
ABSTRACT
Colorectal cancer (CRC) is the most common malignant tumor and one of the leading causes of cancer-related death worldwide. Oncogene KRAS is a commonly mutated in CRC and plays crucial roles in the colorectal tumorigenesis. Emerging evidence suggests that increased expression levels of c-Myc are critical for KRAS-mediated CRC progression. However, it is unclear exactly how c-Myc expression is regulated in CRC. In the present study, we found that blockade of 5/12-lipoxygenase (5/12-LO) or leukotriene B4 receptor (BLT2) markedly reduced c-Myc expression in KRAS-mutant LOVO cells, while the control COLO 320DM cells were not affected. When the 5/12-LO products LTB4 and 12(S)-HETE, ligands of BLT2, were added to LOVO cells, c-Myc expression levels were restored, together suggesting that ‘5/12-LO-BLT2 cascade’ regulates c-Myc expression in KRAS-mutant CRC cells. We also observed that ‘5/12-LO-BLT2’-mediated c-Myc upregulation contributes to cell proliferation by regulating the expression of cyclin D1 in LOVO cells. Moreover, ‘5/12-LO-BLT2-cMyc’ cascade regulates the expression of EMT-related proteins in KRAS-mutant CRC cells, and depletion of both c-Myc and BLT2 using siRNA significantly enhanced the level of E-cadherin and reduced the level of Vimentin in LOVO cells. Taken together, our findings suggest that the BLT2-linked cascade promotes KRAS-mutant CRC cell proliferation and migration through the elevated expression of c-Myc. Thus, our results suggest that BLT2 appears to be an effective therapeutic target for KRAS-mutant CRCs.
Introduction
Colorectal cancer (CRC) is the world’s most common malignant tumor and the leading cause of cancer-related death worldwide (Ferlay et al. Citation2015; Shayimu et al. Citation2021). KRAS mutations constitute approximately 40% of all CRC cases, making it a common oncogene mutation in CRC patients (Dienstmann et al. Citation2020; Zhu et al. Citation2021). Mutations in KRAS activate the KRAS protein, which increases cell proliferation and survival, thereby accelerating cancer development (Roth et al. Citation2010; Dienstmann et al. Citation2017; Zhu et al. Citation2021). Previous studies have shown that c-Myc promotes proliferation and migration of KRAS-mutant CRC cells (Liao et al. Citation2007; Hosseini et al. Citation2019; Ruan et al. Citation2020). Moreover, overexpression of c-Myc in CRC cells increases KRAS-mediated oncogenesis, suggesting an important role for c-Myc in the progression of KRAS-mutant cancers (Soucek et al. Citation2008; Soucek et al. Citation2013; Dews et al. Citation2014). However, the signaling mechanism by which mutant KRAS influences c-Myc expression in CRC cells has not been fully elucidated.
The low-affinity leukotriene B4 receptor (BLT2) and its ligands are associated with progression in various types of cancer, including pancreatic, bladder, breast, and ovarian cancer (Hennig et al. Citation2008; Kim et al. Citation2010b; Kim et al. Citation2013; Park et al. Citation2016). In the tumor microenvironment, BLT2, a G protein-coupled receptor (GPCR) on the cell surface, is activated by inflammatory lipid moieties, such as leukotriene B4 (LTB4) and 12-hydroxyeicosatetraenoic acid [12(S)-HETE] (Yokomizo et al. Citation2001). The 5-lipoxygenase (5-LO) and 12-lipoxygenase (12-LO) are the enzymes responsible for the synthesis LTB4 and 12(S)-HETE, respectively, from arachidonic acid (AA). According to recent studies, the BLT2 pathway is affected by the presence of oncogenic RAS specifically. For example, BLT2 and the levels of LTB4 or 12(S)-HETE are markedly increased in cells harboring oncogenic RAS, and these cellular components help drive the transforming activity of RAS (Yoo et al. Citation2004; Kim et al. Citation2010a; Kim et al. Citation2014). Further, our previous study showed that BLT2 mediates proliferation of CRC cells with mutated KRAS downstream of oncogenic KRAS (Park et al. Citation2019). However, it has yet to be established whether BLT2 and c-Myc are integrated each other in KRAS-mutant CRC cells. Based on the results of this study, BLT2 was shown to promote c-Myc expression, which leads to proliferation and migration of CRC cells with KRAS mutations.
Materials and methods
Materials
RPMI 1640 medium and Fetal Bovine Serum (FBS) were purchased from Corning (Coring, NY, USA). LTB4, 12(S)-HETE and LY255283 were purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). Dimethyl sulfoxide (DMSO) was acquired from Sigma-Aldrich (St. Louis, MO, USA). MK886 and baicalein were acquired from Calbiochem (La Jolla, CA, USA). Antibodies against 5-LO (sc-136195), 12-LO (sc-365194), c-Myc (sc-40), cyclin D1 (sc-450), E-cadherin (sc-8426) and vimentin (sc-6260) were acquired from Santa Cruz Biotechnology (Santa Cruz, CA, USA); Antibodies against β-actin (#4967) were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibodies against BLT2 (Q924U0) were obtained from Enzo Life Sciences (Farmingdale, NY, USA).
Cell culture
Human CRC cell lines from the Korean Cell Line Bank (Seoul, Korea) were used in this study, LOVO and COLO 320DM. A 10% FBS-supplemented RPMI 1640 medium was used for maintaining LOVO and COLO 320DM cells at 37°C in a humidified atmosphere containing 5% CO2.
RNA or DNA transfection
KRAS-specific (5′-CUGUCUUGUGUUUUCAUGU-3′; siKRAS), BLT2-specific (5′-CCACGCAGUCAACCUUCUG-3′; siBLT2), c-Myc-specific (predesigned), and control (scrambled) siRNAs were obtained from Bioneer (Daejeon, Korea). Oligofectamine reagent and Lipofectamine reagent were obtained from Invitrogen (Waltham, MA, USA). Opti-MEM was obtained from Life Technologies (Carlsbad, CA, USA). Transfecting siRNAs and scrambled siRNA into cells in Opti-MEM for the indicated amounts of time was performed using Oligofectamine reagent. For transfection into cells in Opti-MEM, lipofectamine reagent was used to transfect both pCGN-KRAS-G12V and control (pCGN) vectors for indicated periods of time.
Semiquantitative RT–PCR analysis
An easy-BLUE RNA extraction kit provided by Intron (Gyeonggi-do, Korea) was used to isolate total RNA from cells. Following that, the concentration of RNA was determined by measuring the absorbance at 260 nm after it had been dissolved in diethylpyrocarbonate-treated water. With an RT–PCR PreMix kit purchased from Intron, the reverse transcription (RT) of one gram of RNA was followed by PCR amplification of BLT2, KRAS, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA. The primer sequences used were as follows: human BLT2 (forward, 5′-AGCCTGGAGACTCTGACCGCTTTCG-3′; reverse, 5′-GACGTAGCACCGGGTTGACGCTA-3′), human KRAS (forward, 5′-TACAGTGCAATGAGGGACCA-3′; reverse, 5′-AGGCATCATCAACACCCTGT-3′) and GAPDH (forward, 5′-CTGCACCACCAACTGCTTAGC-3′; reverse, 5′-CTTCACCACCTTCTTGATGTC-3′). Purified PCR products were electrophoresed on 1.5% agarose gels and visualized with ethidium bromide.
Immunoblot analysis
A lysis buffer containing 20 mM Tris-HCl (pH 7.5), 5 mM ethylenediaminetetraacetic acid (EDTA), 150 mM NaCl, 0.5% NP-40, 1% Triton X-100, and protease inhibitors was used to collect and lyse the cells. The proteins were separated and transferred to polyvinylidene difluoride (PVDF) membranes using sodium dodecyl sulfate-polyacrylamide gel electro-phoresis (SDS-PAGE). A blocking solution containing 5% nonfat dry milk in Tris-buffered saline was used to block and the membranes were incubated with primary antibodies followed by peroxidase-conjugated secondary antibodies. Eenhanced chemiluminescence (ECL) reagents were purchased from GE Healthcare (Chicago, IL, USA) to visualize the protein bands.
Enzyme-linked immunosorbent assay (ELISA)
Human LTB4 and 12(S)-HETE ELISA kits were obtained from Cayman Chemical Co. As recommended by the manufacturer, LTB4 and 12(S)-HETE concentrations were measured.
Bromodeoxyuridine (BrdU) assay
As directed by the manufacturer, the incorporation of BrdU was detected by ELISA using a BrdU cell proliferation assay kit from Cell Signaling Technology.
Cell proliferation assay
The proliferation rate of cells was assayed by counting viable cells plated in 12-well plates in RPMI 1640 medium for 24 h at a density of 1 × 105 cells/well. For the indicated periods of time, cells were incubated in RPMI 1640 medium with 1% FBS at 37°C after transfecting with siRNA or treating with reagent. After collecting cells with trypsin-EDTA, viable cells were counted with trypan blue staining.
Cell migration assay
A six-well culture plate was seeded with 5 × 105 cells/well and cultured in RPMI 1640 medium in a standard CO2 incubator until 70% confluent. After generating wounds using a 200P pipette tip, unattached cells and cell debris were removed by washing twice and then subsequently transfected or treated with siRNA, vectors or drugs in serum-free RPMI 1640 medium. For each sample, four fields were imaged in the same position immediately following scratching and 24 h later. A duplicate of each sample was performed three times.
Statistical data analysis
The results are presented as the mean ± SD. Statistical analyses were conducted using Student's t-test in SigmaPlot 8.0 software, and P-values of <0.05 were deemed significant.
Results
BLT2 is downstream of KRAS and affects c-Myc expression in KRAS-mutant CRC cells
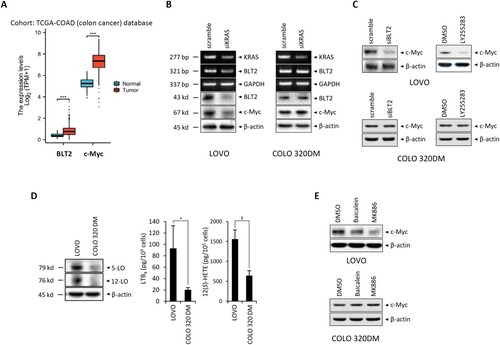
First, we wanted to demonstrate the contributory roles of BLT2 and c-Myc in colorectal cancer, and thus we analyzed the patient database stratified by colorectal cancer patients and normal colon samples. Using the TCGA-COAD (colon cancer) database to analyze the specificity of BLT2 and c-Myc gene expression, we found that the colorectal cancer patients showed significantly higher levels of BLT2 and c-Myc gene amplification compared with the normal colon groups ((A)). Then, we analyzed whether KRAS blockade affected the expression levels of BLT2 and c-Myc in KRAS-mutant CRC cells. As shown by the by RT–PCR and immunoblot analysis results in (B), in LOVO cells harboring mutant KRAS, the expression levels of BLT2 were significantly reduced by siKRAS blockade compared to COLO 320DM cells, which harbor wild-type KRAS. Similarly, the levels of c-Myc were markedly reduced by siKRAS, as measured by immunoblot analysis ((B)). To evaluate whether BLT2 mediates the expression of c-Myc in KRAS-mutant CRC cells, we examined the effects of BLT2 blockade via siBLT2 knockdown or treatment with the antagonist LY255283 and found that BLT2 blockade markedly reduced c-Myc expression in LOVO cells, while COLO 320DM cells were not affected ((C)). In KRAS-mutant CRC cells, we also investigated whether c-Myc expression is dependent on the synthesis of BLT2 ligands LTB4 and 12(S)-HETE. These ligands are synthesized by 5- and 12-lipoxygenase (5- and 12-LO) from arachidonic acid (AA), respectively. Clearly, a marked increase in 5-LO and 12-LO expression levels was observed in LOVO cells compared to COLO 320DM cells ((D)). Also, LTB4 and 12(S)-HETE levels were markedly higher in LOVO cells than in COLO 320DM cells ((D)). We then examined the effects of inhibiting 5-LO or 12-LO with MK886 or baicalein. As shown in (E), treatment with MK886 or baicalein markedly reduced c-Myc expression in LOVO cells, but did not affect COLO 320DM cells. Together, these data suggest that BLT2 regulates c-Myc expression in KRAS-mutant CRC cells.
Figure 1. BLT2 lies downstream of KRAS and influences c-Myc expression in KRAS-mutant CRC cells. (A) mRNA levels of BLT2 and c-Myc are higher in colon adenocarcinoma tissues (n = 480) compared with normal colon tissues (n = 41). The RNAseq data in TPM form and clinical information were obtained from TCGA-COAD (colon cancer) data. (B) Semiquantitative RT-PCR of KRAS, BLT2 and GAPDH (internal control) mRNA levels (top half), and immunoblotting of BLT2, c-Myc, and β-actin (internal control) protein expression (bottom half), in LOVO and COLO 320DM cells transfected with siKRAS or control siRNA for 24 h. (C) Immunoblotting of c-Myc and β-actin protein expression in LOVO and COLO 320DM cells transfected with siBLT2 or control siRNA for 24 h (left), or treated with LY255283 (10 μM) or DMSO for 24 h (right). (D) Immunoblotting of 5-LO, 12-LO and β-actin protein expression in LOVO and COLO 320DM cells. For the ELISAs, LOVO (5 × 105) and COLO 320DM (5 × 105) cells were plated in 35-mm dishes and cultured for 24 h; then, LTB4 and 12(S)-HETE levels in the culture supernatants were measured. (E) LOVO and COLO 320DM cells were treated with DMSO, baicalein (10 μM), or MK886 (2.5 μM) for 24 h, and immunoblotting was performed to measure c-Myc and β-actin protein expression. All quantitative data are presented as the mean ± SD of three independent experiments. *P < 0.05, ‡P < 0.005, ***P < 0.001.
Diminished BLT2 ligand synthesis by 5/12-LO leads to decreased cyclin D1 expression and proliferation in KRAS-mutant CRC cells
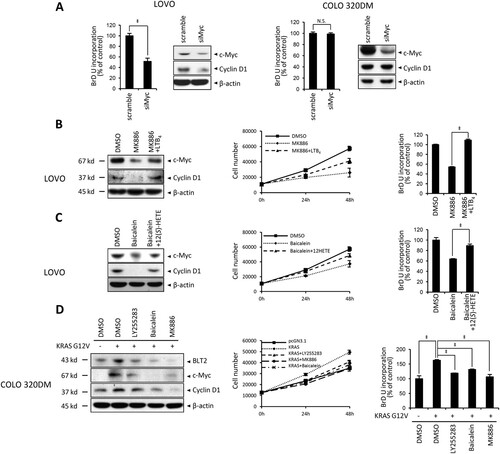
Next, we evaluated whether BLT2-mediated c-Myc expression contributes to cell proliferation in LOVO cells. Previous studies suggested that c-Myc promotes KRAS-mutant CRC cells proliferation (Liao et al. Citation2007; Song et al. Citation2020). Consistent with these reports, the proliferation of LOVO cells was clearly reduced by c-Myc depletion with siMyc ((A)). Additionally, we observed decreased levels of cyclin D1 in LOVO cells. However, siMyc transfection did not affect proliferation or cyclin D1 expression in control COLO 320DM cells ((A)). In addition, we observed that both cyclin D1 expression and proliferation are dependent on the synthesis of BLT2 ligands in LOVO cells. By inhibiting 5-LO or 12-LO with MK886 or baicalein, cyclin D1 expression and proliferation were significantly reduced in LOVO cells. When LTB4 or 12(S)-HETE was added to MK886- or baicalein-treated LOVO cells, cyclin D1 expression and proliferation were significantly recovered. Adding these ligands also restored c-Myc expression levels ((B and C)). Next, we examined whether forced KRAS mutant expression could stimulate the BLT2–c-Myc–cyclin D1-linked cascade and proliferation in control COLO 320DM cells. Transient transfecting COLO 320DM cells with pCGN-KRAS-G12 V clearly enhanced BLT2, c-Myc, and cyclin D1 expression. When we inhibited BLT2 or 5-/12-LO with each specific inhibitor, both c-Myc and cyclin D1 expression, along with proliferation, were significantly reduced in pCGN-KRAS-G12V-transfected cells ((D)). Taken together, these data indicate that 5-/12-LO–BLT2–c-Myc-linked cascade mediates the proliferation and expression of cyclin D1 in KRAS-mutant CRC cells.
Figure 2. Diminished BLT2 ligand synthesis by 5/12-LO leads to decreased cyclin D1 expression and proliferation in KRAS-mutant CRC cells. (A) LOVO and COLO 320DM cells were transfected with siMyc or control siRNA (scramble) for 24 h. Then, BrdU assays were performed as an indicator of cell proliferation, and immunoblotting was conducted to detect c-Myc, cyclin D1 and β-actin protein expression. (B) LOVO cells were pretreated with DMSO or MK886 (2.5 μM) for 3 h before LTB4 (300 nM) treatment for 21 h. Then, immunoblotting was performed to detect c-Myc, cyclin D1 and β-actin protein levels (left panel), and cell proliferation was assessed by cell counting (middle panel) and BrdU assays (right panel). (C) LOVO cells were pretreated with DMSO or baicalein (10 μM) for 3 h before 12(S)-HETE (300 nM) treatment for 21 h. Then, immunoblotting was performed to detect c-Myc, cyclin D1, and β-actin protein levels (left panel), and cell proliferation was assessed by cell counting (middle panel) and BrdU assays (right panel). (D) COLO 320DM cells were transfected with pCGN-KRAS-G12 V or a control vector for 24 h and then treated with DMSO, LY255283 (10 μM), baicalein (10 μM), or MK886 (2.5 μM) for 24 h. Immunoblotting was performed to detect BLT2, c-Myc, cyclin D1, β-actin protein levels (left panel), and cell proliferation was assessed by cell counting (middle panel) and BrdU assays (right panel). All quantitative data are presented as the mean ± SD of three independent experiments. *P < 0.05, ‡P < 0.005.
5/12-LO/BLT2 and c-Myc contribute to the migration of KRAS-mutant CRC cells
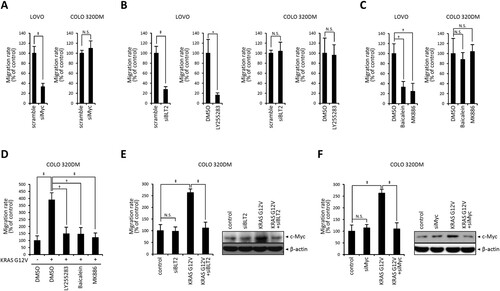
A previous report implicated c-Myc in promoting KRAS-mutant CRC cell migration and invasion (Hosseini et al. Citation2019). Consistent with this report, c-Myc depletion markedly reduced the migration of LOVO cells, while COLO 320DM cells were not affected ((A)). Next, the 5-/12-LO–BLT2–c-Myc cascade was tested for its effect on KRAS-mutant CRC cell migration. First, we examined whether BLT2 inhibition affected LOVO and COLO 320DM cell migration. BLT2 depletion clearly suppressed the migration of LOVO cells, while COLO 320DM cells were not affected ((B)). Additionally, 5-/12-LO depletion significantly reduced the migration of KRAS-mutant LOVO cells, while COLO 320DM cells were not affected ((C)). To further investigate whether the 5-/12-LO–BLT2–c-Myc cascade mediates the migration of CRC cells, we assessed the migration of oncogenic KRAS-transfected COLO 320DM cells. Transfection of pCGN-KRAS-G12 V enhanced the migration of COLO 320DM cells. When the pCGN-KRAS-G12V-transfected cells were treated with BLT2 or 5-/12-LO inhibitors, migration was significantly suppressed ((D)). Next, we examined whether BLT2 depletion via siBLT2 affected pCGN-KRAS-G12V-transfected COLO 320DM cell migration. BLT2 depletion significantly reduced migration and c-Myc expression in pCGN-KRAS-G12V-transfected COLO 320DM cells ((E)). Similarly, c-Myc depletion significantly reduced migration and the c-Myc expression level in pCGN-KRAS-G12 V transfected COLO 320DM cells ((F)). Together, these data suggest that the BLT2–c-Myc cascade is responsible for KRAS-mutant CRC cell migration.
Figure 3. 5/12-LO/BLT2 and c-Myc contribute to the migration of KRAS-mutant CRC cells. (A) LOVO and COLO 320DM cells were transfected with siMyc or control siRNA (scramble) for 24 h. Then, wound healing assays were performed to measure the migration rate over 24 h. (B) LOVO and COLO 320DM cells were transfected with siBLT2 or control siRNA (scramble) for 24 h or treated with DMSO or LY255283 (10 μM) for 24 h. Then, wound healing assays were performed as described in A to measure the migration rate. (C) LOVO and COLO 320DM cells were treated with DMSO, baicalein (10 μM), or MK886 (2.5 μM) for 24 h, and wound healing assays were performed as described in A to measure the migration rate. (D) COLO 320DM cells were transfected with pCGN-KRAS-G12 V or a control vector for 24 h and then treated with DMSO, LY255283 (10 μM), baicalein (10 μM), or MK886 (2.5 μM) for 24 h. Wound healing assays were performed as described in A to measure the migration rate. (E) COLO 320DM cells were transfected with pCGN-KRAS-G12 V or a control vector for 3 h and then transfected with siBLT2 or control siRNA for 21 h. Wound healing assays were performed as described in A to measure the migration rate, and immunoblotting was conducted to detect c-Myc and β-actin protein levels. (F) COLO 320DM cells were transfected with pCGN-KRAS-G12 V or a control vector for 3 h and then transfected with siMyc or control siRNA for 21 h. Wound healing assays were performed as described in A to measure the migration rate, and immunoblotting was conducted to detect c-Myc and β-actin protein levels. All quantitative data are shown as the mean ± SD of three independent experiments. *P < 0.05, †P < 0.01, ‡P < 0.005.
5/12-LO/BLT2 And c-Myc promote epithelial–mesenchymal transition (EMT), as indicated by E-cadherin downregulation and vimentin upregulation, in KRAS-mutant CRC cells
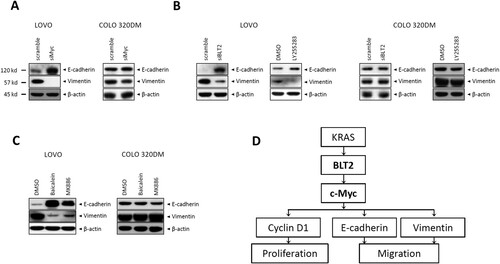
The migration of tumor cells is usually associated with processes of the malignant transformation, such as epithelial–mesenchymal transition (EMT) (Wei et al. Citation2015; Lee et al. Citation2021). Therefore, we examined whether BLT2-mediated c-Myc expression is related to the expression of EMT-related proteins. As shown by the immunoblot analysis results in (A), c-Myc depletion with siMyc in LOVO cells resulted in an increased E-cadherin levels and a decreased Vimentin levels compared with COLO 320DM cells. Similarly, BLT2 depletion markedly enhanced E-cadherin levels and decreased Vimentin levels in LOVO cells, while COLO 320DM cells were not affected ((B)). Next, we examined the effect of 5-/12-LO inhibition. Immunoblot analysis revealed that 5-/12-LO depletion significantly enhanced the E-cadherin level and reduced the Vimentin level in LOVO cells ((C)). Together, these data suggest that E-cadherin and Vimentin expression in KRAS-mutant CRC cells is regulated downstream of the 5-/12-LO–BLT2–c-Myc cascade.
Figure 4. 5/12-LO/BLT2 and c-Myc promote EMT, as indicated by E-cadherin downregulation and vimentin upregulation, in KRAS-mutant CRC cells. (A) LOVO and COLO 320DM cells were transfected with siMyc or control siRNA for 24 h, and then, immunoblotting was performed to detect E-cadherin, vimentin, and β-actin protein levels. (B) LOVO and COLO 320DM cells were transfected with siBLT2 or control siRNA for 24 h or treated with DMSO or LY255283 (10 μM) for 24 h. Then, immunoblotting was performed to detect E-cadherin, vimentin, and β-actin protein levels. (C) LOVO and COLO 320DM cells were treated with DMSO, baicalein (10 μM), or MK886 (2.5 μM) for 24 h, and immunoblotting was then performed to detect E-cadherin, vimentin, and β-actin protein levels. (D) Scheme showing the proposed involvement of BLT2 and c-Myc in KRAS-mutant CRC cells.
Discussion
Despite the evidence that increased c-Myc expression promotes proliferation and migration of KRAS-mutant CRC cells, the mechanisms regulating c-Myc expression remained unclear. Our study demonstrated that the 5/12-LO/BLT2 cascade regulates c-Myc expression in KRAS-mutant LOVO cells to mediate the proliferation and migration of these cells. Additionally, when mutant KRAS was overexpressed in COLO 320DM colorectal cells, which harbor wild-type KRAS, the expression of BLT2 and c-Myc was clearly upregulated, thus mediating proliferation and migration. Taken together, our findings suggest that the BLT2-linked cascade may promote KRAS-mutant CRC cell proliferation and migration through increased c-Myc expression.
Inflammation is generally accepted as a factor contributing to CRC proliferation and metastasis (Sun et al. Citation2016; Korniluk et al. Citation2017). Additionally, emerging evidence suggests that c-Myc overexpression modulates the inflammatory microenvironment of tumors (Zhong et al. Citation2018; Zhang et al. Citation2019b). In 44% of CRC cases, c-Myc is overexpressed, which may play a crucial role in its development and progression (Goel and Boland Citation2012; Li et al. Citation2018). c-Myc regulates the expression of genes important for proliferation, survival, differentiation, metabolism, apoptosis, angiogenesis, and other biological functions in both normal and cancerous cells (Sears and Nevins Citation2002; Magudia et al. Citation2012; Kazi et al. Citation2018; Knight et al. Citation2021). In particular, c-Myc-associated inflammation is well established to promote cancer in patients with CRC (Xie et al. Citation2018; Yang et al. Citation2019). In KRAS-mutant CRC, however, the mechanism underlying c-Myc’s effect on inflammation is poorly understood. According to this study, we propose that the BLT2-linked cascade lies downstream of KRAS acts as a bridge between KRAS and c-Myc, which is necessary for CRC progression. BLT2 is a highly induced GPCR for inflammatory lipid mediators in the tumor microenvironment (Cho et al. Citation2013). Indeed, we observed that mutant-KRAS upregulated BLT2 expression in LOVO cells and blocking of BLT2 or 5-/12-LO markedly reduced c-Myc expression in LOVO cells, as well as the proliferation and migration of these cells ( and ). To demonstrate that the BLT2–c-Myc cascade is involved in KRAS-mutant CRC cells, we transiently overexpressed mutant KRAS in control COLO 320DM colorectal cells and found that BLT2 and c-Myc expression levels were increased, resulting in enhanced proliferation and migration of these cells ( and ). In addition to BLT2, leukotriene B4 receptor 1 (BLT1) or cyclooxygenase (COX) may play roles in the progression of CRC (Gao et al. Citation2005; Ihara et al. Citation2007). Thus, we investigated whether BLT1 or COX2 inhibition could affect KRAS-mutant CRC cells. However, we could not observe any inhibitory effects of the BLT1 inhibitor U75302 or the COX2 inhibitor NS398 on c-Myc expression, proliferation, and migration of KRAS-mutant CRC cells (data not shown), excluding the possibility that BLT1 and COX2 contribute to c-Myc expression in KRAS-mutant CRC. As a result of our findings, BLT2 plays an important role in regulating c-Myc expression in KRAS-mutant CRC.
In stem cell biology, c-Myc has recently been discovered to play an important role (Takahashi and Yamanaka Citation2006). Accumulating evidence supports the existence of a subset of cells, referred to as cancer stem cells, within CRC that possess stem cell-like characteristics and contribute to tumor development, metastasis, and resistance to therapy (O’Brien et al. Citation2007; Ricci-Vitiani et al. Citation2007; Ieta et al. Citation2008; Zhang et al. Citation2011). Moreover, Zhang et al. demonstrated the significant involvement of c-Myc in regulating the self-renewal and resistance to chemotherapy in CRC cells with KRAS mutations (Zhang et al. Citation2019a). In our previous study, we established that the PI3 K/Akt contributes to the synthesis of cyclin D1, which is essential for KRAS-mutant CRC cells proliferation via the KRAS-BLT2 pathway (Park et al. Citation2019). Consistent with prior research, our findings indicate that the induction of c-Myc expression by KRAS-BLT2 is crucial for cyclin D1 expression and proliferation of LOVO cells (). In addition, Tang et al. have previously reported the involvement of c-Myc in EMT of CRC cells (Tang et al. Citation2020). Based on the findings of this study, BLT2 upregulation occurs downstream of KRAS and leads to the expression of EMT-related proteins in KRAS-mutant CRC cells. Notably, we observed that the depletion of both c-Myc and BLT2 using siRNA significantly enhanced the level of E-cadherin and reduced the level of Vimentin in LOVO cells ((A and B)). Similarly, LOVO cells treated MK886 or baicalein selectively inhibited 5-LO and 12-LO showed enhanced E-cadherin level and reduced Vimentin level ((C)). These findings support a role for the 5-/12-LO–BLT2–c-Myc cascade in regulating EMT-related protein expression, which is critical for cell migration of KRAS-mutant CRC cells. Based on our observations, stemness genes, such as slug, were up-regulated and they functioned as downstream elements of the BLT2-c-Myc cascade in LOVO KRAS-mutant cancer cells (data not shown). Additional investigations are required to fully comprehend the downstream mechanism of the BLT2–c-Myc cascade in KRAS-mutant CRC cells. These results led us to propose a model in which BLT2 and c-Myc promote the proliferation and migration of KRAS-mutant CRC cells ((D)).
In conclusion, our results indicate that BLT2 is a novel factor contributing to c-Myc expression in KRAS-mutant CRC. In addition, our study is expected to enhance comprehension of CRC progression mechanisms and facilitate the identification of potential therapeutic targets specifically for KRAS-mutant CRCs.
Acknowledgments
This work was supported by a Bio and Medical Technology Development Program grant (2017M3A9D8063317) and a Mid-Career Researcher Program grant (2017R1A2B4002203) through the National Research Foundation (NRF) funded by the Ministry of Science, Information and Communication Technologies (ICT) and Future Planning of the Republic of Korea, the Scientific Research Project of Taizhou Municipal Science and Technology Bureau (grant number 23gyb09 to JDW). This work was also supported by a Korea University Grant.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Cho NK, Joo YC, Wei JD, Park JI, Kim JH. 2013. BLT2 is a pro-tumorigenic mediator during cancer progression and a therapeutic target for anti-cancer drug development. Am J Cancer Res. 3:347–355.
- Dews M, Tan GS, Hultine S, Raman P, Choi J, Duperret EK, Lawler J, Bass A, Thomas-Tikhonenko A. 2014. Masking epistasis between MYC and TGF-beta pathways in antiangiogenesis-mediated colon cancer suppression. J Natl Cancer Inst. 106:dju043. doi:10.1093/jnci/dju043.
- Dienstmann R, Connor K, Byrne AT, Consortium C. 2020. Precision therapy in RAS mutant colorectal cancer. Gastroenterology. 158:806–811. doi:10.1053/j.gastro.2019.12.051.
- Dienstmann R, Mason MJ, Sinicrope FA, Phipps AI, Tejpar S, Nesbakken A, Danielsen SA, Sveen A, Buchanan DD, Clendenning M, et al. 2017. Prediction of overall survival in stage II and III colon cancer beyond TNM system: a retrospective, pooled biomarker study. Ann Oncol. 28:1023–1031. doi:10.1093/annonc/mdx052.
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. 2015. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. doi:10.1002/ijc.29210.
- Gao XQ, Han JX, Huang HY, Song B, Zhu B, Song CZ. 2005. Effect of NS398 on metastasis-associated gene expression in a human colon cancer cell line. World J Gastroenterol. 11:4337–4343. doi:10.3748/wjg.v11.i28.4337.
- Goel A, Boland CR. 2012. Epigenetics of colorectal cancer. Gastroenterology. 143:1442–1460 e1441. doi:10.1053/j.gastro.2012.09.032.
- Hennig R, Osman T, Esposito I, Giese N, Rao SM, Ding XZ, Tong WG, Buchler MW, Yokomizo T, Friess H, Adrian TE. 2008. BLT2 is expressed in PanINs, IPMNs, pancreatic cancer and stimulates tumour cell proliferation. Br J Cancer. 99:1064–1073. doi:10.1038/sj.bjc.6604655.
- Hosseini SA, Zand H, Cheraghpour M. 2019. The influence of curcumin on the downregulation of MYC, insulin and IGF-1 receptors: a possible mechanism underlying the anti-growth and anti-migration in chemoresistant colorectal cancer cells. Medicina (Kaunas). 55(4):90. doi:10.3390/medicina55040090.
- Ieta K, Tanaka F, Haraguchi N, Kita Y, Sakashita H, Mimori K, Matsumoto T, Inoue H, Kuwano H, Mori M. Biological and genetic characteristics of tumor-initiating cells in colon cancer. Ann Surg Oncol. 15:638–648. doi:10.1245/s10434-007-9605-3.
- Ihara A, Wada K, Yoneda M, Fujisawa N, Takahashi H, Nakajima A. 2007. Blockade of leukotriene B4 signaling pathway induces apoptosis and suppresses cell proliferation in colon cancer. J Pharmacol Sci. 103:24–32. doi:10.1254/jphs.FP0060651.
- Kazi A, Xiang S, Yang H, Delitto D, Trevino J, Jiang RHY, Ayaz M, Lawrence HR, Kennedy P, Sebti SM. 2018. GSK3 suppression upregulates beta-catenin and c-Myc to abrogate KRas-dependent tumors. Nat Commun. 9:5154. doi:10.1038/s41467-018-07644-6.
- Kim EY, Seo JM, Cho KJ, Kim JH. 2010a. Ras-induced invasion and metastasis are regulated by a leukotriene B4 receptor BLT2-linked pathway. Oncogene. 29:1167–1178. doi:10.1038/onc.2009.412.
- Kim EY, Seo JM, Kim C, Lee JE, Lee KM, Kim JH. 2010b. BLT2 promotes the invasion and metastasis of aggressive bladder cancer cells through a reactive oxygen species-linked pathway. Free Radic Biol Med. 49:1072–1081. doi:10.1016/j.freeradbiomed.2010.06.023.
- Kim H, Choi JA, Kim JH. 2014. Ras promotes transforming growth factor-beta (TGF-beta)-induced epithelial-mesenchymal transition via a leukotriene B4 receptor-2-linked cascade in mammary epithelial cells. J Biol Chem. 289:22151–22160. doi:10.1074/jbc.M114.556126.
- Kim H, Park GS, Lee JE, Kim JH. 2013. A leukotriene B4 receptor-2 is associated with paclitaxel resistance in MCF-7/DOX breast cancer cells. Br J Cancer. 109:351–359. doi:10.1038/bjc.2013.333.
- Knight JRP, Alexandrou C, Skalka GL, Vlahov N, Pennel K, Officer L, Teodosio A, Kanellos G, Gay DM, May-Wilson S, et al. 2021. Mnk inhibition sensitizes KRAS-mutant colorectal cancer to mTORC1 inhibition by reducing eIF4E phosphorylation and c-MYC expression. Cancer Discov. 11:1228–1247. doi:10.1158/2159-8290.CD-20-0652.
- Korniluk A, Koper O, Kemona H, Dymicka-Piekarska V. 2017. From inflammation to cancer. Ir J Med Sci. 186:57–62. doi:10.1007/s11845-016-1464-0.
- Lee JE, Lim YH, Kim JH. 2021. UCH-L1 and UCH-L3 regulate the cancer stem cell-like properties through PI3 K/Akt signaling pathway in prostate cancer cells. Anim Cells Syst (Seoul). 25:312–322. doi:10.1080/19768354.2021.1987320.
- Li W, Zhu W, Lv C, Qu H, Xu K, Li H, Du Y, Liu G, Wang Y, Wei HJ, Zhao HY. 2018. Low-dose paclitaxel downregulates MYC proto-oncogene bHLH transcription factor expression in colorectal carcinoma cells. Oncol Lett. 15:1881–1887. doi:10.3892/ol.2017.7525.
- Liao DJ, Thakur A, Wu J, Biliran H, Sarkar FH. 2007. Perspectives on c-Myc, Cyclin D1, and their interaction in cancer formation, progression, and response to chemotherapy. Crit Rev Oncog. 13:93–158. 016f7acc754baaa2,6431569536aac216.
- Magudia K, Lahoz A, Hall A. 2012. K-Ras and B-Raf oncogenes inhibit colon epithelial polarity establishment through up-regulation of c-myc. J Cell Biol. 198:185–194. doi:10.1083/jcb.201202108.
- O’Brien CA, Pollett A, Gallinger S, Dick JE. 2007. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 445:106–110. doi:10.1038/nature05372.
- Park J, Jang JH, Kim JH. 2019. Mediatory role of BLT2 in the proliferation of KRAS mutant colorectal cancer cells. Biochim Biophys Acta Mol Cell Res. 1866:329–336. doi:S0167-4889(18)30285-4.
- Park J, Park SY, Kim JH. 2016. Leukotriene B4 receptor-2 contributes to chemoresistance of SK-OV-3 ovarian cancer cells through activation of signal transducer and activator of transcription-3-linked cascade. Biochim Biophys Acta. 1863:236–243. doi:10.1016/j.bbamcr.2015.11.011.
- Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. 2007. Identification and expansion of human colon-cancer-initiating cells. Nature. 445:111–115. doi:10.1038/nature05384.
- Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, Dietrich D, Biesmans B, Bodoky G, Barone C, et al. 2010. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 28:466–474. doi:10.1200/JCO.2009.23.3452.
- Ruan H, Li X, Xu X, Leibowitz BJ, Tong J, Chen L, Ao L, Xing W, Luo J, Yu Y, et al. 2020. eIF4E S209 phosphorylation licenses myc- and stress-driven oncogenesis. Elife. 9. doi:10.7554/eLife.60151.
- Sears RC, Nevins JR. 2002. Signaling networks that link cell proliferation and cell fate. J Biol Chem. 277:11617–11620. doi:10.1074/jbc.R100063200.
- Shayimu P, Yusufu A, Rehemutula A, Redati D, Jiapaer R, Tuerdi R. 2021. MTBP promoted the proliferation, migration and invasion of colon cancer cells by activating the expression of ZEB2. Anim Cells Syst (Seoul). 25:152–160. doi:10.1080/19768354.2021.1938218.
- Song J, Park S, Oh J, Kim D, Ryu JH, Park WC, Baek IJ, Cheng X, Lu X, Jin EJ. 2020. NUDT7 loss promotes Kras(G12D) CRC development. Cancers (Basel). 12: 576. doi:10.3390/cancers12030576.
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. 2008. Modelling Myc inhibition as a cancer therapy. Nature. 455:679–683. doi:10.1038/nature07260.
- Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, Swigart LB, Evan GI. 2013. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 27:504–513. doi:10.1101/gad.205542.112.
- Sun Y, Zhao Y, Wang X, Zhao L, Li W, Ding Y, Kong L, Guo Q, Lu N. 2016. Wogonoside prevents colitis-associated colorectal carcinogenesis and colon cancer progression in inflammation-related microenvironment via inhibiting NF-kappaB activation through PI3 K/Akt pathway. Oncotarget. 7:34300–34315. doi:10.18632/oncotarget.8815.
- Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126:663–676. doi:S0092-8674(06)00976-7
- Tang Q, Chen J, Di Z, Yuan W, Zhou Z, Liu Z, Han S, Liu Y, Ying G, Shu X, Di M. TM4SF1 promotes EMT and cancer stemness via the Wnt/beta-catenin/SOX2 pathway in colorectal cancer. J Exp Clin Cancer Res. 39:232. doi:10.1186/s13046-020-01690-z.
- Wei SC, Fattet L, Yang J. 2015. The forces behind EMT and tumor metastasis. Cell Cycle. 14:2387–2388. doi:10.1080/15384101.2015.1063296.
- Xie QK, Chen P, Hu WM, Sun P, He WZ, Jiang C, Kong PF, Liu SS, Chen HT, Yang YZ, et al. 2018. The systemic immune-inflammation index is an independent predictor of survival for metastatic colorectal cancer and its association with the lymphocytic response to the tumor. J Transl Med. 16:273. doi:10.1186/s12967-018-1638-9.
- Yang J, Guo X, Wu T, Niu K, Ma X. 2019. Prognostic significance of inflammation-based indexes in patients with stage III/IV colorectal cancer after adjuvant chemoradiotherapy. Medicine (Baltimore). 98:e14420. doi:10.1097/MD.0000000000014420.
- Yokomizo T, Kato K, Hagiya H, Izumi T, Shimizu T. 2001. Hydroxyeicosanoids bind to and activate the low affinity leukotriene B4 receptor, BLT2. J Biol Chem. 276:12454–12459. doi:10.1074/jbc.M011361200.
- Yoo MH, Song H, Woo CH, Kim H, Kim JH. 2004. Role of the BLT2, a leukotriene B4 receptor, in Ras transformation. Oncogene. 23:9259–9268. doi:10.1038/sj.onc.1208151.
- Zhang H, Li W, Nan F, Ren F, Wang H, Xu Y, Zhang F. 2011. MicroRNA expression profile of colon cancer stem-like cells in HT29 adenocarcinoma cell line. Biochem Biophys Res Commun. 404:273–278. doi:10.1016/j.bbrc.2010.11.106.
- Zhang HL, Wang P, Lu MZ, Zhang SD, Zheng L. 2019a. c-Myc maintains the self-renewal and chemoresistance properties of colon cancer stem cells. Oncol Lett. 17:4487–4493. doi:10.3892/ol.2019.10081.
- Zhang S, Li J, Xie P, Zang T, Shen H, Cao G, Zhu Y, Yue Z, Li Z. 2019b. Stat3/c-Myc axis-mediated metabolism alternations of inflammation-related glycolysis involve with colorectal carcinogenesis. Rejuvenation Res. 22:138–145. doi:10.1089/rej.2018.2089.
- Zhong X, Lee HN, Surh YJ. 2018. RvD1 inhibits TNFalpha-induced c-Myc expression in normal intestinal epithelial cells and destabilizes hyper-expressed c-Myc in colon cancer cells. Biochem Biophys Res Commun. 496:316–323. doi:S0006-291X(17)32583-4.
- Zhu G, Pei L, Xia H, Tang Q, Bi F. 2021. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol Cancer. 20:143. doi:10.1186/s12943-021-01441-4.