
ABSTRACT
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) has chemotherapeutic potential as a regulator of an extrinsic apoptotic ligand, but its effect as a drug is limited by innate and acquired resistance. Recent findings suggest that an intermediate drug tolerance could mediate acquired resistance, which has made the main obstacle for limited utility of TRAIL as an anti-cancer therapeutics. We propose miRNA-dependent epigenetic modification drives the drug tolerant state in TRAIL-induced drug tolerant (TDT). Transcriptomic analysis revealed miR-29 target gene activation in TDT cells, showing oncogenic signature in lung cancer. Also, the restored TRAIL-sensitivity was associated with miR-29ac and 140-5p expressions, which is known as tumor suppressor by suppressing oncogenic protein RSK2 (p90 ribosomal S6 kinase), further confirmed in patient samples. Moreover, we extended this finding into 119 lung cancer cell lines from public data set, suggesting a significant correlation between TRAIL-sensitivity and RSK2 mRNA expression. Finally, we found that increased RSK2 mRNA is responsible for NF-κB activation, which we previously showed as a key determinant in both innate and acquired TRAIL-resistance. Our findings support further investigation of miR-29ac and -140-5p inhibition to maintain TRAIL-sensitivity and improve the durability of response to TRAIL in lung cancer.
Introduction
Lung cancer is the leading cause of cancer-related death, and it is often incurable when diagnosed at advanced stages. Over 70% of lung cancers are defined as a non-small cell lung cancer (NSCLC) type, and the rest of them are mainly classified as small cell lung cancer (SCLC) type. The reasons for the high mortality of lung cancer are multifactorial including late diagnosis, but the exact mechanism underlying cancer progression is poorly understood (Molina et al. Citation2008; Patel et al. Citation2011; Siegel et al. Citation2018). TNF-related apoptosis-inducing ligand (TRAIL) known as an extrinsic apoptotic ligand is a promising anti-cancer agent, which has been attractive because of high cancer specificity with minimal cytotoxicity (Wiley et al. Citation1995; Pan et al. Citation1997; Ashkenazi et al. Citation1999; Ichikawa et al. Citation2001). Unlike the murine system, two distinct receptors, DR4 and DR5 mediate TRAIL-induced cell death signal, and there are either or both expressions in most types of stage III NSCLC tumors, suggesting that there is high therapeutic potential using TRAIL and/or its agonistic antibodies to the receptors (Ashkenazi et al. Citation1999; Ichikawa et al. Citation2001; Spierings et al. Citation2003; Herbst et al. Citation2010).
Despite its promising therapeutic prospects, preclinical and clinical research indicates that resistance to TRAIL, resulting from either inherent or developed mutations, restricts its effectiveness in chemotherapy. Dulanermin, an early recombinant version of TRAIL, faced failure in clinical trials due to resistance emerging from the activation of pro-survival proteins such as NF-κB after 62 months, confirmed in other observations in vitro and in vivo (Chaudhary et al. Citation1997; Schneider et al. Citation1997; Ashkenazi et al. Citation1999; Plantivaux et al. Citation2009; Subbiah et al. Citation2012; Jeon et al. Citation2015). Similarly, Conatumumab, a monoclonal antibody that targets human TRAIL-R2, demonstrated potential in inhibiting tumor growth across various types of tumors both in vivo and in vitro (Kaplan-Lefko et al. Citation2010). However, even with the preliminary positive effects of these agonists in preclinical studies, antibodies targeting TRAIL-R did not lead to significant improvements in either the objective response rate or overall survival of patients in clinical trials alongside other treatments for different cancers (Kindler et al. Citation2012; Fuchs et al. Citation2013; Skelton et al. Citation2020).
Cancer heterogeneity has been considered a source of drug resistance, and the small subpopulation of cancer cells is resistant to chemotherapeutic agents without obvious actionable mutations (Ke et al. Citation2022; Li et al. Citation2022). The intermediate resistant stage has been characterized as a reversible phenotype when the signal is withdrawn to the cells. This drug-tolerant stage possibly gives time to the cells acquiring mutations under such the cell death pressure, which consequently fuels to acquisition of the resistance against a chemotherapeutic agent (Roesch et al. Citation2010; Sharma et al. Citation2010; Lee et al. Citation2014). It has been known that the epigenetic regulation of an oncogenic pathway is responsible for maintaining the intermediate state in response to targeted therapeutic drugs. Moreover, a recent study suggested that O-GlcNacylation on DR4 receptor was dynamically regulated in the drug-tolerant state compared to its naïve control cells (Hata et al. Citation2016; Smith et al. Citation2016; Guler et al. Citation2017). However, the cause and consequence of the drug-persistent stage are largely unknown.
The p90 ribosomal protein s6 kinases encoded by RPS6KA gene are serine/threonine protein kinase, which consists of four different isoforms. Although they exhibited functional redundancy with high sequence homology, the expression and roles of each isoform are context-specific in mediating diverse subcellular processes (Lara et al. Citation2013). A major type of pro-survival MAPK protein, ERK1/2 directly phosphorylates and activates RSKs in response to several external stimuli such as EGFR activation. Like the other isoforms, RSK2-mediated phosphorylation on diverse substrates enhanced cell cycle progression, cell survival, and cell proliferation (Poomakkoth et al. Citation2016). However, little is known about the oncogenic role of RSK2 in TRAIL-resistance.
Most abundantly expressed small non-coding RNA better known as miRNA plays an important role in cancer progression by targeting subcellular proteins at post-transcriptional or -translational levels (Calin and Croce Citation2006; Hwang et al. Citation2023). One of the most interesting features of a miRNA exhibited a context-dependency via regulating multiple targets even in the same cancer types (Genc et al. Citation2023). MiR-29 family consists of three members, encoded by two different genomic loci at chromosome 1 and 7. The miR-29 family was found to be suppressed in lung cancer, which consequently enhanced cancer progression by the loss of suppressing ability to their target oncogenes including DNMT3s (Volinia et al. Citation2006; Fabbri et al. Citation2007; Plaisier et al. Citation2012). Compared to miR-29 family, the roles of miR-140 in lung cancer have been less elucidated despite a few studies consistently showing its robust tumor suppressive role in lung cancer. MiR-140 located on chromosome 16q was found to be suppressed miRNA in NSCLC patients, and its overexpression decreased the metastatic potential of lung cancer cells in vitro and in vivo (Yuan et al. Citation2013). In particular, the overexpression of miR-140-5p induced resistance of lung cancer cells in response to tyrosine kinase inhibitors or platinum-based chemotherapy (Flamini et al. Citation2017). However, the roles of both miRNAs in TRAIL-induced persistent resistance are largely unknown.
Materials and methods
Cell culture, cell line generation, and gene delivery
Human lung cancer cells, H460, H292, and A549 cells were purchased from ATCC, and the cell lines were maintained in DMEM supplemented with 10% FBS and 1% Penicillin–streptomycin (Sigma). The generation of TRAIL-resistant H460R cells was described in a previous study (Jeon et al. Citation2015). The H460 revertant was generated by abolishing TRAIL stimulation for weeks. TRAIL-sensitivity was determined weekly basis to determine revertant phenotype. pcDNA-RPS6KA3 plasmid was introduced using Lipofectamine 3000 reagent (ThermoFisher Scientific), and miRNAs and siRNAs used in the current study were transfected by Lipofectamine RNAi Max reagent (ThermoFisher Scientific) according to the manufacturer’s protocols.
Antibodies
Anti-GAPDH (#5174), anti-PARP-1 (#9532), anti-phosphor p65 (#3033), anti-RSK2 (#5528), and anti-Vinculin (#13901) antibodies were purchased from Cell Signaling Technology. Anti-p65 (#ABE136) was purchased from Sigma-Aldrich. Anti-tubulin (#sc-8035) antibody was purchased from Santa Cruz Biotechnology.
Cell cytotoxicity assay
The cells (1.0×103) were cultured into 96 well plates. After 24h, the cells were exposed to TRAIL and/or other drugs as indicated concentration shown in figures or figure legend. Subsequently, the cell survival rate was determined by CellTiter-Glo® Luminescent Cell Viability Assay kit (Promega, #G7572) according to the manufacturer’s protocol.
NF-κB promoter assay activity
A pGL4.3-luc2p/NF-κB construct harboring five copies of NF-κB binding sites (Promega, #E8491) was co-transfected either alone or with the corresponding miRNAs and/or pcDNA-RPS6KA3 plasmid. After 24–48h of transfection, the cells were harvested and subject to Dual Luciferase® (Promega, #E1910) as followed by the manufacturer’s protocol.
Public data analysis for the expression analysis of miR-29 and -140-associated signatures
The miRNA (GSE55821) and gene expression profiles (GSE55859) of TRAIL-sensitive and -resistant H460 cells were obtained from a previous study (Jeon et al.). A robust multi-array average (RMA) algorithm was used to normalize the data using R package. The hierarchical- and principal component analysis based on 2500 top variable genes were performed using hclust and prcomp function in R package. Differential expression analysis was performed by fitting linear models using the empirical Bayes method as implemented in the limma R package (Smyth Citation2004) and P values were adjusted for multiple testing using FDR method. Genes with an adjusted P value smaller than 0.05 and fold change greater than two-fold selected as significant differentially expressed genes (DEGs). DEGs from limma were used as input for the implementation in The Molecular Signatures Database (Subramanian et al. Citation2005) to calculate the overlap for highlighting common processes, pathways, and underlying biological processes. Gene ontology terms with P value smaller than 0.05 were selected as significant and used subsequently for visualizations.
The miRNA, RNA-seq expression profiles and corresponding clinical information of Lung Squamous Cell Carcinoma (LUSC) and Lung Adenocarcinoma (LUAD) were obtained from the TCGA data portal (https://portal.gdc.cancer.gov/) with a total of 1143 LUAD samples (1035 tumor- and 108 normal tissue) and 523 LUSC samples (478 tumor- and 45 normal tissue). The data were downloaded in April 2021. Kaplan-Meier curves were plotted for two distinct groups of patients based on miRNA expression, defined by quantile, using the survfit function, and the P values (log-rank test) were calculated using the survdiff function in the survival R package.
Co-expression analyses NF-κB and TRAIL-sensitivity
We collected data on the TRAIL-sensitivity of various lung cancer cell lines from previous literature (Shivapurkar et al. Citation2004; Wagner et al. Citation2007; Zitzmann et al. Citation2011; Yang et al. Citation2013; Lu et al. Citation2014; Jeon et al. Citation2015; Joshi et al. Citation2015; Li et al. Citation2017). Additionally, the remaining lung cancer cell lines used in the current study were mined from the Genomics of Drug Sensitivity in Cancer database (Yang et al. Citation2013). We also acquired data on the RPS6KA3 mRNA expression in the corresponding lung cancer cell lines from the Cancer Cell Line Encyclopedia (CCLE) (Barretina et al. Citation2012). We have finally utilized 119 lung cancer cell lines to investigate the relationship between TRAIL sensitivity and RPS6KA3 expression (Supplementary Table S1). We categorized the TRAIL sensitivity based on existing literature. Additionally, cell lines exhibiting IC50 values higher than the median value (0.634nM) were classified as TRAIL-resistant, while the remaining cell lines were categorized as TRAIL-sensitive. The significance was determined by Wilcoxon rank sum test.
Migration and invasion assays
The migration and invasion kits were purchased from Calbiochem. The cells were subcultured into 6-well plates. After 24h, the cells were transfected with pre-miR-29a or -140-5p for 24h, and the media containing reduced serum (1%) was subsequently changed for an additional 24h. The cells subsequently plated into migration (5*10^5 cells/ml) and invasion chamber (1*10^6 cells/ml), and the bottom chambers remained with the serum-reduced media. After 24–48h, the migrated or invasive cells were quantified Calcein AM staining, followed by obtaining fluorescence at an excitation wavelength of 485nm and an emission wavelength of 520nm.
Statistical analysis
G-I, B-E, and and were confirmed by at least 2 indicated biological replicates. The figures in this manuscript are the representative ones based on technical replicates. Student’s t-test or Fisher exact t-test was used to address significance. Error bars are expressed as mean ± standard deviation (SD). Statistical significance assessed by calculating p-value was less than 0.05.
Figure 1. Downregulation of oncogenic miR-29 and -140-5p causes TRAIL-induced Drug-tolerant (TDT) state. (A) A schematic diagram showing the strategy to generate TRAIL-revertant from the resistant cells. (B) Gene Ontology terms associated genes differentially expressed in TRAIL-resistant cell (H460R) compared with sensitive cell. Left panel is a principal component analysis showing clustering between sensitive and resistant quadruplicate. (C) Heatmap showing upregulated miR-29 target genes in H460R cell. (D) Heatmap of miR-29 target genes upregulated in TRAIL-resistant cells. Kaplan-Meier curve depicting the survival probability in the patients with LUAD associated with (E) miR-29 or (F) miR-140 expression. (G) The restored expression of miR-29 a/c and -140-5p in H460R in response to drug holiday. (H) Cell survival rate using CellTiter-Glo assay. Error bars indicate mean ±SD (n = 4) and the p-values were calculated by one-way ANOVA test (**p < 0.01). (I) Chamber invasion assay of H460R cell reconstituted by miR-29 and/or -140-5p. Error bars show mean ±SD (n = 6) and the p-values were calculated by two-tailed student t-test (* p < 0.05, **p < 0.01).
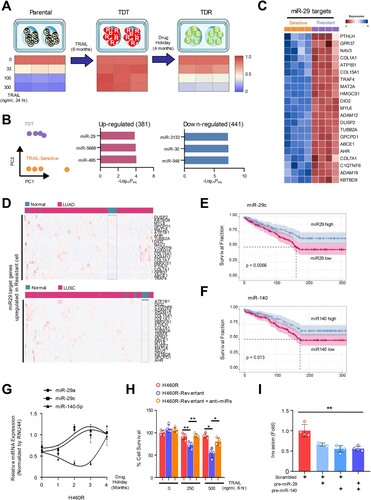
Figure 2. MiR-29a/c and -140-5p directly suppressed RSK2 protein expression causing TRAIL-sensitivity (A) A schematic diagram showing seed sequence of miR-29/-140-5p and the target sequence of RPS6KA3 3’UTR. (B) Luciferase reporter assay using 3’UTR of RPS6KA3 or 3’UTR harboring deletion mutations of the miR-29a/c and -140-5p in HEK293 cells over-expressing miR-29a/c or-140-5p. Error bars indicate mean ±SD (n = 3). P-values were calculated by two-tailed student t-test. (C) Western blot analysis indicating suppressed RSK2 protein level in lung cancer cells. Indicated cells were transfected by indicated miRNAs, respectively. After 48h, cells were harvested and subjected to Western blotting with indicated antibodies. (D) Expressional comparison of the miR-29a and -140-5p in lung cancer tissues compared to their adjunct normal tissues. Taqman-based qRT-PCR analysis was applied to analyze expression of the target miRNAs, and the expression of the target miRNAs in each cancer sample (n = 15) was normalized by their paired control sample (n = 15), respectively. P-values were obtained by one-way ANOVA test (* p < 0.05). (E) Correlation analysis of miR-29a/-140-5p and RSK2 protein expression in cancer. Expression of RSK2 protein was quantitated by measuring band intensities using Image J software. The intensity in cancer sample was normalized by paired normal sample. Likewise, relative value for miR-29a or miR-140-5p was calculated by comparing qRT-PCR values between cancer sample (n = 15) and its paired control sample (n = 15). P-value was obtained by one-way ANOVA test (**p < 0.01). (F) Expressional correlation of RPS6KA3 and TRAIL-sensitivity in NSCLC. TRAIL-sensitivity for each cell line was obtained from previous literatures and colored by their sensitivity as indicated. For statistical analysis, cell lines were divided into two groups with median RPS6KA3 expression, annotated by TRAIL-sensitivity from literatures, Wilcoxon rank sum test was applied to obtain significance. Detailed information about mRNA expression of RPS6KA3 gene and TRAIL-sensitivity is described in Material and Methods section. Gene Ontology analysis using upregulated and downregulated genes in H460R cells showing differentially regulated pathways. (*p < 0.05, **p < 0.01)
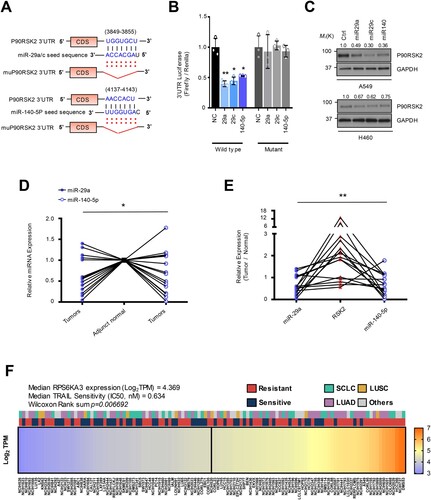
Figure 3. RSK2 is responsible for H460 revertant phenotype. (A and B) Western blot analysis showing the alteration of RSK2 expression in H460R revertant cell. (C) Western blot analysis showing increased PARP-1 cleavage by targeting RSK2. H292 TRAIL-sensitive cells were treated with anti-miR-29 / -140 with siRSK2 siRNAs for 48h. Subsequently the cells were treated by TRAIL, followed by performing Western blot analysis as indicated antibodies. (D) Cell survival of the H460R cells in RSK2-dependent manner. The precursor of miR-29a or -140-5p was co-transfected with either empty vector or pcDNA-RPS6KA3 plasmid for 48h, and the cells were subsequently stimulated by TRAIL as indicated. After that, the CellTiter-Glo assay was performed to determine cell survival rate. P-value was calculated by two tailed student t-test (*p < 0.05, **p <0.01). (E) Enhanced TRAIL-sensitivity by pharmacologic inhibition of RSK2 activity. BI-D1870 was treated to H460R as indicated, and cell survival rate was determined by CellTiter-Glo assay. Bars shows mean ±SD (n = 6) and the p-values were calculated by two-tailed student t-test (* p < 0.01, **p < 0.001). (F) The re-sensitized TRAIL-sensitivity upon RSK2 suppression in cells harboring anti-miR-29 and -140-5p. Bars shows mean ±SD (n = 4) and the p-values were calculated by two-tailed student t test (*p < 0.01, **p < 0.001).
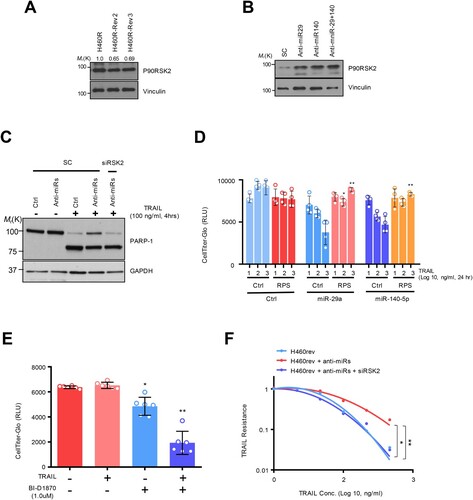
Figure 4. The NF-κB regulation by miRNA-dependent RSK2 suppression: (A) NF-κB promoter measurement using luciferase reporter assay. (B) Restored NF-κB activity by RSK2 overexpression in miR-29/-140-5p expressing A549. The cells were co-transfected by miR-29a/-140-5p and pcDNA-RPS6KA3 plasmid for 48h. Subsequently the promoter activity was measured by NF-κB promoter luciferase assay. Error bars indicate mean ±SD (n = 3) and the p-value was obtained by fisher exact t-test (*p < 0.05, **p < 0.01). (C) The phosphorylation of P65 subunit of NF-κB protein was analyzed by Western blot analysis. (D) Subcellular fractionation assay indicating nuclear localization of p65 depending on RSK2. Enhanced NF-κB activity by miR-29/-140 suppression. The H460 revertant was co-transfected by anti-miR-29/-140 and siRPS6KA3 siRNAs for 48h, and the promoter activities were addressed by (E) luciferase assay and (F) Western blot analysis. P-values were calculated by paired student t-test (*p < 0.05, **p < 0.005, ***p < 0.0005).
Results
Dysregulation of oncogenic miR-29ac and -140-5p is responsible for TRAIL-induced drug-tolerant (TDT) state
To characterize the mechanism underlying TRAIL-resistance, we previously generated TRAIL-resistant cells by treating a subtoxic range of TRAIL to H460 and H292-TRAIL sensitive cells (H460S and H292S) for 6 months, and that resistance was abolished by giving those cells drug-holiday, suggesting that the resistant cells we previously generated showed drug tolerant phenotype referred as TRAIL-induced drug tolerance (TDT) ((A)) (Jeon et al. Citation2015; Joshi et al. Citation2015). Although we previously characterized how the TDT cell was mechanistically established and its significance in lung cancer progression, it has been not sufficiently analyzed as a transcriptome-wide. From the result of our previous study, we found around 400 differentially expressed genes in H460R compared to H460S (Jeon et al. Citation2015). Subsequent Gene Ontology analysis using those genes suggested that the miRNA signatures were successfully identified. Of those, the enhanced miR-29 target genes in H460R brought our interest to further analysis because we initially identified miR-29 and -140-5p as suppressed miRNAs in TDT cells that we established ((B); Supplementary Figure S1A). Moreover, the upregulated miR-29 target genes exhibited lung cancer-specific signatures ((C–D)). Additionally, TCGA data showed that miR-29 was found to be decreased in lung squamous cell carcinoma (LUSC), and miR-140 was suppressed in both LUSC and lung adenocarcinoma (LUAD), associated with favorable survival prediction of the patients with LUAD (E-F; Supplementary Figure S1B-S1C). These data suggest that miR-29 pathway is suppressed in TDT cells, which could play a significant role in lung cancer progression. To analyze whether these miRNAs could be associated with the reversible sensitivity in TDT cells, we gave 4 months of drug holiday to H460R cells and found both miR-29ac and -140-5p expression restored in response to drug holiday, referring to it as a TRAIL holiday-induced drug revertant (TDR) cell ((G)). Moreover, the revertant was sensitive to TRAIL-induced cell death, rescued by the genetic inhibition of miR-29ac and -140-5p with anti-miRNA inhibitor treatment (H; Supplementary Figure S2A). Conversely, the overexpression of miR-29 and/or miR-140 in TRAIL-sensitive H460 and H292 cells decreased cell survival rate, consistently showed an increase in caspase substrate activity and cleaved PARP-1 protein (Supplementary Figure S1D and S2B-S2D). Moreover, these miR-29 and -140-dependent TRAIL-sensitivity seems to be conserved in the Calu-1 cell innately resistant to TRAIL (Supplementary Figure S2E-S2G). Consequently, the H460R overexpressing pre-miR-29ac and/or -140-5p decreased in migratory and invasive abilities (I; Supplementary Figure S3C). These data strongly suggest that the TDT cells exhibit a reversible phenotype referring to a drug-tolerant persistent stage, which could be mainly driven by alteration of miR-29ac and -140-5p expression.
RPS6KA3 gene is a direct target for both miR-29ac and -140
To elucidate the mechanism underlying the miR-29/-140-dependent TRAIL resistance, we investigated the potential target genes using in silico tools and found that RPS6KA3 (RSK2 for protein) could be a potential target for both miRNAs (A) (McGeary et al. Citation2019). The enforced expression of miR-29ac and -140-5p in HEK293 cells reduced the 3’UTR luciferase activity, whereas the mutants harboring deletion mutations on the binding sites for the miRNAs remained unchanged in the same context (Figure 2B). Consistently, Western blot analysis using two lung cancer cells showed that RSK2 protein was suppressed by the overexpression of the miRNAs ((C)). However, we did not see any significant suppression of RPS6KA3 mRNA level in both qRT-PCR and microarray the same context, suggesting that miR-29 and -140-5p regulate RPS6KA3 at translational level without inducing RPS6KA3 mRNA decay (Supplementary Figure S4A-S4B). To analyze whether the miR-29ac/-140-5p-dependent RPS6KA3 suppression could be conserved in vivo, we employed 15 cases of LUAD samples and their adjunct normal lung cells. As a result, we found that the miR-29a expression was decreased in 10 cases of tumor samples compared to adjunct tissue samples, whereas 2 cases showed increases and the rest changed minimally. In the analysis of miR-140, the 9 cases are found to be decreased, but the remaining 6 cases showed either unchanged or enhanced expression in lung cancer sample (D). Next, we analyzed the expression of RSK2 protein and found that RSK2 protein was upregulated in 10 cases of the lung tumor tissues, suggesting that RSK2 protein expression was significantly increased in tumor tissues compared to adjunct control cells (Supplementary Figure S4C-S4D). Collectively, there was a significant inverse correlation between both miRNAs and RSK2 protein in lung cancer, suggesting that miR-29ac/-140-5p-dependent RPS6KA3 suppression is conserved in the patient samples ((E)).
Given our current observations of the direct relation between miR-29/-140 and RPS6KA3 expression, we hypothesized that RPS6KA3 expression contributes to the establishment of TRAIL persistent phenotype. To evaluate this hypothesis, we employed public data to compare the bona fide nature of TRAIL sensitivity in lung cancer cells and analyzed any correlation between the expression of RPS6KA3 gene and TRAIL sensitivity. We identified lung cancer cell lines annotated by TRAIL sensitivity using IC50 score. Also, the lung cancer cell lines containing the feature of TRAIL resistance were additionally analyzed and added to the lists from the previous works of literature. As a result, 119 lung cancer cell lines were classified as either TRAIL-sensitive or TRAIL-resistant cell lines derived from SCLC and two major subtypes of NSCLC such as LUAD and LUSC and others (Shivapurkar et al. Citation2004; Wagner et al. Citation2007; Zitzmann et al. Citation2011; Yang et al. Citation2013; Lu et al. Citation2014; Jeon et al. Citation2015; Joshi et al. Citation2015; Li et al. Citation2017). In addition, RPS6KA3 expression in the corresponding lung cancer cell lines was analyzed from Broad Institute Cancer Cell Line Encyclopedia (CCLE), and was ranked by its abundance (Barretina et al. Citation2012). As a result, we found that the cells with more RPS6KA3 expression were more resistant to TRAIL-induced cell death ((F)). To clarify the transcriptional changes in H460R compared to H460S, differently expressed genes were analyzed and annotated to KEGG and Gene Ontology database (Kanehisa and Goto Citation2000; Thomas et al. Citation2022). ‘MAPK signaling pathway’ signature was significantly identified in both up- and down-regulated genes (Supplementary Figure S4E-S4F). All things considered, we not only identified the inverse correlation between miR-29/ -140 and RSK2, but also figured out there is a relation between RSK2 and TRAIL-resistance.
The expression of RPS6KA3 is responsible for TRAIL-induced drug tolerance
To get better insight into the TRAIL resistance, we first analyzed how the expression of the RSK2 protein is changed in accordance with TRAIL sensitivity. The RSK2 protein abundance was decreased in TDR cells, which showed a rescued expression upon miR-29 and/or -140 suppression in the TDR cells, suggesting that miR-29 and -140-dependent RPS6KA3 regulation is associated with the drug-tolerant status ((A,B)). We next sought to determine if the axis of miR-29/-140-5p and RPS6KA3 regulates TRAIL-sensitivity. The suppression of miR-29a /-140-5p expression by corresponding anti-miRNA oligonucleotides increased TRAIL resistance in parental H460 and H292 cells with decreased PARP-1 cleavage and/or caspase-3/-7 activation. However, the sensitivity was restored by siRPS6KA3 treatment in the same context (C; Supplementary Figure S5A-S5C). Conversely, decreased cell survival rate and increased caspase-3/-7 substrate activity were observed in TDT H460R cells by overexpressing miR-29 and miR-140-5p, whereas the reconstituted RPS6KA3 restored cell survival rate in response to TRAIL treatment (D; Supplementary Figure S5D). Moreover, pharmacological inhibition of RSK2 activity using a specific inhibitor, BI-D1870 synergistically suppressed cell survival rate with TRAIL in H460R cells (E) (Sapkota et al. Citation2007). Finally, anti-miR-29 and -140-5p treatment to H460 revertant cell enhanced TRAIL-resistance, but the cells were re-sensitized upon RPS6KA3 suppression (F). Taken together, these data strongly suggest that miR-29ac and -140-5p-dependent RPS6KA3 suppression is conserved in TRAIL-resistant lung cancer cell, which plays a critical role in establishing TRAIL-persistent phenotype.
RPS6KA3 induces NF-κB activation in TRAIL-induced drug-tolerant (TDT) cell
It has been known that the resistant phenotype could be associated with an aggressive cancer phenotype, mediated by NF-κB activation (Schneider et al. Citation1997; Kim et al. Citation2011; Grunert et al. Citation2012; Jeon et al. Citation2015; Joshi et al. Citation2015; Wang et al. Citation2024). Moreover, our group previously confirmed that NF-κB activation is one of the established resistant markers in innate TDT cells by establishing TDT cells with long-term exposure to TRAIL, showing higher NF-κB activity, which seems to be involved in aggressive cancer phenotype (Sheridan et al. Citation1997; Wang et al. Citation1998; Chen et al. Citation2003; Hall and Cleveland Citation2007; Plantivaux et al. Citation2009; Jeon et al. Citation2015). Moreover, RSK2 induced phosphorylation-dependent proteasomal degradation of IκB-a in TNF receptor pathway (Peng et al. Citation2010). Therefore, we examined whether this oncogenic pathway could be conserved in the TDR cells. Regarding this, we first sought to analyze the aggressiveness of TRAIL-resistance with NF-κB pathway activation. A series of luciferase reporter assays showed that NF-κB promoter activity was reactivated by miR-29ac/-140-5p inhibition ((A)). Conversely, the overexpression of the precursor miRNAs diminished TRAIL-dependent NF-κB activation in both H292R and Calu-1 cells (Supplementary Figure S3A-S3B). We then employed a pGL4.3-luc2p/ NF-κB construct to monitor NF-KB promoter activity and found that a decrease in NF-κB promoter activity was observed after overexpressing miR-29 and -140-5p. However, the ectopic expression of RPS6KA3 rehabilitated the promoter activity ((B)). Also, a major subunit of NF-κB, p65 protein was less phosphorylated in the miR-29 and -140-5p expressing H460R cell, which consistently upregulated in response to RPS6KA3 overexpression ((C)). Moreover, nuclear localization of p65 protein inhibited in miR-29 and -140 overexpression condition was dependent on RPS6KA3 expression ((D)) Importantly, H460 revertant has conserved mechanism by which miR-29ac/-140-5p-dependent RPS6KA3 suppression exhibited a decrease in NF-κB promoter activity, similarly conserved in TRAIL sensitive H460 cell ((E)). Accordingly, suppressive effect of p65 protein phosphorylation enforced by siRSK2 siRNA treatment was rescued by miR-29ac suppression ((F)). Taken these together, RSK2-NF-kB axis is responsible for TDT state.
Discussion
Drug resistance to cancer is a multi-step process, a major obstacle in chemotherapeutic approach. To identify the molecular mechanism underlying acquired TRAIL-resistance, we previously established the resistant cells by treating TRAIL to the sensitive H460 cells for 6 months and reported that the positive feedback loop between miRNAs (miR-21, -30c and -140) and NF-κB activation is responsible for acquired TRAIL-resistance, consistent with the studies for innate TRAIL-resistance in that NF-κB is major player for both types of resistance (Chaudhary et al. Citation1997; Schneider et al. Citation1997; Plantivaux et al. Citation2009; Jeon et al. Citation2015) Despite more robust analysis to identify any mutation associated with TRAIL-resistance, we did not find any actionable mutation in the genes involved in TRAIL-induced signaling pathway (Jeon et al. Citation2015), which initially motivated us to characterize any mutation or pathway involved in the resistant pathway in the current study. Our current observation suggests that the restored expression of the miR-29 and -140-5p in H460R revertant is responsible for TRAIL-sensitivity, and their novel target, RSK2 protein could mediate miR-29/-140-5p-dependent TRAIL-resistance.
Cancer evolution has been thought to be a mechanism underlying drug resistance. In particular, recent studies have suggested that the drug-tolerant stage was driven by epigenetic regulation of the oncogenic pathway, and could mediate the acquired resistance driven by emerging mutations, being consequently an important issue to reduce the chance for the resistance by blocking the intermediated stage (Roesch et al. Citation2010; Sharma et al. Citation2010; Lee et al. Citation2014; Hata et al. Citation2016; Smith et al. Citation2016; Guler et al. Citation2017). Regarding this, we initially observed that miR-29a/c and -140-5p were restored, associated with recovering TRAIL-sensitivity, suggesting that the established TDT cells were drug-tolerant persister (; Supplementary Figure 1-3). Despite the suggested tumor suppressive roles of both miR-29 family and miR-140, recent analysis of circulating plasma miRNAs interestingly suggested that serum miR-29 family were significantly elevated in the patients with NSCLC, associated with poor 5-year overall survival, raised the feasibility of the miR-29 as an oncogenic modifier in lung cancer (Yang et al. Citation2019). However, our current analysis using TCGA data set confirms previous findings that miR-29 is exclusively suppressed in both LUAD and LUSC patient samples, and its suppression is closely associated with cancer survival probability (). Moreover, we also confirmed that miR-29 overexpression suppressed cell survival rate and NF-κB activity in TRAIL-resistant cells, whereas the inhibition of miR-29 sensitizes TDR to TRAIL-induced cell death, suggesting miR-29 as a tumor suppressive miRNA in our system. Therefore, the discrepancy in the studies between lung cancer tissues and plasma should be further analyzed and characterized.
RSK2 is a member of p90 ribosomal S6 kinase, a highly conserved serine/threonine kinase mediating Ras-MAPK signaling pathway. Given their oncogenic roles, interestingly, genetic mutation and amplification were rarely found in many cancers (Roberts and Der Citation2007; Lara et al. Citation2013). However, RSK2 encoded by RPS6KA3 gene activated by ERK1/2 protein enhanced cancer progression through destabilizing a proapoptotic protein, BAD and caspase-8, and by transcriptionally activating anti-apoptotic Bcl-2 as well as inducing DR5 expression, suggesting that RSK2 expression and its kinase activity is likely to be associated with TRAIL resistance (Bonni et al. Citation1999; Oh et al. Citation2010; Im et al. Citation2018). Our current data shows that miR-29 and -140-5p regulate oncogenic RSK2 protein, which consequently plays a significant role in restoring the sensitivity from TDT cells ( and ; Supplementary Figure 4). Given RSK2 expression and annotated TRAIL-sensitivity in 119 different lung cancer cell lines, we observed that there is a significant correlation between RSK2 expression and TRAIL resistance. Moreover, MAPK, as an upstream regulator to RSK2 protein, was dysregulated in TDT cells. Additionally, Gene ontology data following transcriptome analysis suggests that the target genes of the miR-29 are enriched in TDT cells, consistent with suppressed miR-29 expression in our qRT-PCR analysis (). Consistent with previous observations, NF-κB activation was regulated by miR-29 and miR-140, which seems to be dependent on RSK2 regulation (Figure 4) (Peng et al. Citation2010). Furthermore, NF-κB activation is one of the major determinants of TRAIL resistance (Chaudhary et al. Citation1997; Schneider et al. Citation1997; Plantivaux et al. Citation2009; Jeon et al. Citation2015). All these data strongly suggest that miR-29/-140-RSK2 axis plays an important role in TDT status, which will provide an important insight for chemotherapy using TRAIL in lung cancer.
Conclusions
TRAIL has shown potential as a therapeutic agent for lung cancer, but its efficacy is hindered by the early development of both innate and acquired resistance. To enhance its therapeutic utility, it is crucial to understand the resistance and restoring mechanisms of TRAIL resistance. Our study has revealed that miR-29/-140-depdent RSK2 regulation is a reversal in terms of TRAIL-resistance, which could be an important feature for a TRAIL-tolerant state, suggesting that the inhibition of this pathway plays a critical role in maximizing clinical utility using TRAIL in lung cancer.
Furthermore, TRAIL and its receptors are implicated in various cancer types, underscoring the potential applicability of our findings. For instance, higher expression levels of TRAIL-R1 have been identified as prognostic markers in colon cancer (Strater et al. Citation2002), hepatocellular carcinoma (Kriegl et al. Citation2010), and skin cancer (Omran and Ata Citation2014). Similarly, increased expression of TRAIL-R2 has been associated with improved survival outcomes in glioblastoma (Kuijlen et al. Citation2006), renal cell carcinoma (Macher-Goeppinger et al. Citation2009), breast cancer (Labovsky et al. Citation2017), and pancreatic cancer. These examples highlight the potential of TRAIL as a universal anticancer agent, capable of targeting a wide spectrum of malignancies.
The implications of our study extend beyond lung cancer, suggesting that the mechanisms of TRAIL resistance we have uncovered could be relevant across a diverse range of cancers. However, our findings are based solely on lung cancer models and in vitro studies. Therefore, further investigation, particularly in vivo studies, is necessary to validate our conclusions and fully understand the therapeutic potential of TRAIL in other cancer types. This future research could pave the way for developing more effective and universally applicable TRAIL-based cancer therapies.
Supplemental Material
Download MS Word (41.1 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, et al. 1999. Safety and antitumor activity of recombinant soluble Apo2 ligand. Journal of Clinical Investigation. 104(2):155–162. doi:10.1172/JCI6926.
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. 2012. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. Nature. 483(7391):603–607. eng. doi:10.1038/nature11003.
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. 1999. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.]. Science. 286(5443):1358–1362. eng. doi:10.1126/science.286.5443.1358.
- Calin GA, Croce CM. 2006. MicroRNA signatures in human cancers [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't Review]. Nature Reviews Cancer. 6(11):857–866. eng. doi:10.1038/nrc1997.
- Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. 1997. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway [Research Support, Non-U.S. Gov't]. Immunity. 7(6):821–830. eng. doi:10.1016/S1074-7613(00)80400-8.
- Chen X, Kandasamy K, Srivastava RK. 2003. Differential roles of RelA (p65) and c-Rel subunits of nuclear factor kappa B in tumor necrosis factor-related apoptosis-inducing ligand signaling [Research Support, Non-U.S. Gov't]. Cancer Research. 63(5):1059–1066. eng.
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, et al. 2007. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B [Research Support, N.I.H., Extramural]. PNAS. 104(40):15805–15810. eng. doi:10.1073/pnas.0707628104.
- Flamini V, Jiang WG, Cui Y. 2017. Therapeutic role of MiR-140-5p for the treatment of non-small cell lung cancer. Anticancer Research. 37(8):4319–4327. eng.
- Fuchs CS, Fakih M, Schwartzberg L, Cohn AL, Yee L, Dreisbach L, Kozloff MF, Hei YJ, Galimi F, Pan Y, et al. 2013. TRAIL receptor agonist conatumumab with modified FOLFOX6 plus bevacizumab for first-line treatment of metastatic colorectal cancer: a randomized phase 1b/2 trial. Cancer. 119(24):4290–4298. doi:10.1002/cncr.28353.
- Genc S, Yagci T, Vageli DP, Dundar R, Doukas PG, Doukas SG, Tolia M, Chatzakis N, Tsatsakis A, Taghizadehghalehjoughi A. 2023. Exosomal microRNA-223, microRNA-146, and microRNA-21 profiles and biochemical changes in laryngeal cancer. ACS Pharmacology & Translational Science. 6(5):820–828. doi:10.1021/acsptsci.3c00038.
- Grunert M, Gottschalk K, Kapahnke J, Gundisch S, Kieser A, Jeremias I. 2012. The adaptor protein FADD and the initiator caspase-8 mediate activation of NF-kappaB by TRAIL [Research Support, Non-U.S. Gov't]. Cell Death & Disease. 3:e414. eng. doi:10.1038/cddis.2012.154.
- Guler GD, Tindell CA, Pitti R, Wilson C, Nichols K, KaiWai Cheung T, Kim HJ, Wongchenko M, Yan Y, Haley B, et al. 2017. Repression of stress-induced LINE-1 expression protects cancer cell subpopulations from lethal drug exposure. Cancer cell. 32(2):221–237. e213. eng. doi:10.1016/j.ccell.2017.07.002.
- Hall MA, Cleveland JL. 2007. Clearing the TRAIL for cancer therapy [comment review]. Cancer cell. 12(1):4–6. eng. doi:10.1016/j.ccr.2007.06.011.
- Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V, et al. 2016. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, Non-P.H.S.]. Nature Medicine. 22(3):262–269. eng. doi:10.1038/nm.4040.
- Herbst RS, Kurzrock R, Hong DS, Valdivieso M, Hsu CP, Goyal L, Juan G, Hwang YC, Wong S, Hill JS, et al. 2010. A first-in-human study of conatumumab in adult patients with advanced solid tumors [Clinical Trial, Phase I Research Support, Non-U.S. Gov't]. Clinical cancer research : an official journal of the American Association for Cancer Research. 16(23):5883–5891. eng. doi:10.1158/1078-0432.CCR-10-0631.
- Hwang H, Chang HR, Baek D. 2023. Determinants of functional microRNA targeting. Molecules and Cells. 46(1):21–32. doi:10.14348/molcells.2023.2157.
- Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T, Zhang H, Mountz JD, Koopman WJ, Kimberly RP, et al. 2001. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.]. Nature Medicine. 7(8):954–960. eng. doi:10.1038/91000.
- Im JY, Kim BK, Lee JY, Park SH, Ban HS, Jung KE, Won M. 2018. DDIAS suppresses TRAIL-mediated apoptosis by inhibiting DISC formation and destabilizing caspase-8 in cancer cells [Research Support, Non-U.S. Gov't]. Oncogene. 37(9):1251–1262. eng. doi:10.1038/s41388-017-0025-y.
- Jeon YJ, Middleton J, Kim T, Lagana A, Piovan C, Secchiero P, Nuovo GJ, Cui R, Joshi P, Romano G, et al. 2015. A set of NF-kappaB-regulated microRNAs induces acquired TRAIL resistance in lung cancer. PNAS. 112(26):E3355–E3364. eng.
- Joshi P, Jeon YJ, Lagana A, Middleton J, Secchiero P, Garofalo M, Croce CM. 2015. MicroRNA-148a reduces tumorigenesis and increases TRAIL-induced apoptosis in NSCLC. PNAS. 112(28):8650–8655. eng. doi:10.1073/pnas.1500886112.
- Kanehisa M, Goto S. 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research. 28(1):27–30. doi:10.1093/nar/28.1.27.
- Kaplan-Lefko PJ, Graves JD, Zoog SJ, Pan Y, Wall J, Branstetter DG, Moriguchi J, Coxon A, Huard JN, Xu R, et al. 2010. Conatumumab, a fully human agonist antibody to death receptor 5, induces apoptosis via caspase activation in multiple tumor types. Cancer Biology & Therapy. 9(8):618–631. doi:10.4161/cbt.9.8.11264.
- Ke J, Chen J, Liu X. 2022. Analyzing and validating the prognostic value and immune microenvironment of clear cell renal cell carcinoma. Animal Cells and Systems. 26(2):52–61. doi:10.1080/19768354.2022.2056635.
- Kim JY, Lee JY, Kim DG, Koo GB, Yu JW, Kim YS. 2011. TRADD is critical for resistance to TRAIL-induced cell death through NF-kappaB activation [Research Support, Non-U.S. Gov't]. FEBS letters. 585(14):2144–2150. eng. doi:10.1016/j.febslet.2011.05.034.
- Kindler HL, Richards DA, Garbo LE, Garon EB, Stephenson Jr JJ, Rocha-Lima CM, Safran H, Chan D, Kocs DM, Galimi F, et al. 2012. A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Annals of Oncology. 23(11):2834–2842. doi:10.1093/annonc/mds142.
- Kriegl L, Jung A, Engel J, Jackstadt R, Gerbes AL, Gallmeier E, Reiche JA, Hermeking H, Rizzani A, Bruns CJ, et al. 2010. Expression, cellular distribution, and prognostic relevance of TRAIL receptors in hepatocellular carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 16(22):5529–5538. doi:10.1158/1078-0432.CCR-09-3403.
- Kuijlen JM, Mooij JJ, Platteel I, Hoving EW, van der Graaf WT, Span MM, Hollema H, den Dunnen WF. 2006. TRAIL-receptor expression is an independent prognostic factor for survival in patients with a primary glioblastoma multiforme. Journal of Neuro-Oncology. 78(2):161–171. doi:10.1007/s11060-005-9081-1.
- Labovsky V, Martinez LM, Davies KM, de Lujan Calcagno M, Garcia-Rivello H, Wernicke A, Feldman L, Matas A, Giorello MB, Borzone FR, et al. 2017. Prognostic significance of TRAIL-R3 and CCR-2 expression in tumor epithelial cells of patients with early breast cancer. BMC Cancer. 17(1):280. doi:10.1186/s12885-017-3259-8.
- Lara R, Seckl MJ, Pardo OE. 2013. The p90 RSK family members: common functions and isoform specificity [Review]. Cancer Research. 73(17):5301–5308. eng. doi:10.1158/0008-5472.CAN-12-4448.
- Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. 2014. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer cell. 26(2):207–221. eng. doi:10.1016/j.ccr.2014.05.019.
- Li B, Gu X, Zhang H, Xiong H. 2022. Comprehensive analysis of the prognostic value and immune implications of the TTK gene in lung adenocarcinoma: a meta-analysis and bioinformatics analysis. Animal Cells and Systems. 26(3):108–118. doi:10.1080/19768354.2022.2079718.
- Li X, You M, Liu YJ, Ma L, Jin PP, Zhou R, Zhang ZX, Hua B, Ji XJ, Cheng XY, et al. 2017. Reversal of the apoptotic resistance of non-small-cell lung carcinoma towards TRAIL by natural product Toosendanin [Research Support, Non-U.S. Gov't]. Scientific Reports. 7:42748. eng. doi:10.1038/srep42748.
- Lu M, Marsters S, Ye X, Luis E, Gonzalez L, Ashkenazi A. 2014. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Molecular Cell. 54(6):987–998. eng. doi:10.1016/j.molcel.2014.04.029.
- Macher-Goeppinger S, Aulmann S, Tagscherer KE, Wagener N, Haferkamp A, Penzel R, Brauckhoff A, Hohenfellner M, Sykora J, Walczak H, et al. 2009. Prognostic value of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors in renal cell cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 15(2):650–659. doi:10.1158/1078-0432.CCR-08-0284.
- McGeary SE, Lin KS, Shi CY, Pham TM, Bisaria N, Kelley GM, Bartel DP. 2019. The biochemical basis of microRNA targeting efficacy. Science. 366(6472):p. eaav174110.1126/science.aav1741.
- Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. 2008. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship [Review]. Mayo Clinic Proceedings. 83(5):584–594. eng. doi:10.1016/S0025-6196(11)60735-0.
- Oh YT, Liu X, Yue P, Kang S, Chen J, Taunton J, Khuri FR, Sun SY. 2010. ERK/ribosomal S6 kinase (RSK) signaling positively regulates death receptor 5 expression through co-activation of CHOP and Elk1 [Research Support, N.I.H., Extramural Research Support, U.S. Gov't, Non-P.H.S.]. Journal of Biological Chemistry. 285(53):41310–41319. eng. doi:10.1074/jbc.M110.153775.
- Omran OM, Ata HS. 2014. Expression of tumor necrosis factor-related apoptosis-inducing ligand death receptors DR4 and DR5 in human nonmelanoma skin cancer. The American Journal of Dermatopathology. 36(9):710–717. doi:10.1097/DAD.0b013e3182a3d31d.
- Pan G, O'Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM. 1997. The receptor for the cytotoxic ligand TRAIL [Research Support, U.S. Gov't, P.H.S.]. Science. 276(5309):111–113. eng. doi:10.1126/science.276.5309.111.
- Patel AR, Wedzicha JA, Hurst JR. 2011. Reduced lung-cancer mortality with CT screening [Letter Comment]. The New England Journal of Medicine. 365(21):2035. author reply 2037-2038. eng. doi:10.1056/NEJMc1110293.
- Peng C, Cho YY, Zhu F, Xu YM, Wen W, Ma WY, Bode AM, Dong Z. 2010. RSK2 mediates NF-{kappa}B activity through the phosphorylation of IkappaBalpha in the TNF-R1 pathway [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. The FASEB journal : the journal of the Federation of American Societies for Experimental Biology. 24(9):3490–3499. eng. doi:10.1096/fj.09-151290.
- Plaisier CL, Pan M, Baliga NS. 2012. A miRNA-regulatory network explains how dysregulated miRNAs perturb oncogenic processes across diverse cancers [Research Support, N.I.H., Extramural Research Support, U.S. Gov't, Non-P.H.S.]. Genome Research. 22(11):2302–2314. eng. doi:10.1101/gr.133991.111.
- Plantivaux A, Szegezdi E, Samali A, Egan L. 2009. Is there a role for nuclear factor kappaB in tumor necrosis factor-related apoptosis-inducing ligand resistance? [Review]. Annals of the New York Academy of Sciences. 1171:38–49. eng. doi:10.1111/j.1749-6632.2009.04725.x.
- Poomakkoth N, Issa A, Abdulrahman N, Abdelaziz SG, Mraiche F. 2016. p90 ribosomal S6 kinase: a potential therapeutic target in lung cancer. Journal of Translational Medicine. 14.(1410.1186/s12967-016-0768-1.
- Roberts PJ, Der CJ. 2007. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer [Research Support, Non-U.S. Gov't Review]. Oncogene. 26(22):3291–3310. eng. doi:10.1038/sj.onc.1210422.
- Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M. 2010. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. Cell. 141(4):583–594. eng. doi:10.1016/j.cell.2010.04.020.
- Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frodin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, et al. 2007. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo [Research Support, Non-U.S. Gov't]. Biochemical Journal. 401(1):29–38. eng. doi:10.1042/BJ20061088.
- Schneider P, Thome M, Burns K, Bodmer JL, Hofmann K, Kataoka T, Holler N, Tschopp J. 1997. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB [Research Support, Non-U.S. Gov't]. Immunity. 7(6):831–836. eng. doi:10.1016/S1074-7613(00)80401-X.
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. 2010. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. Cell. 141(1):69–80. eng. doi:10.1016/j.cell.2010.02.027.
- Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, Ramakrishnan L, Gray CL, Baker K, Wood WI, et al. 1997. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 277(5327):818–821. eng. doi:10.1126/science.277.5327.818.
- Shivapurkar N, Toyooka S, Toyooka KO, Reddy J, Miyajima K, Suzuki M, Shigematsu H, Takahashi T, Parikh G, Pass HI, et al. 2004. Aberrant methylation of trail decoy receptor genes is frequent in multiple tumor types [Research Support, U.S. Gov't, P.H.S.]. International Journal of Cancer. 109(5):786–792. eng. doi:10.1002/ijc.20041.
- Siegel RL, Miller KD, Jemal A. 2018. Cancer statistics, 2018. CA: A Cancer Journal for Clinicians. 68(1):7–30. eng. doi:10.3322/caac.21442.
- Skelton W, Turba E, Sokol L. 2020. Durable complete response to AMG 655 (Conatumumab) and Vorinostat in a patient with relapsed classical Hodgkin Lymphoma: extraordinary response from a phase 1b clinical protocol. Clinical Lymphoma, Myeloma and Leukemia. 20(12):e944–e946. doi:10.1016/j.clml.2020.07.012.
- Smith MP, Brunton H, Rowling EJ, Ferguson J, Arozarena I, Miskolczi Z, Lee JL, Girotti MR, Marais R, Levesque MP, et al. 2016. Inhibiting drivers of non-mutational drug tolerance is a salvage strategy for targeted melanoma therapy [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. Cancer cell. 29(3):270–284. eng. doi:10.1016/j.ccell.2016.02.003.
- Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology. 3:Article3. doi:10.2202/1544-6115.1027.
- Spierings DC, de Vries EG, Timens W, Groen HJ, Boezen HM, de Jong S. 2003. Expression of TRAIL and TRAIL death receptors in stage III non-small cell lung cancer tumors [Research Support, Non-U.S. Gov't]. Clinical cancer research : an official journal of the American Association for Cancer Research. 9(9):3397–3405. eng.
- Strater J, Hinz U, Walczak H, Mechtersheimer G, Koretz K, Herfarth C, Moller P, Lehnert T. 2002. Expression of TRAIL and TRAIL receptors in colon carcinoma: TRAIL-R1 is an independent prognostic parameter. Clinical cancer research : an official journal of the American Association for Cancer Research. 8(12):3734–3740.
- Subbiah V, Brown RE, Buryanek J, Trent J, Ashkenazi A, Herbst R, Kurzrock R. 2012. Targeting the apoptotic pathway in chondrosarcoma using recombinant human Apo2L/TRAIL (dulanermin), a dual proapoptotic receptor (DR4/DR5) agonist. Molecular Cancer Therapeutics. 11(11):2541–2546. doi:10.1158/1535-7163.MCT-12-0358.
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 102(43):15545–15550. doi:10.1073/pnas.0506580102.
- Thomas PD, Ebert D, Muruganujan A, Mushayahama T, Albou LP, Mi H. 2022. PANTHER: making genome-scale phylogenetics accessible to all. Protein Science. 31(1):8–22. doi:10.1002/pro.4218.
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al. 2006. A microRNA expression signature of human solid tumors defines cancer gene targets [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. PNAS. 103(7):2257–2261. eng. doi:10.1073/pnas.0510565103.
- Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K, Lee D, von Goetz M, Yee SF, Totpal K, et al. 2007. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nature Medicine. 13(9):1070–1077. eng. doi:10.1038/nm1627.
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin Jr AS. 1998. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.]. Science. 281(5383):1680–1683. eng. doi:10.1126/science.281.5383.1680.
- Wang M, Li G, Jiang G, Cai J, Liu Z, Huang R, Huang X, Wang H. 2024. Novel NF-kappaB inhibitor-conjugated Pt(IV) prodrug to enable cancer therapy through ROS/ER stress and mitochondrial dysfunction and overcome multidrug resistance. Journal of Medicinal Chemistry.
- Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA, et al. 1995. Identification and characterization of a new member of the TNF family that induces apoptosis [Research Support, Non-U.S. Gov't]. Immunity. 3(6):673–682. eng. doi:10.1016/1074-7613(95)90057-8.
- Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, et al. 2013. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells [Research Support, Non-U.S. Gov't]. Nucleic Acids Research. 41(Database issue):D955–D961. eng.
- Yang X, Zhang Q, Zhang M, Su W, Wang Z, Li Y, Zhang J, Beer DG, Yang S, Chen G. 2019. Serum microRNA signature is capable of early diagnosis for non-small cell lung cancer [Research Support, Non-U.S. Gov't]. International Journal of Biological Sciences. 15(8):1712–1722. eng. doi:10.7150/ijbs.33986.
- Yuan Y, Shen Y, Xue L, Fan H. 2013. miR-140 suppresses tumor growth and metastasis of non-small cell lung cancer by targeting insulin-like growth factor 1 receptor. PLoS One. 8(9):e73604. eng. doi:10.1371/journal.pone.0073604.
- Zitzmann K, de Toni E, von Ruden J, Brand S, Goke B, Laubender RP, Auernhammer CJ. 2011. The novel Raf inhibitor Raf265 decreases Bcl-2 levels and confers TRAIL-sensitivity to neuroendocrine tumour cells [Research Support, Non-U.S. Gov't]. Endocrine-Related Cancer. 18(2):277–285. eng. doi:10.1530/ERC-10-0108.