
ABSTRACT
Invasion of periodontal pathogens into periodontal tissues is an important step that can cause tissue destruction in periodontal diseases. Porphyromonas gingivalis is a keystone pathogen and its gingipains are key virulence factors. Fusobacterium nucleatum is a bridge organism that mediates coadhesion of disease-causing late colonizers such as P. gingivalis and early colonizers during the development of dental biofilms. The aim of this study was to investigate how P. gingivalis, in particular its gingipains, influences the invasion of coinfecting F. nucleatum into gingival epithelial cells. When invasion of F. nucleatum was analyzed after 4 h of infection, invasion of F. nucleatum was suppressed in the presence of P. gingivalis compared with during monoinfection. However, coinfection with a gingipain-null mutant of P. gingivalis did not affect invasion of F. nucleatum. Inhibition of PI3K reduced invasion of F. nucleatum. P. gingivalis inactivated the PI3K/AKT pathway, which was also dependent on gingipains. Survival of intracellular F. nucleatum was promoted by P. gingivalis with Arg gingipain mutation. The results suggest that P. gingivalis, in particular its gingipains, can affect the invasion of coinfecting F. nucleatum through modulating intracellular signaling of the host cells.
Introduction
Chronic periodontitis is not only one of the leading causes of tooth loss, but it is also an important risk factor for multiple systemic diseases, such as rheumatoid arthritis and cardiovascular disease [Citation1]. Polymicrobial biofilms present in subgingival crevices are the most important etiological factor in the pathogenesis of periodontitis. Polymicrobial communities develop through interspecies interactions and adaptation within the surrounding microenvironments, and more than 400 species of bacteria have been detected in subgingival biofilms [Citation2,Citation3].
Porphyromonas gingivalis, a Gram-negative obligatory anaerobe, has been classified into a disease-causing ‘red complex’ [Citation4]. Recently it was demonstrated that P. gingivalis acts as a keystone pathogen in the pathogenesis of periodontitis [Citation5]. Subversion of host immune surveillance by P. gingivalis creates an environment that facilitates dysbiosis of subgingival microbiota, and the dysbiotic microbiota with increased pathogenicity overactivate inflammation in periodontal tissues [Citation5]. Gingipains (Arg-X-specific Arg gingipains [RgpA and RgpB] and Lys-X-specific Lys gingipain [Kgp]), major proteases of P. gingivalis, are essential for its pathogenic potential [Citation6–Citation8] and are often considered to be potential therapeutic targets. Along with other virulence factors, such as lipopolysaccharide, fimbriae, and several other proteases, gingipains contribute to the ability of P. gingivalis to dysregulate host immune systems [Citation9,Citation10].
Fusobacterium nucleatum is a spindle-shaped, Gram-negative anaerobe frequently and abundantly found in the oral cavity under both healthy and diseased conditions [Citation11]. The importance of F. nucleatum in the development of polymicrobial biofilms has long been recognized. F. nucleatum binds to early colonizers and acts as a bridging organism that mediates coadherence of disease-causing late colonizers, including P. gingivalis, to dental biofilms [Citation12]. Several adhesins, including FomA, aid1, Fap2, and RadD, are involved in interactions with various oral bacteria, such as oral streptococci and P. gingivalis [Citation13–Citation15]. Other virulence factors such as GroEL and FadA contribute to the ability of F. nucleatum to cause several systemic diseases, such as atherosclerosis and colorectal cancer [Citation16,Citation17].
Invasion of periodontal pathogens into periodontal tissues is an important step that can initiate periodontal diseases [Citation18]. Gingival epithelium is the first cell layer that bacteria in subgingival biofilms have to pass through in order to invade into the deeper parts of periodontal tissues. P. gingivalis invades into gingival epithelial cells via interactions of its fimbriae with β1 integrins of the cells [Citation19], while invasion of F. nucleatum into gingival epithelial cells is mediated by its FadA [Citation20]. F. nucleatum has been shown to enhance the invasion of P. gingivalis [Citation21,Citation22]. However, the effect of P. gingivalis on F. nucleatum invasion has not yet been studied. This study reports that P. gingivalis coinfection decreases F. nucleatum invasion into gingival epithelial cells in a gingipain-dependent manner, possibly via modulating the PI3K/AKT pathway.
Methods
Bacteria and cell culture
F. nucleatum ATCC 25586 was grown in brain heart infusion (BHI) broth supplemented with 5 μg/mL of hemin and 1 μg/mL of vitamin K under anaerobic conditions (10% H2, 10% CO2, and 80% N2) at 37°C. Gingipain mutants of P. gingivalis ATCC 33277, KDP129 (kgp–), KDP133 (rgpA– rgpB–), and KDP136 (kgp– rgpA– rgpB–) were kindly provided by Dr Koji Nakayama of Nagasaki University, Nagasaki, Japan. Wild-type P. gingivalis (ATCC 33277) and the mutant strains were grown anaerobically at 37°C in enriched BHI broth supplemented with 5 μg/mL of hemin and 1 μg/mL of vitamin K, and the following antibiotics, as described previously [Citation6]: chloramphenicol (20 μg/mL), erythromycin (10 μg/mL), and tetracycline (0.7 μg/mL).
Human Oral Keratinocyte-16B (HOK-16B) cells were kindly provided by Dr NH Park, University of California, Los Angeles, CA, and were immortalized by transfecting keratinized gingival epithelium from excised retromolar gingival tissues with human papilloma virus type 16 [Citation23]. The cells were cultured in keratinocyte growth medium containing a supplementary growth factor bullet kit (KGM; Clonetics, San Diego, CA) in a humidified 5% CO2 atmosphere at 37°C.
Intracellular invasion by F. nucleatum
HOK-16B cells (1 × 105 cells/well in 24-well plates) were infected with carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Eugene, OR)–labeled F. nucleatum in the presence or absence of unlabeled P. gingivalis at the indicated multiplicities of infection (MOIs). Invasion by F. nucleatum into the cells was analyzed after 4 h of infection because a previous study [Citation24] reported that the percentage of cells containing F. nucleatum decreased slightly after 5 h of infection. In the preliminary experiments of F. nucleatum monoinfection, the bacterial invasion into the cells increased in a MOI-dependent manner, and a significant increase was observed at a MOI of 500 (data not shown). Accordingly, F. nucleatum at a MOI of 500 was used for coinfection with P. gingivalis. To determine the role of gingipains in the effect of coinfecting P. gingivalis on F. nucleatum invasion, wild-type and gingipain mutant strains of P. gingivalis were used. To examine the effect of inhibition of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), the cells were preincubated with the indicated concentration of wortmannin (Sigma–Aldrich, St. Louis, MO) for 30 min and then infected with CFSE-labeled F. nucleatum at a MOI of 500 in the presence or absence of P. gingivalis.
Flow cytometry
Invasion into HOK-16B cells by F. nucleatum was analyzed by flow cytometry, as described previously [Citation24–Citation26], with a slight modification. After 4 h of infection, the cells were detached with trypsin/ethylenediaminetetraacetic acid, and the extracellular fluorescence from attached but not internalized bacteria was quenched using 400 μg/mL of trypan blue to measure fluorescence signal only from intracellular bacteria. The internalization of F. nucleatum was then analyzed via flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA). After HOK-16B cells were gated based on forward scatter and side scatter of uninfected cells, the fluorescence intensity of cells in fluorescence channel FL1-H (for green fluorescence) was determined. At least 10,000 cells of each group were analyzed and, compared with uninfected cells (a negative control), cells with intracellular F. nucleatum exhibited increased fluorescence intensity. The percentage of cells with intracellular F. nucleatum and the mean fluorescence intensity were determined.
Confocal microscopy
HOK-16B cells (2 × 104 cells/well in 24-well plates) were infected with CFSE-labeled F. nucleatum in the presence or absence of unlabeled P. gingivalis at a MOI of 500 for 4 h. The infected cells were washed with phosphate-buffered saline (PBS; pH 7.4) and fixed with 4% paraformaldehyde. After permeabilization, the actin cytoskeleton and nuclei were stained with rhodamine phalloidin (Sigma–Aldrich) and Hoechst 33342 (Molecular Probes), respectively. Invasion of F. nucleatum into the cells was examined on a confocal laser scanning microscope (LSM 700; Carl Zeiss, Jena, Germany) and ZEN software (Carl Zeiss) with z-sections (10 z-stack images with 1 µm step size) using 405, 488, and 555 nm lasers for Hoechst 33342, CFSE, and rhodamine, respectively.
Immunoblot
HOK-16B cells (5 × 105 cells/well in six-well plates) were infected with P. gingivalis in the presence or absence of F. nucleatum at a MOI of 500 for the indicated time. The cells were washed with cold PBS, harvested, and lysed with radioimmunoprecipitation assay buffer. The lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA). After the membranes were incubated in blocking buffer (Tris-buffered saline containing 0.1% Tween 20 and 5% non-fat dried milk) for 1 h, the blots were probed with the following primary antibodies: rabbit polyclonal anti-phospho-PI3K p85 (Tyr458)/p55 (Tyr199), rabbit polyclonal anti-PI3K p85, rabbit polyclonal anti-phospho-AKT (Ser473), and rabbit polyclonal anti-AKT from Cell Signaling Technology (Beverly, MA), and mouse monoclonal anti-actin from BD Biosciences. The blots were incubated with horseradish peroxidase–conjugated anti-rabbit immunoglobulin G (IgG) and anti-mouse IgG secondary antibodies (R&D Systems, Minneapolis, MN), and binding of the proteins was visualized using ECL Western blotting substrate (SUPEX; Dyne-Bio, Sungnam, Korea).
Intracellular survival
HOK-16B cells (1 × 105 cells/well in 24-well plates) were infected with F. nucleatum in the presence or absence of P. gingivalis at a MOI of 500 for 4 h. The infected cells were washed twice with PBS and incubated in media containing 300 μg/mL of gentamicin and 200 μg/mL of metronidazole for 1 h to kill extracellular bacteria. Then, the cells were washed twice with PBS and further incubated in fresh culture media for 12, 18, 24, and 48 h. At each time point, the cells were lysed with 0.5% saponin in PBS for 10 min at room temperature, and the lysates containing viable F. nucleatum were 10-fold serially diluted and plated on a BHI agar plate containing 5 μg/mL of hemin, 1 μg/mL of vitamin K, 5% sheep blood, and 15.7 μg/mL of vancomycin. In preliminary experiments, 15.7 μg/mL of vancomycin inhibited the growth of wild-type and gingipain mutant strains of P. gingivalis, but the growth of F. nucleatum was not affected. Colonies of F. nucleatum and P. gingivalis on BHI blood agar plates were clearly distinguishable by their color and morphology, and no colony of P. gingivalis was observed on the agar plates containing 15.7 μg/mL of vancomycin. After incubation for 4–5 days under anaerobic conditions, the colonies were counted and expressed as percentages of the number of intracellular F. nucleatum in monoinfection.
Statistical analysis
Experiments were carried out in triplicate and independently repeated at least three times. A standard two-tailed t-test was used for statistical analysis, and p-values of <0.05 were considered significant.
Results
Coinfection with P. gingivalis curtails invasion of F. nucleatum into gingival epithelial cells
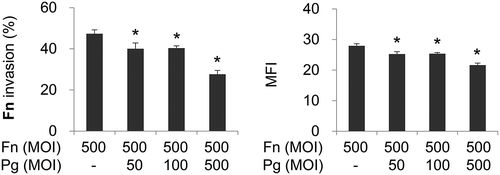
To examine whether P. gingivalis affects invasion of coinfecting F. nucleatum, HOK-16B cells were infected with CFSE-labeled F. nucleatum in the presence or absence of P. gingivalis. In monoinfection, when analyzed by flow cytometry, F. nucleatum invaded into >40% of HOK-16B cells (). This result is consistent with a previous study that reported the high invasive capacity of F. nucleatum into gingival epithelial cells [Citation24]. When coinfected with P. gingivalis, invasion of F. nucleatum into HOK-16B cells significantly decreased in a MOI-dependent manner (). When it was tested whether P. gingivalis also inhibited invasion of other oral bacteria, such as Tannerella forsythia and Streptococcus oralis, invasion of T. forsythia was enhanced by P. gingivalis coinfection, whereas invasion of S. oralis was not affected by the presence of P. gingivalis (Supplementary Fig. 1). These results suggest that P. gingivalis differentially affects the invasion of co-infecting bacteria and that the suppression of F. nucleatum invasion by P. gingivalis might result from specific interactions among F. nucleatum, P. gingivalis, and the gingival epithelial cells.
Figure 1. Porphyromonas gingivalis infection reduces the invasion of coinfecting Fusobacterium nucleatum into gingival epithelial cells. Human Oral Keratinocyte-16B (HOK-16B) cells were infected with carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled F. nucleatum and unlabeled P. gingivalis at the indicated multiplicities of infection (MOIs) for 4 h. The cells were analyzed using flow cytometry after quenching the extracellular fluorescence from bacteria attached to the cell surface with trypan blue. The percentage of cells containing F. nucleatum (left panel) and the mean fluorescence intensity (MFI; right panel) are shown as the mean ± standard deviation. Representative data from three independent experiments are shown. Fn, F. nucleatum; Pg, P. gingivalis. *p < 0.05 compared with F. nucleatum monoinfection.
Gingipains of coinfecting P. gingivalis are required for the suppression of F. nucleatum invasion
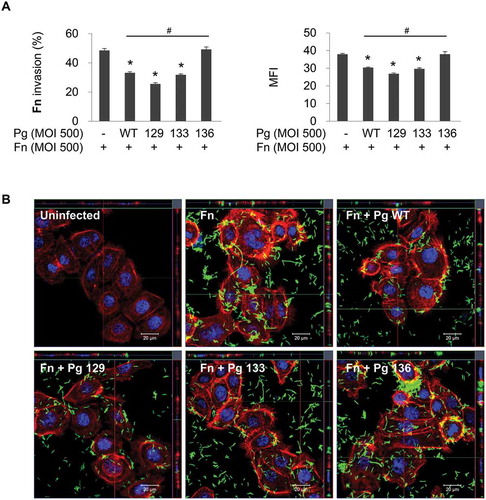
To investigate the role of gingipains in the inhibitory effect of P. gingivalis on F. nucleatum invasion, gingipain mutant strains of P. gingivalis were used for coinfection. Kgp- and Rgp-deficient mutants (KDP129 and KDP133, respectively) exhibited inhibitory effects, similar to the wild-type, on F. nucleatum invasion. However, the gingipain-null mutant (KDP136) did not decrease the invasion of F. nucleatum into HOK-16B cells ()). Confocal microscopy also revealed that deletion of all gingipains abrogates the inhibition of F. nucleatum invasion by P. gingivalis ()). These results indicate that the presence of either Kgp or Rgp in P. gingivalis exerts an inhibitory action on F. nucleatum invasion. In contrast, in HGFs and macrophages, P. gingivalis enhanced F. nucleatum invasion and phagocytosis, respectively, and gingipains contributed to the enhancement (Supplementary Fig. 2), suggesting that the effect of P. gingivalis and gingipains on the internalization of F. nucleatum depends on the cell type.
Figure 2. Gingipain deficiency reverses the suppression of F. nucleatum invasion by P. gingivalis. HOK-16B cells were infected with CFSE-labeled F. nucleatum and unlabeled wild-type P. gingivalis or gingipain mutants at a MOI of 500 for 4 h. (A) Cells were analyzed using flow cytometry after quenching the extracellular fluorescence from the bacteria attached to the cell surface with trypan blue. The percentage of cells containing F. nucleatum (left panel) and the MFI (right panel) are shown as the mean ± standard deviation. Representative data from three independent experiments are shown. (B) The cells were examined under a confocal laser scanning microscope after staining for F-actin (red) and nuclei (blue). CFSE-labeled F. nucleatum is displayed in green. Fn, F. nucleatum; Pg, P. gingivalis; WT, P. gingivalis ATCC 33277; 129, KDP129 (kgp−); 133, KDP133 (rgpA− rgpB−); 136, KDP136 (kgp− rgpA− rgpB−). *p < 0.05 compared with F. nucleatum monoinfection; #p < 0.05 compared with coinfection with wild-type P. gingivalis.
Inhibition of PI3K attenuates F. nucleatum invasion
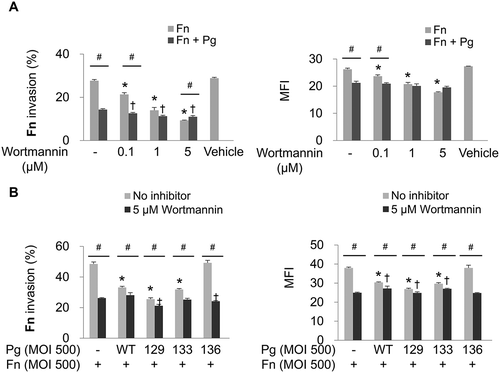
In addition to the numerous cellular functions of the PI3K/AKT signaling pathway, such as cell survival, proliferation, apoptosis, and protein synthesis [Citation27], the pathway has been reported to be involved in the internalization of some pathogens into host cells [Citation28–Citation31]. To determine whether the PI3K signaling pathway is involved in F. nucleatum invasion, HOK-16B cells were infected with F. nucleatum in the presence of wortmannin, a commonly used inhibitor of PI3K. In monoinfection, wortmannin significantly inhibited invasion of F. nucleatum into HOK-16B cells in a dose-dependent manner, whereas when coinfected with wild-type P. gingivalis, the inhibitory effect of wortmannin on F. nucleatum invasion was negligible ()). Upon coinfection with Kgp- or Rgp-deficient P. gingivalis (KDP129 or KDP133, respectively), only a slight decrease in F. nucleatum invasion by wortmannin was observed. However, invasion of F. nucleatum was significantly decreased during coinfection with the gingipain-null mutant (KDP136) to the same degree as the decrease in the monoinfection resuling from wortmannin treatment ()).
Figure 3. Invasion of F. nucleatum is suppressed by PI3K inhibition. (A) HOK-16B cells were preincubated with wortmannin at the indicated concentration for 30 min and then infected with CFSE-labeled F. nucleatum and unlabeled wild-type P. gingivalis at a MOI of 500 for 4 h. The percentage of cells containing F. nucleatum (left panel) and the MFI (right panel) are shown as the mean ± standard deviation. *p < 0.05 compared with cells without preincubation with wortmannin in F. nucleatum monoinfection; †p < 0.05 compared with cells without preincubation with wortmannin in mixed infection with F. nucleatum and P. gingivalis; #p < 0.05 compared between the two groups. (B) HOK-16B cells were preincubated with 5 μM of wortmannin for 30 min and infected with CFSE-labeled F. nucleatum and unlabeled wild-type P. gingivalis or gingipain mutants at a MOI of 500 for 4 h. *p < 0.05 compared with F. nucleatum monoinfection in the absence of wortmannin; †p < 0.05 compared with F. nucleatum monoinfection in the presence of wortmannin; #p < 0.05 compared between the two groups. Representative data from three independent experiments are shown. Fn, F. nucleatum; Pg, P. gingivalis; WT, P. gingivalis ATCC 33277; 129, KDP129 (kgp−); 133, KDP133 (rgpA− rgpB−); 136, KDP136 (kgp− rgpA− rgpB−).
P. gingivalis degrades PI3K and AKT, thereby abrogating basal activation of the PI3K/AKT pathway
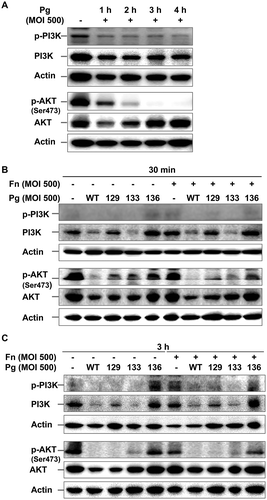
Because the inhibitory effect of wortmannin on F. nucleatum invasion was minute during coinfection with P. gingivalis expressing gingipains, it was hypothesized that P. gingivalis, in particular its gingipains, could affect the PI3K signaling pathway. To test this hypothesis, the phosphorylation status of PI3K and AKT in HOK-16B cells was examined after infection with P. gingivalis. Infection with wild-type P. gingivalis led to a decrement in the level of phosphorylated PI3K, as well as its total form for up to 4 h post infection ()). The level of total AKT level also markedly decreased after 1 h of P. gingivalis infection and recovered 4 h post infection. However, AKT phosphorylation remained abated for 4 h of P. gingivalis infection ()). P. gingivalis–induced decreases in phospho-PI3K, phospho-AKT, total PI3K, and total AKT were observed as early as 30 min after infection with wild-type P. gingivalis ()). The levels of the phosphorylated and total forms of PI3K and AKT were also reduced after both 30 min and 3 h of infection with Kgp- and Rgp-deficient mutants (KDP129 and KDP133, respectively), but not after infection with the gingipain-null mutant (KDP136; ) and (C)). Infection with F. nucleatum did not affect the phosphorylated or total form of PI3K and AKT during monoinfection, and changes in their levels after coinfection with F. nucleatum and P. gingivalis were consistent with monoinfection with P. gingivalis () and (C)).
Figure 4. P. gingivalis inactivates the PI3K/AKT pathway in a gingipain-dependent manner. (A) HOK-16B cells were infected with wild-type P. gingivalis at a MOI of 500 for the indicated time. (B) and (C) HOK-16B cells were infected with wild-type P. gingivalis or gingipain mutants in the presence or absence of F. nucleatum at a MOI of 500 for 30 min (B) or 3 h (C). The levels of phosphorylated and total forms of PI3K and AKT were analyzed by immunoblot analysis. Fn, F. nucleatum; Pg, P. gingivalis; WT, P. gingivalis ATCC 33277; 129, KDP129 (kgp−); 133, KDP133 (rgpA− rgpB−); 136, KDP136 (kgp− rgpA− rgpB−).
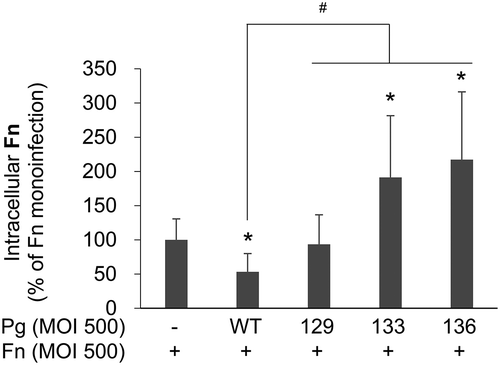
P. gingivalis with Rgp mutation promoted the intracellular survival of F. nucleatum
Next, the study examined whether intracellular survival of F. nucleatum is affected by coinfecting P. gingivalis. Twelve hours after the antibiotic protection, the number of intracellular F. nucleatum in the monoinfection group remained higher than in the group coinfected with wild-type P. gingivalis (). However, after coinfection with Kgp-deficient mutant KDP129, the number of intracellular F. nucleatum was similar to that after monoinfection (). Interestingly, after coinfection with Rgp-deficient mutant KDP133 or the gingipain null mutant KDP136, more F. nucleatum remained viable within the infected cells than after monoinfection (). Considering that KDP129 and KDP133 suppressed F. nucleatum invasion and that during coinfection with KDP 136 F. nucleatum invasion was similar to monoinfection, this result suggests that the intracellular survival of F. nucleatum is facilitated by coinfecting gingipain mutants of P. gingivalis. Few F. nucleatum survived after 18 h of infection, regardless of coinfection with P. gingivalis (data not shown).
Figure 5. More intracellular F. nucleatum remains viable after coinfection with P. gingivalis with Rgp mutation than in monoinfection. After HOK-16B cells were infected with F. nucleatum in the presence or absence of wild-type P. gingivalis or gingipain mutants at a MOI of 500 for 4 h, extracellular bacteria were killed by incubation with antibiotics for 1 h. After incubation in fresh media for 12 h, the cells were lysed, and the lysates were plated on brain heart infusion blood agar plates containing vancomycin. The data represent the mean ± standard deviation of four independent experiments. *p < 0.05 compared with F. nucleatum monoinfection; #p < 0.05 compared between the two groups. Fn, F. nucleatum; Pg, P. gingivalis; WT, P. gingivalis ATCC 33277; 129, KDP129 (kgp−); 133, KDP133 (rgpA− rgpB−); 136, KDP136 (kgp− rgpA− rgpB−).
Discussion
The present study demonstrated that P. gingivalis compromised invasion of coinfecting F. nucleatum into gingival epithelial cells in a gingipain-dependent manner. While PI3K inhibition reduced F. nucleatum invasion into gingival epithelial cells, P. gingivalis infection inactivated the PI3K/AKT pathway. P. gingivalis with Rgp mutation seemed to facilitate the survival of intracellular F. nucleatum.
F. nucleatum invades into gingival epithelial cells via a zipper mechanism [Citation32], and a pre-FadA-mature FadA complex mediates attachment and invasion of F. nucleatum into gingival epithelial cells [Citation20]. Infection of gingival epithelial cells by F. nucleatum increases the expression of antimicrobial peptides and cytokines, such as human beta defensin (HBD)-2, HBD-3, interleukin (IL)-6, and IL-8 [Citation22,Citation24,Citation32,Citation33]. In particular, the induction of HBD-2, HBD-3, and IL-8 production by F. nucleatum depends on bacterial invasion and endosomal maturation [Citation24,Citation34]. After invasion, endosomes containing F. nucleatum rapidly fuse with lysosomes, and intracellular F. nucleatum is cleared via endolysosomal degradation [Citation24].
Due to the polymicrobial nature of the pathogenesis of periodontitis, it is important to understand the polymicrobial interactions as well as the virulence mechanisms of each periodontopathogen. Polymicrobial infections of periodontal pathogens often result in synergistic pathogenicity [Citation35,Citation36]. Mixed infection with F. nucleatum and P. gingivalis has been shown to aggravate alveolar bone loss in a mouse model of experimental periodontitis [Citation36] and abscess formation in a murine abscess model [Citation35]. However, the mechanism of synergism between these two bacterial species remains unclear. A recent study demonstrated that coaggregation of F. nucleatum with a capsulated P. gingivalis strain contributes to the augmented virulence of the mixed infection [Citation37]. Previous studies have shown that P. gingivalis inhibits the induction of HBD-2 and IL-8 expression in gingival epithelial cells by F. nucleatum [Citation22,Citation38,Citation39], which might be partly explained by the finding in the present study that P. gingivalis restricts F. nucleatum invasion. A previous study showed that invasion by P. gingivalis into gingival epithelial cells is significantly inhibited by preceding infection with F. nucleatum [Citation40]. Conversely, it may be possible that P. gingivalis invasion into gingival epithelial cells interferes with cellular pathways required for subsequent invasion by F. nucleatum into the cells, although in the present study sequential infection with P. gingivalis followed by F. nucleatum was not tested. In contrast to the present observation, a recent study reported that mixed infection with P. gingivalis increased F. nucleatum invasion into keratinocytes derived from mouse palatal tissues [Citation37]. The discrepancy might result from differences in the cell types and bacterial strains used in the studies.
Gingipains are essential for the establishment of P. gingivalis infection, as well as for immune subversion by P. gingivalis [Citation6–Citation10]. In the process of P. gingivalis invasion into gingival epithelial cells, gingipains seem to play multiple roles with conflicting effects. A previous study reported that inhibition of P. gingivalis proteases results in reduced P. gingivalis invasion. Rgp is required for maturation of P. gingivalis FimA, a major adhesin for gingival epithelial cells [Citation41]. Additionally, adhesin domains of Kgp and RgpA have been shown to participate in attachment of P. gingivalis to oral epithelial cells [Citation42,Citation43]. On the contrary, it also has been reported that Rgp mutation substantially increases the bacterial invasion, suggesting that Rgp may modulate the bacterial attachment by cleaving cell surface receptors [Citation42,Citation44].
The role of gingipains in the interaction between P. gingivalis and F. nucleatum has not been well studied yet. In multispecies biofilms, a deficiency of gingipains in P. gingivalis does not affect the growth of F. nucleatum [Citation45]. A previous study showed that P. gingivalis gingipains attenuate the F. nucleatum–induced IL-8 response by degrading IL-8 [Citation38]. In the present study, while gingipains contributed to the enhancement of the internalization of F. nucleatum into gingival fibroblasts and macrophages, they were responsible for the inhibition of F. nucleatum invasion into gingival epithelial cells by P. gingivalis. These results suggest that P. gingivalis gingipains differentially affect interactions between F. nucleatum and host cells depending on cell types. While receptors for F. nucleatum in gingival fibroblasts and macrophages remain unknown, it has been demonstrated that E-cadherin in colon epithelial cells and VE-cadherin in endothelial cells are required for FadA-mediated intracellular invasion by F. nucleatum [Citation16,Citation46]. Because gingival epithelial cells, including HOK-16B cells, express E-cadherin [Citation47–Citation49], it is likely that FadA-dependent invasion by F. nucleatum into gingival epithelial cells is also mediated by E-cadherin. Inhibition of F. nucleatum invasion into gingival epithelial cells by P. gingivalis might be attributable to degradation of E-cadherin by P. gingivalis gingipains, which has been previously reported [Citation48]. However, further research is required to explore this possibility. Intracellular survival of F. nucleatum seemed to be increased by P. gingivalis with Rgp mutation but not by wild-type P. gingivalis. Because colocalization of intracellular F. nucleatum with lysosomes was not affected by P. gingivalis coinfection (data not shown), further studies are needed to elucidate how Rgp mutation affects the intracellular fate of coinfecting F. nucleatum.
The PI3K/AKT pathway has been reported to be exploited by a number of pathogens, such as group A streptococcus, group B streptococcus, and T. forsythia, for their internalization into host cells [Citation28–Citation30]. Conversely, it has also been reported that the pathway restricts the establishment of certain bacteria, such as Dr + Escherichia coli [Citation31]. The present result demonstrating a decrease in F. nucleatum invasion caused by wortmannin, a PI3K inhibitor, indicates that invasion of F. nucleatum into gingival epithelial cells also requires the PI3K pathway. In addition, gingipain-expressing P. gingivalis inactivated the PI3K/AKT pathway, a result that is consistent with a previous study [Citation50], and the inhibitory effect of the PI3K inhibitor on F. nucleatum invasion was minute in coinfection with gingipain-expressing P. gingivalis. These findings imply that the suppressive effect of P. gingivalis on F. nucleatum invasion is partially mediated by attenuation of the PI3K/AKT pathway by gingipains. Additional studies will be required to investigate whether the effects of gingipains on other cellular receptors, intracellular pathway, or adhesins of F. nucleatum are involved.
In summary, P. gingivalis suppresses the invasion of coinfecting F. nucleatum into gingival epithelial cells, to which inactivation of the PI3K/AKT pathway by its gingipains might partly contribute. Because P. gingivalis degrades intercellular junction complexes in the epithelium [Citation49], P. gingivalis may guide F. nucleatum toward a paracellular pathway in order to avoid intracellular degradation. Furthermore, by inhibiting and degrading antimicrobial peptides and chemokines [Citation22,Citation38,Citation39], P. gingivalis provides a local environment favorable for the survival of F. nucleatum, which might allow F. nucleatum to invade into deeper periodontal tissues. Further research on this possibility might help us understand how P. gingivalis acts as a keystone pathogen.
Supplementary_materials.zip
Download Zip (4.6 MB)Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT and Future Planning (NRF-2015R1A2A2A01002598). The authors have no conflicts of interest to declare.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed here.
Additional information
Funding
Notes on contributors
Young-Jung Jung
Young-Jung Jung, DDS, PhD, from Seoul National University. Currently works as a postdoctoral researcher at Department of Oral Immunology and Infectious Diseases, School of Dentistry, University of Louisville, KY, USA. Primary research interest is pathogenicity of P. gingivalis and oral polymicrobial infections.
Hye-Kyoung Jun
Hye-Kyoung Jun, PhD from Seoul National University. Currently holds a position as a senior research engineer in OSSTEM IMPLANT Co., Seoul, Korea. Primary research interest lies studying pathogenesis of periodontal diseases.
Bong-Kyu Choi
Bong-Kyu Choi, PhD from Albert-Ludwigs University of Freiburg. Currently holds a position as a Professor at Department of Oral Microbiology and Immunology, School of Dentistry, Seoul National University, Seoul, South Korea. Main research interests include innate immunity against periodontal pathogens, biofilm formation and quorum sensing of oral microbes.
References
- Linden GJ, Lyons A, Scannapieco FA. Periodontal systemic associations: review of the evidence. J Periodontol. 2013;84:1–9.
- Dewhirst FE, Chen T, Izard J, et al. The human oral microbiome. J Bacteriol. 2010;192:5002–9.
- Paster BJ, Boches SK, Galvin JL, et al. Bacterial diversity in human subgingival plaque. J Bacteriol. 2001;183:3770–3783.
- Socransky SS, Haffajee AD, Cugini MA, et al. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144.
- Hajishengallis G, Liang S, Payne MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10:497–506.
- Shi Y, Ratnayake DB, Okamoto K, et al. Genetic analyses of proteolysis, hemoglobin binding, and hemagglutination of Porphyromonas gingivalis. Construction of mutants with a combination of rgpA, rgpB, kgp, and hagA. J Biol Chem. 1999;274:17955–17960.
- O’Brien-Simpson NM, Paolini RA, Hoffmann B, et al. Role of RgpA, RgpB, and Kgp proteinases in virulence of Porphyromonas gingivalis W50 in a murine lesion model. Infect Immun. 2001;69:7527–7534.
- Curtis MA, Aduse Opoku J, Rangarajan M, et al. Attenuation of the virulence of Porphyromonas gingivalis by using a specific synthetic Kgp protease inhibitor. Infect Immun. 2002;70:6968–6975.
- Zenobia C, Hajishengallis G. Porphyromonas gingivalis virulence factors involved in subversion of leukocytes and microbial dysbiosis. Virulence. 2015;6:236–243.
- Potempa J, Banbula A, Travis J. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol 2000. 2000;24:153–192.
- Moore WE, Moore LV. The bacteria of periodontal diseases. Periodontol 2000. 1994;5:66–77.
- Kolenbrander PE, Andersen RN, Blehert DS, et al. Communication among oral bacteria. Microbiol Mol Biol Rev. 2002;66:486–505.
- Kaplan A, Kaplan CW, He X, et al. Characterization of aid1, a novel gene involved in Fusobacterium nucleatum interspecies interactions. Microb Ecol. 2014;68:379–387.
- Park J, Shokeen B, Haake SK, et al. Characterization of Fusobacterium nucleatum ATCC 23726 adhesins involved in strain-specific attachment to Porphyromonas gingivalis. Int J Oral Sci. 2016;8:138–144.
- Kinder SA, Holt SC. Localization of the Fusobacterium nucleatum T18 adhesin activity mediating coaggregation with Porphyromonas gingivalis T22. J Bacteriol. 1993;175:840–850.
- Rubinstein MR, Wang X, Liu W, et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195–206.
- Lee H-R, Jun H-K, Kim H-D, et al. Fusobacterium nucleatum GroEL induces risk factors of atherosclerosis in human microvascular endothelial cells and ApoE(-/-) mice. Mol Oral Microbiol. 2012;27:109–123.
- Ji S, Choi YS, Choi Y. Bacterial invasion and persistence: critical events in the pathogenesis of periodontitis? J Periodontal Res. 2015;50:570–585.
- Yilmaz O, Watanabe K, Lamont RJ. Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cell Microbiol. 2002;4:305–314.
- Xu M, Yamada M, Li M, et al. FadA from Fusobacterium nucleatum utilizes both secreted and nonsecreted forms for functional oligomerization for attachment and invasion of host cells. J Biol Chem. 2007;282:25000–25009.
- Saito A, Inagaki S, Kimizuka R, et al. Fusobacterium nucleatum enhances invasion of human gingival epithelial and aortic endothelial cells by Porphyromonas gingivalis. FEMS Immunol Med Microbiol. 2008;54:349–355.
- Li Y, Guo H, Wang X, et al. Coinfection with Fusobacterium nucleatum can enhance the attachment and invasion of Porphyromonas gingivalis or Aggregatibacter actinomycetemcomitans to human gingival epithelial cells. Arch Oral Biol. 2015;60:1387–1393.
- Park NH, Min BM, Li SL, et al. Immortalization of normal human oral keratinocytes with type 16 human papillomavirus. Carcinogenesis. 1991;12:1627–1631.
- Ji S, Shin JE, Kim YC, et al. Intracellular degradation of Fusobacterium nucleatum in human gingival epithelial cells. Mol Cells. 2010;30:519–526.
- Pils S, Schmitter T, Neske F, et al. Quantification of bacterial invasion into adherent cells by flow cytometry. J Microbiol Methods. 2006;65:301–310.
- Dabija-Wolter G, Cimpan M-R, Costea DE, et al. Fusobacterium nucleatum enters normal human oral fibroblasts in vitro. J Periodontol. 2009;80:1174–1183.
- Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4:a011189.
- Burnham CA, Shokoples SE, Tyrrell GJ. Invasion of HeLa cells by group B streptococcus requires the phosphoinositide-3-kinase signalling pathway and modulates phosphorylation of host-cell Akt and glycogen synthase kinase-3. Microbiology. 2007;153:4240–4252.
- Mishima E, Sharma A. Tannerella forsythia invasion in oral epithelial cells requires phosphoinositide 3-kinase activation and clathrin-mediated endocytosis. Microbiology. 2011;157:2382–2391.
- Wang B, Yurecko RS, Dedhar S, et al. Integrin-linked kinase is an essential link between integrins and uptake of bacterial pathogens by epithelial cells. Cell Microbiol. 2006;8:257–266.
- Banadakoppa M, Goluszko P, Liebenthal D, et al. PI3K/Akt pathway restricts epithelial adhesion of Dr + Escherichia coli by down-regulating the expression of decay accelerating factor. Exp Biol Med (Maywood). 2014;239:581–594.
- Han YW, Shi W, Huang GT, et al. Interactions between periodontal bacteria and human oral epithelial cells: fusobacterium nucleatum adheres to and invades epithelial cells. Infect Immun. 2000;68:3140–3146.
- Hasegawa Y, Mans JJ, Mao S, et al. Gingival epithelial cell transcriptional responses to commensal and opportunistic oral microbial species. Infect Immun. 2007;75:2540–2547.
- Kim Y, Jo A-R, Jang DH, et al. Toll-like receptor 9 mediates oral bacteria-induced IL-8 expression in gingival epithelial cells. Immunol Cell Biol. 2012;90:655–663.
- Metzger Z, Lin Y-Y, Dimeo F, et al. Synergistic pathogenicity of Porphyromonas gingivalis and Fusobacterium nucleatum in the mouse subcutaneous chamber model. J Endod. 2009;35:86–94.
- Polak D, Wilensky A, Shapira L, et al. Mouse model of experimental periodontitis induced by Porphyromonas gingivalis/Fusobacterium nucleatum infection: bone loss and host response. J Clin Periodontol. 2009;36:406–410.
- Polak D, Ferdman O, Houri-Haddad Y. Porphyromonas gingivalis capsule-mediated coaggregation as a virulence factor in mixed infection with Fusobacterium nucleatum. J Periodontol. 2016;1–15.
- Huang GT, Kim D, Lee JK, et al. Interleukin-8 and intercellular adhesion molecule 1 regulation in oral epithelial cells by selected periodontal bacteria: multiple effects of Porphyromonas gingivalis via antagonistic mechanisms. Infect Immun. 2001;69:1364–1372.
- Darveau RP, Belton CM, Reife RA, et al. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun. 1998;66:1660–1665.
- Saito A, Kokubu E, Inagaki S, et al. Porphyromonas gingivalis entry into gingival epithelial cells modulated by Fusobacterium nucleatum is dependent on lipid rafts. Microb Pathog. 2012;53:234–242.
- Nakayama K, Yoshimura F, Kadowaki T, et al. Involvement of arginine-specific cysteine proteinase (Arg-gingipain) in fimbriation of Porphyromonas gingivalis. J Bacteriol. 1996;178:2818–2824.
- Chen T, Nakayama K, Belliveau L, et al. Porphyromonas gingivalis gingipains and adhesion to epithelial cells. Infect Immun. 2001;69:3048–3056.
- Boisvert H, Duncan MJ. Clathrin-dependent entry of a gingipain adhesin peptide and Porphyromonas gingivalis into host cells. Cell Microbiol. 2008;10:2538–2552.
- Suwannakul S, Stafford GP, Whawell SA, et al. Identification of bistable populations of Porphyromonas gingivalis that differ in epithelial cell invasion. Microbiology. 2010;156:3052–3064.
- Bao K, Belibasakis GN, Thurnheer T, et al. Role of Porphyromonas gingivalis gingipains in multi-species biofilm formation. BMC Microbiol. 2014;14:258.
- Fardini Y, Wang X, Témoin S, et al. Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol. 2011;82:1468–1480.
- Lee G, Kim HJ, Kim H-M. RhoA-JNK regulates the E-cadherin junctions of human gingival epithelial cells. J Dent Res. 2016;95:284–291.
- Katz J, Yang Q-B, Zhang P, et al. Hydrolysis of epithelial junctional proteins by Porphyromonas gingivalis gingipains. Infect Immun. 2002;70:2512–2518.
- Katz J, Sambandam V, Wu JH, et al. Characterization of Porphyromonas gingivalis-induced degradation of epithelial cell junctional complexes. Infect Immun. 2000;68:1441–1449.
- Nakayama M, Inoue T, Naito M, et al. Attenuation of the phosphatidylinositol 3-kinase/Akt signaling pathway by Porphyromonas gingivalis gingipains RgpA, RgpB, and Kgp. J Biol Chem. 2015;290:5190–5202.