamamoto K. 2018. Rheumatoid arthritis. Nat Rev Dis Primers. 4(1):1–23. doi:10.1038/nrdp.2018.1.
- Song G, Lu Q, Fan H, Zhang X, Ge L, Tian R, Wang S, Feng T, Pan J, Feng J, et al. 2019. Inhibition of hexokinases holds potential as treatment strategy for rheumatoid arthritis. Arthritis Res Ther. 21(1):87. doi:10.1186/s13075-019-1865-3.
- Streeter HB, Wraith DC. 2021. Manipulating antigen presentation for antigen-specific immunotherapy of autoimmune diseases. Curr Opin Immunol. 70:75–81. doi:10.1016/j.coi.2021.03.019.
- Sysi-Aho M, Ermolov A, Gopalacharyulu PV, Tripathi A, Seppänen-Laakso T, Maukonen J, Mattila I, Ruohonen ST, Vähätalo L, Yetukuri L, et al. 2011. Metabolic regulation in progression to autoimmune diabetes. PLoS Comput Biol. 7(10):e1002257. doi:10.1371/journal.pcbi.1002257.
- Tavazzi B, Batocchi AP, Amorini AM, Nociti V, D’Urso S, Longo S, Gullotta S, Picardi M, Lazzarino G. 2011. Serum metabolic profile in multiple sclerosis patients. Mult Scler Int. 2011:1–8. doi:10.1155/2011/167156.
- Tse HM, Milton MJ, Schreiner S, Profozich JL, Trucco M, Piganelli JD. 2007. Disruption of innate-mediated proinflammatory cytokine and reactive oxygen species third signal leads to antigen-specific hyporesponsiveness. J Immunol. 178(2):908–917. doi:10.4049/jimmunol.178.2.908.
- Tsokos GC. 2011. Systemic lupus erythematosus. N Engl J Med. 365(22):2110–2121. doi:10.1056/NEJMra1100359.
- Vadrucci M, Castellani M, Benti R. 2016. Active subcutaneous calcinosis demonstrated by fluorine-18 fluorodeoxyglucose positron emission tomography/computed tomography in a case of limited cutaneous systemic sclerosis. Indian J Nucl Med. 31(2):154. doi:10.4103/0972-3919.178335.
- Vander Heiden MG, DeBerardinis RJ. 2017. Understanding the intersections between metabolism and cancer biology. Cell. 168(4):657–669. doi:10.1016/j.cell.2016.12.039.
- Volkmann ER, Andréasson K, Smith V. 2023. Systemic sclerosis. Lancet. 401(10373):304–318. doi:10.1016/S0140-6736(22)01692-0.
- Wahl D, Petersen B, Warner R, Richardson B, Glick G, Opipari A. 2010. Characterization of the metabolic phenotype of chronically activated lymphocytes. Lupus. 19(13):1492–1501. doi:10.1177/0961203310373109.
- Wang L, Wang F-S, Gershwin ME. 2015. Human autoimmune diseases: a comprehensive update. J Intern Med. 278(4):369–395. doi:10.1111/joim.12395.
- Weyand CM, Zeisbrich M, Goronzy JJ. 2017. Metabolic signatures of T-cells and macrophages in rheumatoid arthritis. Curr Opin Immunol. 46:112–120. doi:10.1016/j.coi.2017.04.010.
- Woo YD, Jeong D, Chung DH. 2021. Development and functions of alveolar macrophages. Mol Cells. 44(5):292–300. doi:10.14348/molcells.2021.0058.
- Xiao W, Wang R-S, Handy DE, Loscalzo J. 2018. Nad(H) and NADP(H). Redox couples and cellular energy metabolism. Antioxid Redox Signal. 28(3):251–272. doi:10.1089/ars.2017.7216.
- Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu R-M, Bernard K, Thannickal VJ, Liu G. 2015. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am J Respir Crit Care Med. 192(12):1462–1474. doi:10.1164/rccm.201504-0780OC.
- Xu J, Jiang C, Wang X, Geng M, Peng Y, Guo Y, Wang S, Li X, Tao P, Zhang F, et al. 2020. Upregulated PKM2 in macrophages exacerbates experimental arthritis via STAT1 signaling. J Immunol. 205(1):181–192. doi:10.4049/jimmunol.1901021.
- Yin Y, Choi S-C, Xu Z, Perry DJ, Seay H, Croker BP, Sobel ES, Brusko TM, Morel L. 2015. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 7(274):274ra18–274ra18. doi:10.1126/scitranslmed.aaa0835.
- Yin Y, Choi S-C, Xu Z, Zeumer L, Kanda N, Croker BP, Morel L. 2016. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol. 196(1):80–90. doi:10.4049/jimmunol.1501537.
- Young SP, Kapoor SR, Viant MR, Byrne JJ, Filer A, Buckley CD, Kitas GD, Raza K. 2013. The impact of inflammation on metabolomic profiles in patients with arthritis. Arthritis Rheum. 65(8):2015–2023. doi:10.1002/art.38021.
- Zhao X, Fu J, Du J, Xu W. 2020. The role of D-3-phosphoglycerate dehydrogenase in cancer. Int J Biol Sci. 16(9):1495–1506. doi:10.7150/ijbs.41051.
- Zhao Y, Yan X, Li X, Zheng Y, Li S, Chang X. 2016. PGK1, a glucose metabolism enzyme, may play an important role in rheumatoid arthritis. Inflamm Res. 65(10):815–825. doi:10.1007/s00011-016-0965-7.
- Zorov DB, Juhaszova M, Sollott SJ. 2014. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 94(3):909–950. doi:10.1152/physrev.00026.2013.
- Zou Y, Zeng S, Huang M, Qiu Q, Xiao Y, Shi M, Zhan Z, Liang L, Yang X, Xu H. 2017. Inhibition of 6-phosphofructo-2-kinase suppresses fibroblast-like synoviocytes-mediated synovial inflammation and joint destruction in rheumatoid arthritis. Br J Pharmacol. 174(9):893–908. doi:10.1111/bph.13762.