
ABSTRACT
Autophagy plays a critical role in regulating the quality and quantity of cellular compartments; however, the mechanisms governing endoplasmic reticulum (ER) autophagy (ER-phagy) are still largely undefined. In this study, we identified several inhibitors of USP14, a ubiquitin-specific protease, including IU1 and b-AP15, as novel ER-phagy inducers through chemical library screening. While USP14 is known to act as an ER stress inducer, its precise role in ER-phagy remains unclear. Our findings demonstrate that treatment with either IU1 or b-AP15 induces ER-phagy by increasing ER stress in HepG2 cells. Similarly, depletion of USP14 augments ER-phagy and the ER stress response. The blockage of autophagy using an ULK1 inhibitor, SBI0206965, impedes ER-phagy. Moreover, inhibition of ER stress with tauroursodeoxycholic acid reverses ER-phagy by alleviating ER stress in HepG2 cells. We also found that suppression of the stress kinase JNK inhibited ER-phagy in IU1-treated cells. In conclusion, the results of this study suggest that the inhibition of USP14 accelerates ER-phagy by enhancing ER stress and JNK activation in HepG2 cells.
Introduction
The endoplasmic reticulum (ER) is a crucial cellular organelle, which is vital for maintaining cell homeostasis. When subjected to external stimulation, ER stress triggers processes such as the unfolded protein response (UPR) and ER-associated degradation (ERAD), which help preserve ER equilibrium (Almanza et al. Citation2019; Chen and Cubillos-Ruiz Citation2021; Reggiori and Molinari Citation2022). Selective autophagy of the ER, also known as ER-phagy, is a fascinating cellular process that plays a crucial role in maintaining ER homeostasis (Yang et al. Citation2021; Reggiori and Molinari Citation2022; Kwon et al. Citation2023). Within eukaryotic cells, the ER is an important organelle involved in various essential functions, such as protein synthesis, lipid metabolism, and calcium storage (Berridge Citation2002; Schroder and Kaufman Citation2005). During ER stress, the ER becomes dysfunctional or stressed due to various internal or external factors, then the affected cells activate ER-phagy as a protective mechanism to selectively target and degrade damaged or stressed ER components (Stephani et al. Citation2020; Chipurupalli et al. Citation2022a; Reggiori and Molinari Citation2022). According to this notion, cancer cells adapt ER-phagy to survive hypoxic stress (Chipurupalli et al. Citation2022b). This intricate process involves specific molecular machinery and regulatory factors that ensure the efficient removal of aberrant ER structures, ultimately contributing to cellular health and adaptation to varying environmental conditions (He et al. Citation2021; Reggiori and Molinari Citation2022).
Ubiquitination, a post-translational modification, emerges as a pivotal regulatory mechanism in selective autophagy of the ER (Gonzalez et al. Citation2023). Substrate ubiquitination marks damaged ER regions, labeling them for recognition and engulfment by autophagic membranes (Gonzalez et al. Citation2023). Notably, ER-phagy receptors, such as FAM134B, SEC62, and RTN3, can interact with ubiquitinated ER substrates, linking them to the autophagy machinery for degradation (Chiramel et al. Citation2016; Fumagalli et al. Citation2016; Grumati et al. Citation2017, Citation2018). Furthermore, several signaling pathways, including the UPR and ERAD pathway, are closely connected with the ubiquitination process to modulate ER-phagy (Schultz et al. Citation2018; Chiritoiu et al. Citation2020). For instance, during ER stress, the UPR can induce the expression of ER-phagy receptors and promote the ubiquitination of specific ER proteins, facilitating their selective removal (Schultz et al. Citation2018).
A group of mitogen-activated protein kinases known as c-Jun N-terminal kinases (JNKs) play a central role in the cellular response to various stress signals including those that induce ER stress as well as UV radiation, oxidative stress, and cytokines (Davis Citation2000; Johnson and Nakamura Citation2007). Consistently, it has been reported that RNF13 is a critical mediator for facilitating ER stress-induced apoptosis through the activation of the IRE1α-TRAF2-JNK signaling pathway (Arshad et al. Citation2013). Furthermore, Wu et al. showed the dependence of selective ER-phagy on the IRE1α-JNK pathway for resolving ER stress during the initial phase of hepatic steatosis (Guner and Ekmekci Citation2019), offering further insights into our understanding of the intricate regulatory mechanisms involved. The growing body of research on ER stress and ER-phagy highlights the significance of this organelle in maintaining cellular integrity and opens avenues for potential therapeutic interventions in various pathological conditions (He et al. Citation2021; Reggiori and Molinari Citation2022).
In this study, we identified several new inhibitors of enzyme, ubiquitin specific peptidase 14 (USP14), which induce of ER-phagy in mammals. USP14 is a proteasome-associated deubiquitinating enzyme (DUB) that reversely associates with the proteasome and negatively regulates proteasome activity. DUBs, including USP14, play vital roles in protein homeostasis (proteostasis) and the regulation of various cellular activities (de Poot et al. Citation2017; Wang et al. Citation2021a). While the molecular mechanism underlying ER-phagy is not fully understood, we found that the inhibition of USP14 significantly promoted both ER stress and ER-phagy in HepG2 cells. This effect could be reversed in HepG2 cells by inhibiting either the ER stress pathway or the JNK stress kinase.
Materials and methods
Reagents
Libraries containing the PPI inhibitor (L9400) and ubiquitination compound (L8600) for drug screening were obtained from TargetMol (Boston, MA, USA). IU1 (HY-13817), SBI-0206965 (HY-16966), and Tauroursodeoxycholic acid (TUDCA) (HY-19696) were obtained from MedChemExpress LLC. (Monmouth Junction, NJ, USA). b-AP15(11324) were obtained from Cayman Chemical Co. (Ann Arbor, MI, USA). Bafilomycin A1 (B1793) and SP600125 (S5567) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Hoechst 33342 (H3570) was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Short interfering RNA (siRNA) targeting USP14 (#1, 5′-GGAGAAAUUUGAAGGUGUA-3′ and #2, 5′-AGAAAUGCCUUGUAUAUCA-3′) and scrambled siRNA (5′-CCUACGCCACCAAUUUCGU-3′) were synthesized by Genolution (Seoul, Korea).
Cell culture and establishment of stable cell lines
HepG2 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). All cells were cultured at 37°C in an incubator containing a 5% CO2 atmosphere and maintained in DMEM supplemented with 10% FBS (S001-07, WELGENE, Gyeongsan, Korea) and 1% penicillin–streptomycin (LS202-02; WELGENE, Gyeongsan, Korea). Monomeric red fluorescent protein (mCherry)-pH-sensitive green fluorescent protein variant protein (pHluorin) lysine-aspartic acid-glutamic acid-leucine (KDEL) (mCherry-pHluorin-KDEL) were generated by subcloning in pcDNA3. To generate stable cell lines, HepG2 cells were transfected with mCherry-pHluorin-KDEL using Lipofectamine 2000 (11668019; Invitrogen), in accordance with the manufacturer’s protocol. Stable transfectants were established by growth in selection medium containing 1 mg/mL G418 (10131035; Invitrogen) for 7 days. After seeding the individual cells, stable clones were identified under a fluorescence microscope (IX71; Olympus, Tokyo, Japan).
Determination of ER-phagic cells
To quantify cells undergoing ER-phagy, HepG2/mCherry-pHluorin-KDEL cells were grown on a glass cover slip and treated with IU1. The cells were then washed with PBS (LB 001–02; WELGENE, Korea), fixed with 4% paraformaldehyde (P2031; Biosesang, Korea) for 20 min, and stained with the indicated dyes or antibodies. Punctate ER structures were visually determined using confocal microscopy. The number of structural puncta was quantified using ImageJ (NIH, Bethesda, MD, USA), and the experiments were performed at least three times for each condition, as indicated in each Figure. When ER-phagy was activated, the RFP (mCherry) fragment appeared as a result of degradation of the reporter inside lysosomes because the pH-sensitive protein pHluorin was degraded first, so that RFP, which is relatively stable in lysosomes, remained.
Western blotting
Cell lysates were prepared in 2× Laemmli sample buffer (1610737; Bio-Rad). Total protein quantity was measured using the Bradford dye (5000001; Bio-Rad) in accordance with the manufacturer’s instructions. The samples were then separated by SDS polyacrylamide gel electrophoresis and transferred to a PVDF membrane (1620177; Bio-Rad). After blocking with 4% skim milk (BD Bioscience, 90002–594) prepared in TBST [Tris base (T9200; GenDEPOT, Baker, TX, USA), NaCl (G0610; GenDEPOT), and Tween® 20 (P7949; Sigma-Aldrich)], the membrane was incubated with the indicated primary antibodies. Anti-HSPA5 (sc-166490) and anti-DDIT3 (sc-7351) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA); anti-Phospho-SAPK/JNK (Thr183/Tyr185) (9255S) and USP14 (11931S) were purchased from Cell Signaling Technology (Danvers, MA, USA); and anti-LC3 (NB100-2220) and anti-ACTA1 (MAB1501) were obtained from NOVUS Biologicals (Centennial, CO, USA) and Sigma-Aldrich, respectively. For protein detection, membranes were incubated with HRP-conjugated secondary antibodies (7076S and 7074S; Cell Signaling Technology).
Statistical analysis
Data were obtained from at least three independent experiments and are presented as means ± SD. Statistical evaluation of the results was performed using Student’s t test and one-way ANOVA followed by the Bonferroni post hoc test, respectively. **p < 0.01 and ***p < 0.001 were considered significant.
Results
USP14 is a novel ER-phagy regulator in HepG2 cells
To identify novel regulators of ER-phagy, we developed a cell-based high-content screening system using HepG2 cells stably expressing mCherry-pHluorin tagged with the ER retention signal peptide (KEDL) (HepG2/mCherry-pHluorin-KDEL). We screened a chemical library comprising small ubiquitination-related chemical inhibitors and identified several ER-phagy-inducing candidates, including IU1 and b-AP15. IU1, a cell-permeable pyrrolyl pyrrolidinyl-ethanone, specifically inhibits USP14 a member of the ubiquitin-specific processing (UBP) family of proteases (Wang et al. Citation2018). To validate the screening results, HepG2/mCherry-pHluorin-KDEL cells were treated with IU1 or b-AP15, and ER-phagy was monitored through fluorescent imaging. In this assay, yellow puncta representing ER were visible in control cells due to double-positive fluorescence [mCherry(+)/pHluorin(+)], while the number of red-only puncta with reduced green signal [mCherry(+)/pHluorin(−)] increased significantly in cells treated with either IU1 or b-AP15 (A–C). This finding suggests that both IU1 and b-AP15 promote ER-phagy in HepG2 cells. We then further investigated the mechanism of IU1 action on ER-phagy. As it has been recently reported that IU1 induces ER stress, thereby promoting ER-phagy in lung cancer cells (Moghadami et al. Citation2020), HepG2 cells were treated with IU1 for 24 h. As shown in D, IU1 treatment increased ER stress marker proteins, such as heat shock protein family A member 5 (HSPA5) and DNA damage inducible transcript 3 (DDIT3), in HepG2 cells. Additionally, IU1 enhanced LC3 accumulation. Since ER-phagy refers to the selective degradation of the ER via autophagy, we examined IU1's effect on autophagy activation in HepG2 cells. The conversion of LC3-I to LC3-II is widely used to measure autophagy activation. As shown in E,F, LC3-II levels were enhanced in IU1-treated cells and further increased in cells co-treated with IU1 and bafilomycin A1, an inhibitor of lysosome fusion, when compared to control cells. These data indicate that IU1 activates the autophagic flux to induce ER-phagy. Additionally, we found that bafilomycin A1 suppressed the IU1-induced increase in red puncta [mCherry(+)/pHluorin(−)] in HepG2/mCherry-pHluorin-KDEL cells (F,G).
Figure 1. Induction of ER-phagy with USP14 inhibitor in HepG2 cells. (A) Schematic diagram of the measurement of autophagic flux of endoplasmic reticulum using mCherry-pHluorin-KDEL. (B–D) HepG2/mCherry-pHluorin-KDEL cells were treated with inhibitors of USP14 [IU1 (100 µM), b-AP15 (100 nM)] for 24 h. The cells were fixed for imaging under a fluorescence microscope (B). Scale bar: 20 um; the number of puncta pHluorin(−) and mCherry(+)-labeled autolysosomes per cell, which displayed the ER reporter mCherry-pHluorin-KDEL due to lysosomal delivery, was counted by assessing approximately 150 cells (C); IU1 treated HepG2 cells were harvested and analyzed by Western blotting with the indicated antibodies (D); (E–G) HepG2/mCherry-pHluorin-KDEL cells were treated IU1 (100 µM) with and without bafilomycin A1 (Baf. A1, 30 nM) for 24 h. The cells were harvested for Western blotting analysis with the indicated antibodies (E); The cells were fixed for imaging under a fluorescence microscope (F); the number of puncta pHluorin(−) and mCherry(+) per cell was counted by assessing approximately 150 cells (G). The data are presented as the mean as the mean ± SD. **p < 0.01 and ***p < 0.001, by one-way ANOVA with the Bonferroni post hoc test.
![Figure 1. Induction of ER-phagy with USP14 inhibitor in HepG2 cells. (A) Schematic diagram of the measurement of autophagic flux of endoplasmic reticulum using mCherry-pHluorin-KDEL. (B–D) HepG2/mCherry-pHluorin-KDEL cells were treated with inhibitors of USP14 [IU1 (100 µM), b-AP15 (100 nM)] for 24 h. The cells were fixed for imaging under a fluorescence microscope (B). Scale bar: 20 um; the number of puncta pHluorin(−) and mCherry(+)-labeled autolysosomes per cell, which displayed the ER reporter mCherry-pHluorin-KDEL due to lysosomal delivery, was counted by assessing approximately 150 cells (C); IU1 treated HepG2 cells were harvested and analyzed by Western blotting with the indicated antibodies (D); (E–G) HepG2/mCherry-pHluorin-KDEL cells were treated IU1 (100 µM) with and without bafilomycin A1 (Baf. A1, 30 nM) for 24 h. The cells were harvested for Western blotting analysis with the indicated antibodies (E); The cells were fixed for imaging under a fluorescence microscope (F); the number of puncta pHluorin(−) and mCherry(+) per cell was counted by assessing approximately 150 cells (G). The data are presented as the mean as the mean ± SD. **p < 0.01 and ***p < 0.001, by one-way ANOVA with the Bonferroni post hoc test.](/cms/asset/dfa66901-705b-465b-a67a-5dda50664266/tacs_a_2285825_f0001_oc.jpg)
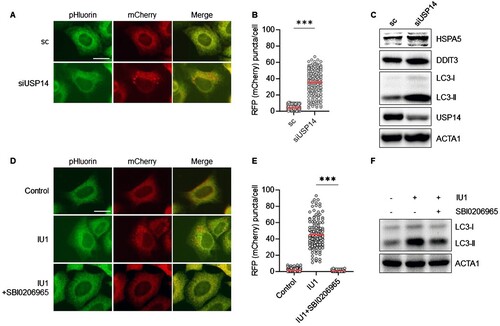
Given that both IU1 and b-AP15 target USP14, we explored the effect of USP14 knockdown on ER-phagy through RNA interference (RNAi). Consistent with the results of chemical inhibition, USP14 depletion using RNAi significantly increased the number of red puncta of mCherry-pHluorin-KDEL (A–C), indicating that the inhibition of USP14 induced ER-phagy in HepG2 cells. Subsequently, we asked whether autophagy affects IU1-induced ER-phagy, using SBI0206965, an inhibitor of ULK1, a highly selective autophagy kinase which plays a crucial role in initiating autophagosome formation in response to autophagy stimuli (Egan et al. Citation2015). HepG2/mCherry-pHluorin-KDEL cells treated with IU1 in the presence or absence of SBI0206965, were screened for ER-phagy. As shown in D,E, the enhanced red puncta of mCherry-pHluorin-KDEL were significantly reduced by co-treatment with SBI0206965 (D–F), suggesting that IU1 promotes ER-phagy through a ULK1-dependent canonical autophagic pathway. Taken together, these results indicate that the inhibition of USP14 selectively induces ER degradation.
Figure 2. Knockdown of USP14 induces ER-phagy in HepG2 cells. (A–C) HepG2/mCherry-pHluorin-KDEL cells were transiently transfected with siRNA targeting USP14 (siUSP14) and were then fixed for imaging under a fluorescence microscope (A) scale bar: 20 um; the numbers of puncta pHluorin(−) and mCherry(+)-labeled autolysosomes per cell were counted by assessing approximately 150 cells (B); reduced expression of USP14 by siRNA was verified by Western blotting with the indicated antibodies (C); (D–F) HepG2/mCherry-pHluorin-KDEL cells were treated IU1 with and without SBI0206965 (10 µM) for 24 h. The cells were fixed for imaging under a fluorescence microscope (D). Scale bar: 20 um; the number of puncta pHluorin(−) and mCherry(+) per cell was counted by assessing approximately 150 cells (E); then, the cells were harvested for Western blotting analysis with the indicated antibodies (F). The data are presented as the mean as the mean ± SD. **p < 0.01 and ***p < 0.001, by the Student’s t test (B) and one-way ANOVA with the Bonferroni post hoc test (E).
IU1 promotes ER-phagy by increasing ER stress and JNK activation in HepG2 cells
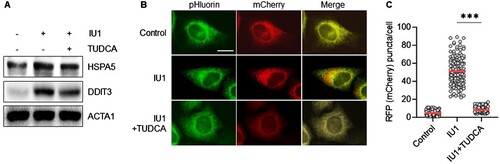
Our group and others have previously demonstrated that the inhibition of USP14 results in ER stress, leading to ER-phagy () (Liao et al. Citation2019). We thus investigated the role of ER stress in ER-phagy in IU1-treated cells by treating them with tauroursodeoxycholic acid (TUDCA). TUDCA is known to protect cells by reducing ER stress, activating various proteins, including HSPA5, EIF2AK3, EIF2A, ATF4, IRE1α, JNK, p38, and DDIT3 (Yoon et al. Citation2016; Chen et al. Citation2022). In line with this understanding, the treatment with TUDCA downregulated the expression of ER stress-response proteins, such as HSPA5 and DDIT3, in IU1-treated HepG2 cells (A). We then further examined the effect of TUDCA on IU1-induced ER-phagy. Specifically, we analyzed ER-phagy in HepG2/mCherry-pHluorin-KDEL cells treated with IU1, either in the presence or absence of TUDCA. As shown in B,C, the increase in red puncta of mCherry-pHluorin-KDEL was significantly reduced by co-treatment with TUDCA. This finding indicates that the inhibition of USP14 triggers ER stress in HepG2 cells, leading to increased ER-phagy.
Figure 3. Inhibition of ER stress suppress ER-phagy in IU1 treated HepG2 cells. (A–C) HepG2/mCherry-pHluorin-KDEL cells were treated with IU1 (100 µM) with and without tauroursodeoxycholic acid (TUDCA, 10 µM) for 24 h. The cells harvested to analyze by Western blotting with indicated antibodies (A); the cells were fixed for imaging under a fluorescence microscope (B). Scale bar: 20 um; the number of puncta pHluorin(−) and mCherry(+) per cell was counted by assessing approximately 150 cells (C). The data are presented as the mean as the mean ± SD. **p < 0.01 and ***p < 0.001, by one-way ANOVA with the Bonferroni post hoc test.
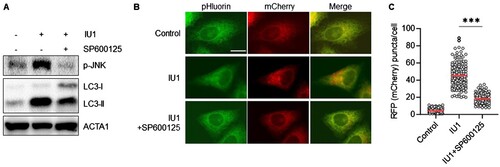
Next, we explored the underlying mechanism of ER stress-mediated ER-phagy induced by IU1. Previous reports have noted that the IRE1α-JNK pathway is activated during ER stress to induce autophagy in HepG2 cells (Wu et al. Citation2021; Lee et al. Citation2022). As JNK can regulate autophagy by phosphorylating regulatory proteins like Bcl-2 and upregulating autophagy-related genes such as Beclin1/ATG6 (Wei et al. Citation2008; Park et al. Citation2009), we assessed the role of JNK in ER-phagy within IU1-treated cells. Our findings show that IU1 treatment increased the substrate phosphorylation of JNK, the process inhibited by the JNK inhibitor SP600125 (A). Notably, the treatment with SP600125 also suppresses the enhanced red puncta of mCherry-pHluorin-KDEL induced by IU1 (B,C). Taken together, these results suggest that IU1 promotes ER-phagy by augmenting ER stress and JNK activation in HepG2 cells.
Figure 4. JNK inhibitor SP600125 blocks IU1-induced ER-phagy in HepG2 cells. (A–C) HepG2/mCherry-pHluorin-KDEL cells were treated with IU1 with and without SP600125 (10 µM) for 24 h. The cells harvested to analyze by Western blotting with indicated antibodies (A); the cells were fixed for imaging under a fluorescence microscope (B). Scale bar: 20 um; the number of puncta pHluorin(−) and mCherry(+) per cell was counted by assessing approximately 150 cells (C). The data are presented as the mean as the mean ± SD. **p < 0.01 and ***p < 0.001, by one-way ANOVA with the Bonferroni post hoc test.
Discussion
ER homeostasis involving the quality and quantity control is critical for the pathology of ER-associated diseases, but the precise molecular mechanism underlying ER-phagy remains largely unknown (Grumati et al. Citation2018). In this study, we identified IU1 and b-AP15 as inducers of ER-phagy. These compounds are small chemical inhibitors that target USP14, a deubiquitinating enzyme (Lee et al. Citation2010; D'Arcy et al. Citation2011). USP14 is overexpressed in cancer cells and is involved in various canonical cellular signaling pathways, including the NF-κB and Wnt/β-catenin signaling pathways (Wang et al. Citation2021b). Dysregulation of USP14 can lead to pathological conditions such as cancer, neurodegenerative diseases, autophagy, immune responses, and viral infections (Wang et al. Citation2021a). Overexpression of USP14 inhibits the ERAD pathway and protects cells from ER stress without affecting proteosomal activity (Nagai et al. Citation2009; Park et al. Citation2020). IRE1a is an ER-resident transmembrane protein that senses ER stress. USP14 has an interaction with IRE1a and detaches from the activated state of IRE1a during ER stress. And, the overexpression of USP14 can hinder the phosphorylation of IRE1a and provide protection against ER stress (Nagai et al. Citation2009; Hyrskyluoto et al. Citation2014). In addition, USP14 is shown to promote tryptophan metabolism and T-cell dysfunction by stabilizing indoleamine 2,3-dioxygenase 1 in colorectal cancer (Shi et al. Citation2022). ER-phagy is a highly regulated process, and its dysregulation may contribute to cancer progression. The role of ER stress in cancer is multifaceted and can lead to both pro-survival and pro-death signals. Thus, identifying USP14 inhibitors represents a strategic approach for the development of anti-cancer drugs.
As an inhibitor of USP14, IU1 has been shown to restore the level and function of protein p53, leading to the death of cancer cells (Ma et al. Citation2020). Additionally, IU1 treatment has been found to reduce MDM2 protein expression in cancer cells (Xu et al. Citation2020). Alongside IU1, b-AP15 inhibits the deubiquitinating activity of both UCHL5 and USP14 in the 19S proteasome. These findings contribute to a comprehensive approach to prevent tumor progression, especially in cases of p53 deficiency (Jiang et al. Citation2022). Notably, it has been recently reported that the inhibition of USP14 induces ER stress-mediated autophagy. This process occurs through the accumulation of ubiquitinated proteins, triggering the ER stress-UPR axis, and the activation of JNK in lung cancer cells (Moghadami et al. Citation2020). In line with this notion, we also found that the inhibition of USP14 by chemical inhibitors or genetic knockdown promotes ER stress responses, leading to ER-phagy in HepG2 cells ( and ). Notably, it was showed that the activation of JNK upregulates the autophagic gene Beclin-1 and phosphorylates Bcl-2 and p53 to increase autophagic cell death (Li et al. Citation2009; Park et al. Citation2009). Consistent with our findings, IU1 is shown to activate JNK in response to ER stress, thereby mediating ER-phagy. The inhibition of JNK abolished ER-phagy, underscoring JNK's role in this process (). However, further studies are needed to explore the role of JNK in IU1-induced ER-phagy. Our findings suggest that the inhibition of USP14 may promote ER-phagy through JNK signaling.
Ubiquitination has recently been recognized as a crucial regulatory mechanism in the clearance of ER proteins through ER-phagy. ER proteins that recognize and select ER fragments, such as FAM134B, RTN3, SEC62, and CCPG1, are targets for ubiquitination. These often contain LC3-interacting regions (LIR), which directly recruit autophagosomes for degradation through ER-phagy (Mochida et al. Citation2015; Grumati et al. Citation2017; Smith et al. Citation2018). While Ji et al. demonstrated that the ER membrane E3 ligase TRIM13 is a ubiquitin-dependent ER-phagy receptor to SQSTM1/p62 (Ji et al. Citation2019), the exact E3 ligase proteins involved in regulating the ubiquitination of ER proteins, including RTN3 and CCPG1, remain unidentified. Both FAM134B and RTN3 are reticulon-type proteins that can remodel the ER network, ensuring basal membrane turnover, whereas SEC62 and CCPG1 are transmembrane ER receptors that function in response to ER stress signals (Grumati et al. Citation2018). Recently, González et al. and Foronda et al. discuss the role of ubiquitin in the autophagic capture of ER by ER-phagy and propose that ubiquitylation of the ER-phagy receptor FAM134B and ER-shaping protein ARL61PL1 encourages receptor clustering in nanodomains, leading to membrane curvature and facilitating autophagosomal capture (Foronda et al. Citation2023; Gonzalez et al. Citation2023). Firstly discovered that the E3 ligase autocrine motility factor receptor (AMFR, also known as gp78) within multimeric ER-phagy receptor clusters directly catalyzes FAM134B ubiquitination, thereby regulating the dynamic flux of ER-phagy (Gonzalez et al. Citation2023). Indeed, E3 ligases and deubiquitinases (DUBs) have a complex relationship, playing contrasting roles in controlling protein ubiquitination and deubiquitination. For example, in mitophagy, parkin functions as an E3 ligase targeting damaged mitochondria to promote the ubiquitination of proteins such as Mfn2 and VDAC (Geisler et al. Citation2010; Bingol et al. Citation2014). Conversely, USP30, a deubiquitinating enzyme, counters parkin action by removing ubiquitin from mitochondrial proteins, thus acting as a negative regulator of mitophagy (Geisler et al. Citation2010; Bingol et al. Citation2014). In this study, we found that the inhibition of USP14 promoted ER-phagy. In this context, USP14 may interact with an E3 ligase such as AMFR to regulate ER-phagy in response to ER stress. Therefore, understanding the precise balance between E3 ligases and DUBs in ER stress and their regulation may provide valuable insights into the role of ER in both human physiology and pathophysiology of various diseases accompanied by disrupted ER homeostasis, such as cancer.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, Luis A, McCarthy N, Montibeller L, More S, et al. 2019. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. 286(2):241–278. doi:10.1111/febs.14608.
- Arshad M, Ye Z, Gu X, Wong CK, Liu Y, Li D, Zhou L, Zhang Y, Bay WP, Yu VC, Li P. 2013. RNF13, a RING finger protein, mediates endoplasmic reticulum stress-induced apoptosis through the inositol-requiring enzyme (IRE1alpha)/c-Jun NH2-terminal kinase pathway. J Biol Chem. 288(12):8726–8736. doi:10.1074/jbc.M112.368829.
- Berridge MJ. 2002. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 32(5-6):235–249. doi:10.1016/S0143416002001823.
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. 2014. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 510(7505):370–375. doi:10.1038/nature13418.
- Chen F, Ge Z, Li N, Yu Z, Wu R, Zhao Y, He X, Cai G. 2022. TUDCA protects against tunicamycin-induced apoptosis of dorsal root ganglion neurons by suppressing activation of ER stress. Exp Ther Med. 24(2):509. doi:10.3892/etm.2022.11436.
- Chen X, Cubillos-Ruiz JR. 2021. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 21(2):71–88. doi:10.1038/s41568-020-00312-2.
- Chipurupalli S, Desiderio V, Robinson N. 2022a. Analysis of ER-phagy in cancer drug resistance. In: Baiocchi M, editor. Cancer drug resistance: methods and protocols. New York, NY: Springer US; p. 211–220.
- Chipurupalli S, Ganesan R, Martini G, Mele L, Reggio A, Esposito M, Kannan E, Namasivayam V, Grumati P, Desiderio V, Robinson N. 2022b. Cancer cells adapt FAM134B/BiP mediated ER-phagy to survive hypoxic stress. Cell Death Dis. 13(4):357. doi:10.1038/s41419-022-04813-w.
- Chiramel AI, Dougherty JD, Nair V, Robertson SJ, Best SM. 2016. FAM134B, the selective autophagy receptor for endoplasmic reticulum turnover, inhibits replication of ebola virus strains Makona and Mayinga. J Infect Dis. 214(suppl 3):S319–S325. doi:10.1093/infdis/jiw270.
- Chiritoiu M, Chiritoiu GN, Munteanu CVA, Pastrama F, Ivessa NE, Petrescu SM. 2020. EDEM1 drives misfolded protein degradation via ERAD and exploits ER-phagy as back-up mechanism when ERAD Is impaired. Int J Mol Sci. 21(10):3468. doi:10.3390/ijms21103468.
- D'Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, Perego P, Sadeghi B, Hassan M, Larsson R, Linder S. 2011. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med. 17(12):1636–1640. doi:10.1038/nm.2536.
- Davis RJ. 2000. Signal transduction by the JNK group of MAP kinases. Cell. 103(2):239–252. doi:10.1016/S0092-8674(00)00116-1.
- de Poot SAH, Tian G, Finley D. 2017. Meddling with fate: the proteasomal deubiquitinating enzymes. J Mol Biol. 429(22):3525–3545. doi:10.1016/j.jmb.2017.09.015.
- Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, et al. 2015. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 59(2):285–297. doi:10.1016/j.molcel.2015.05.031.
- Foronda H, Fu Y, Covarrubias-Pinto A, Bocker HT, Gonzalez A, Seemann E, Franzka P, Bock A, Bhaskara RM, Liebmann L, et al. 2023. Heteromeric clusters of ubiquitinated ER-shaping proteins drive ER-phagy. Nature. 618(7964):402–410. doi:10.1038/s41586-023-06090-9.
- Fumagalli F, Noack J, Bergmann TJ, Cebollero E, Pisoni GB, Fasana E, Fregno I, Galli C, Loi M, Solda T, et al. 2016. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol. 18(11):1173–1184. doi:10.1038/ncb3423.
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. 2010. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 12(2):119–131. doi:10.1038/ncb2012.
- Gonzalez A, Covarrubias-Pinto A, Bhaskara RM, Glogger M, Kuncha SK, Xavier A, Seemann E, Misra M, Hoffmann ME, Brauning B, et al. 2023. Ubiquitination regulates ER-phagy and remodelling of endoplasmic reticulum. Nature. 618(7964):394–401. doi:10.1038/s41586-023-06089-2.
- Grumati P, Dikic I, Stolz A. 2018. ER-phagy at a glance. J Cell Sci. 131(17):jcs217364. doi:10.1242/jcs.217364.
- Grumati P, Morozzi G, Holper S, Mari M, Harwardt MI, Yan R, Muller S, Reggiori F, Heilemann M, Dikic I. 2017. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife. 6:e25555. doi:10.7554/eLife.25555.
- Guner MD, Ekmekci PE. 2019. A survey study evaluating and comparing the health literacy knowledge and communication skills used by nurses and physicians. Inquiry. 56:46958019865831.
- He L, Qian X, Cui Y. 2021. Advances in ER-phagy and its diseases relevance. Cells. 10(9):2328. doi:10.3390/cells10092328.
- Hyrskyluoto A, Bruelle C, Lundh SH, Do HT, Kivinen J, Rappou E, Reijonen S, Waltimo T, Petersen A, Lindholm D, Korhonen L. 2014. Ubiquitin-specific protease-14 reduces cellular aggregates and protects against mutant huntingtin-induced cell degeneration: involvement of the proteasome and ER stress-activated kinase IRE1alpha. Hum Mol Genet. 23(22):5928–5939. doi:10.1093/hmg/ddu317.
- Ji CH, Kim HY, Heo AJ, Lee SH, Lee MJ, Kim SB, Srinivasrao G, Mun SR, Cha-Molstad H, Ciechanover A, et al. 2019. The N-degron pathway mediates ER-phagy. Mol Cell. 75(5):1058–1072. e1059. doi:10.1016/j.molcel.2019.06.028.
- Jiang ZY, Hong J, Zhang JH, Wang XF, Ma YS, Xiong ZX, Sun HR, Cheng C, Xie BZ, Liu JB, et al. 2022. Treatment with b-AP15 to inhibit UCHL5 and USP14 deubiquitinating activity and enhance p27 and cyclin E1 for tumors with p53 deficiency. Technol Cancer Res Treat. 21:15330338221119745.
- Johnson GL, Nakamura K. 2007. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta. 1773(8):1341–1348. doi:10.1016/j.bbamcr.2006.12.009.
- Kwon J, Kim J, Kim KI. 2023. Crosstalk between endoplasmic reticulum stress response and autophagy in human diseases. Anim Cells Syst (Seoul). 27(1):29–37. doi:10.1080/19768354.2023.2181217.
- Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, et al. 2010. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 467(7312):179–184. doi:10.1038/nature09299.
- Lee Y, Kwon J, Jeong JH, Ryu JH, Kim KI. 2022. Kazinol C from Broussonetia kazinoki stimulates autophagy via endoplasmic reticulum stress-mediated signaling. Anim Cells Syst (Seoul). 26(1):28–36. doi:10.1080/19768354.2021.2023628.
- Li DD, Wang LL, Deng R, Tang J, Shen Y, Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al. 2009. The pivotal role of c-Jun NH2-terminal kinase-mediated beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene. 28(6):886–898. doi:10.1038/onc.2008.441.
- Liao Y, Duan B, Zhang Y, Zhang X, Xia B. 2019. Excessive ER-phagy mediated by the autophagy receptor FAM134B results in ER stress, the unfolded protein response, and cell death in HeLa cells. J Biol Chem. 294(52):20009–20023. doi:10.1074/jbc.RA119.008709.
- Ma YS, Wang XF, Zhang YJ, Luo P, Long HD, Li L, Yang HQ, Xie RT, Jia CY, Lu GX, et al. 2020. Inhibition of USP14 deubiquitinating activity as a potential therapy for tumors with p53 deficiency. Mol Ther Oncolytics. 16:147–157. doi:10.1016/j.omto.2019.12.013.
- Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, Nakatogawa H. 2015. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 522(7556):359–362. doi:10.1038/nature14506.
- Moghadami AA, Aboutalebi Vand Beilankouhi E, Kalantary-Charvadeh A, Hamzavi M, Mosayyebi B, Sedghi H, Ghorbani Haghjo A, Nazari Soltan Ahmad S. 2020. Inhibition of USP14 induces ER stress-mediated autophagy without apoptosis in lung cancer cell line A549. Cell Stress Chaperones. 25(6):909–917. doi:10.1007/s12192-020-01125-w.
- Nagai A, Kadowaki H, Maruyama T, Takeda K, Nishitoh H, Ichijo H. 2009. USP14 inhibits ER-associated degradation via interaction with IRE1alpha. Biochem Biophys Res Commun. 379(4):995–1000. doi:10.1016/j.bbrc.2008.12.182.
- Park JE, Tran TXT, Park N, Yeom J, Kim K, Kang MJ. 2020. The function of Drosophila USP14 in endoplasmic reticulum stress and retinal degeneration in a model for autosomal dominant retinitis pigmentosa. Biology (Basel). 9(10):332. doi:10.3390/biology9100332.
- Park KJ, Lee SH, Lee CH, Jang JY, Chung J, Kwon MH, Kim YS. 2009. Upregulation of beclin-1 expression and phosphorylation of Bcl-2 and p53 are involved in the JNK-mediated autophagic cell death. Biochem Biophys Res Commun. 382(4):726–729. doi:10.1016/j.bbrc.2009.03.095.
- Reggiori F, Molinari M. 2022. ER-phagy: mechanisms, regulation, and diseases connected to the lysosomal clearance of the endoplasmic reticulum. Physiol Rev. 102(3):1393–1448. doi:10.1152/physrev.00038.2021.
- Schroder M, Kaufman RJ. 2005. The mammalian unfolded protein response. Annu Rev Biochem. 74(1):739–789. doi:10.1146/annurev.biochem.73.011303.074134.
- Schultz ML, Krus KL, Kaushik S, Dang D, Chopra R, Qi L, Shakkottai VG, Cuervo AM, Lieberman AP. 2018. Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat Commun. 9(1):3671. doi:10.1038/s41467-018-06115-2.
- Shi D, Wu X, Jian Y, Wang J, Huang C, Mo S, Li Y, Li F, Zhang C, Zhang D, et al. 2022. USP14 promotes tryptophan metabolism and immune suppression by stabilizing IDO1 in colorectal cancer. Nat Commun. 13(1):5644. doi:10.1038/s41467-022-33285-x.
- Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A, Behrends C, Wilkinson S. 2018. CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev Cell. 44(2):217–232. e211. doi:10.1016/j.devcel.2017.11.024.
- Stephani M, Picchianti L, Gajic A, Beveridge R, Skarwan E, Sanchez de Medina Hernandez V, Mohseni A, Clavel M, Zeng Y, Naumann C, et al. 2020. A cross-kingdom conserved ER-phagy receptor maintains endoplasmic reticulum homeostasis during stress. Elife. 9:e58396. doi:10.7554/eLife.58396.
- Wang D, Ma H, Zhao Y, Zhao J. 2021a. Ubiquitin-specific protease 14 is a new therapeutic target for the treatment of diseases. J Cell Physiol. 236(5):3396–3405. doi:10.1002/jcp.30124.
- Wang F, Ning S, Yu B, Wang Y. 2021b. USP14: structure, function, and target inhibition. Front Pharmacol. 12:801328. doi:10.3389/fphar.2021.801328.
- Wang Y, Jiang Y, Ding S, Li J, Song N, Ren Y, Hong D, Wu C, Li B, Wang F, et al. 2018. Small molecule inhibitors reveal allosteric regulation of USP14 via steric blockade. Cell Res. 28(12):1186–1194. doi:10.1038/s41422-018-0091-x.
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. 2008. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 30(6):678–688. doi:10.1016/j.molcel.2008.06.001.
- Wu P, Tian T, Zhao J, Song Q, Wu X, Guo Y, Yu Y, Tan S, Xia H. 2021. IRE1alpha-JNK pathway-mediated autophagy promotes cell survival in response to endoplasmic reticulum stress during the initial phase of hepatic steatosis. Life Sci. 264:118668. doi:10.1016/j.lfs.2020.118668.
- Xu L, Wang J, Yuan X, Yang S, Xu X, Li K, He Y, Wei L, Zhang J, Tian Y. 2020. IU1 suppresses proliferation of cervical cancer cells through MDM2 degradation. Int J Biol Sci. 16(15):2951–2963. doi:10.7150/ijbs.47999.
- Yang M, Luo S, Wang X, Li C, Yang J, Zhu X, Xiao L, Sun L. 2021. ER-Phagy: a new regulator of ER homeostasis. Front Cell Dev Biol. 9:684526. doi:10.3389/fcell.2021.684526.
- Yoon YM, Lee JH, Yun SP, Han YS, Yun CW, Lee HJ, Noh H, Lee SJ, Han HJ, Lee SH. 2016. Tauroursodeoxycholic acid reduces ER stress by regulating of Akt-dependent cellular prion protein. Sci Rep. 6:39838. doi:10.1038/srep39838.