
ABSTRACT
Background: Ticks are primary vectors for many well-known disease-causing agents that affect human and animal populations globally such as tick-borne encephalitis, Crimean-Congo hemorrhagic fever and African swine fever. In this study, viral metagenomics was used to identify what viruses are present in Rhipicephalus spp. ticks collected in the Zambezi Valley of Mozambique.
Methods: The RNA was amplified with sequence-independent single primer amplification (SISPA) and high-throughput sequencing was performed on the Ion Torrent platform. The generated sequences were subjected to quality check and classfied by BLAST. CodonCode aligner and SeqMan were used to assemble the sequences.
Results: The majority of viral sequences showed closest sequence identity to the Orthomyxoviridae family, although viruses similar to the Parvoviridae and Coronaviridae were also identified. Nearly complete sequences of five orthomyxoviral segments (HA, NP, PB1, PB2, and PA) were obtained and these showed an amino acid identity of 32–52% to known quaranjaviruses. The sequences were most closely related to the Wellfleet Bay virus, detected and isolated from common eider during a mortality event in the USA.
Conclusions: In summary, this study has identified a highly divergent virus with in the Orthomyxoviridae family associated with Rhipicephalus ticks from Mozambique. Further genetic and biological studies are needed in order to investigate potential pathogenesis of the identified orthomyxovirus.
Introduction
Arthropods can act as biological vectors that transmit infectious agents and thereby cause diseases in humans and animals. After mosquitoes, ticks are the most common arthropod vector for viruses, bacteria, and other parasites causing different vector-borne diseases. Ticks from different parts of the world have been shown to carry viruses belonging to, for example, the Bunyaviridae, Flaviviridae, Asfaviridae and Orthomyxoviridae families [Citation1]. Additionally, ticks carry many non-pathogenic microbes, and some of these microbes have formed symbiotic relationships with their hosts and transmit by vertical transmission [Citation2–Citation5].
The genus Rhipicephalus, belonging to the Ixodidae family (family of hard ticks), is widely distributed worldwide and is considered a potential vector of several emerging pathogens. Different studies have shown that viral pathogens, such as Thogoto viruses, Wad Medani virus, Nairobi sheep disease virus, Crimean-Congo hemorrhagic fever virus, African swine fever virus and Tick-borne encephalitis virus [Citation1,Citation6–Citation8], can be found in Rhipicephalus ticks. Borrelia, Anaplasma, Rickettsia, Ehrlichia, and Babesia are a few examples of the pathogenic bacteria that have been identified in these types of ticks [Citation9,Citation10].
With the advent of metagenomic approaches, the limitations of culture-based methods have been overcome and have enabled the characterization of the entire microbiota associated with the host. Numerous studies have used metagenomics to explore viral communities in different arthropod species and have in these identified viruses associated with a broad range of animals, plants and insects. Many of the identified viruses have been novel, for example, highly divergent viruses belonging to nairoviruses and phleboviruses were reported in Amblyomma and Ixodes ticks from the USA [Citation11–Citation13] and in Rhipicephalus ticks [Citation14].
Previous reports indicate that many tick-borne disease-causing agents exist in Mozambique, such as Nairobi sheep disease virus, Theleiria and Anaplasma [Citation15,Citation16]. Since ticks are known vectors for potentially pathogenic and non-pathogenic viruses and the virome of ticks is understudied in this region, the current study used viral metagenomics to identify viruses associated with Rhipicephalus ticks collected in the Zambezi Valley of Mozambique.
Material and methods
Tick collection and identification
Fifty-one adult ticks were collected in October 2014 from small ruminants in the Cuacua village of the Zambezia province in central Mozambique (Geographical coordinates S 17°48.043ʹ E 035°24.730ʹ). Tick collection was carried out on private land with the permission from the landowner and the local farmers. The collected ticks were stored in RNAlater (Invitrogen), transported to the laboratory, and stored at −80°C until further use. Individual ticks were identified morphologically to determine the genus; however, the sex of the ticks was not determined. The ticks were surface-sterilized with 95% ethanol and rinsed twice in water before nucleic acid extraction.
Nucleic acid extraction
Each tick pool (up to 3 ticks/pool) was mechanically homogenized using the Tissuelyser II (Qiagen) for 30 cycles/sec with 1 ml of TRIzol LS reagent (Invitrogen) and two 5 mm stainless steel beads. The supernatant was collected after centrifugation at 13,000 x g for 10 min at 4°C. Total RNA was extracted from the homogenate according to the manufacturer’s instructions, and the RNA pellet was dissolved in 40 µl of nuclease-free water. After extracting RNA from the aqueous phase of the TRIzol homogenate, the remaining slurry containing the interphase/organic phase was saved at −80°C. Next, 5 µl of RNA from each tube was combined into a single pool, and the RNA was treated with DNase from the RNase-free DNase set (Qiagen) and purified with the RNeasy MinElute Cleanup kit (Qiagen). Ribosomal RNA was depleted using the RiboZero kit (RiboZero Gold Human/Mouse/Rat, Illumina) according to the manufacturer’s protocol, and the RNA was again concentrated using the RNeasy MinElute Cleanup kit (Qiagen).
cDNA labeling, amplification and sequencing
First strand cDNA synthesis, followed by labeling of cDNA and random amplification, was performed with 10 µl of RNA, as described by Cholleti et al. 2016 [Citation17]. The final amplified product were submitted to the National Genomics Centre (SciLife Lab, Uppsala, Sweden) for library preparation and sequencing. High-throughput sequencing was performed with the Ion Torrent PGM sequencing platform using an Ion 318TM chip (v2) and 400 bp read length chemistry. The raw sequencing data are accessible through NCBI’s Sequence Read Archive, SRA: SRP109282.
Sequence processing and taxonomy assignment
The sequences produced from the Ion Torrent platform were quality checked by filtering reads with low quality scores (Q < 20), removing exact duplicate reads and trimming the ends of the reads with PRINSEQ [Citation18]. Good quality reads were mapped to different tick genomes Ixodes scapularis (GenBank assembly accession no. GCF_000208615.1) and Rhipicephalus sanguineus complete mitochondrial genome (GenBank accession no. NC_002074.1), using the default settings of Bowtie2 [Citation19]. Unmapped reads were subjected to BLAST searches querying against NCBI nucleotide (nt) and protein sequence (nr) databases with an e-value cutoff of 1e-03. Reads that were classified as viruses were further assembled to generate longer sequences using the de novo assembler in CodonCode Aligner 6.02 (CodonCode Corporation) and SeqMan 11.2.1 (DNASTAR).
Confirmation of sequences and recovery of genomic ends
The viral contigs were used as a reference for designing specific primers to confirm and analyze the sequences of terminal ends, performed by the rapid amplification of cDNA ends (RACE). All of the primers used in this study are listed in Supplementary Table S2. PCR amplification was performed at the following conditions: 95°C for 10 min, followed by 35 cycles of 95°C for 30 sec, 55–60°C for 30 sec, 72°C for 1 min and finally holding at 72°C for 7 min. RACE products were purified with a GeneJet PCR purification kit (ThermoFisher Scientific) and sequenced at Macrogen Europe (Macrogen Inc.).
Phylogenetic analysis
The phylogenetic tree was constructed using amino acid sequences of selected viruses. For this, viral sequences were downloaded from GenBank and multiple sequence alignment (MSA) was performed using ClustalW with default parameters. The gaps and missing data were eliminated prior to phylogenetic analysis and the tree was generated using maximum likelihood (ML) method using MEGA 7 (version 7.0.26). The statistical significance of the tree topologies was evaluated by 1000 bootstrap replicates.
Results
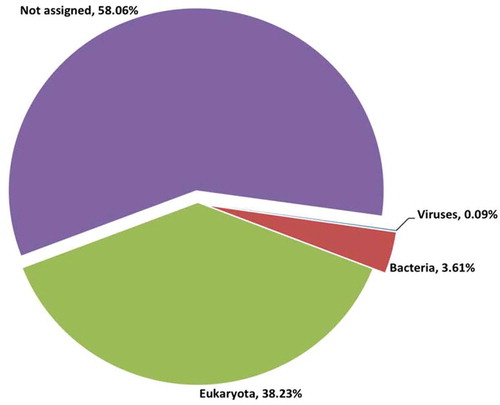
The ticks were morphologically identified to the genus level, and it was determined that all of the collected ticks belonged to the genus Rhipicephalus. Ion Torrent sequencing of the viral RNA metagenome produced 5.4 million reads. After quality control, 88.3% of the reads were classified as good quality, with an average read length of 261 bp. The reads were mapped to two tick genomes (Ixodes and Rhipicephalus), which excluded 5.6% of the reads (). To perform taxonomic profiling of the tick viral metagenome, unmapped reads were classified by BLASTn and BLASTx searches. The majority of the reads were found to be unassigned (58%), and 38% of the reads were eukaryotic sequences derived primarily from the arthropod genome (). The proportion of reads that were derived from bacteria was 3.6% (163,025 reads), and only 0.09% (4092 reads) were derived from viruses. The viral reads were categorized at the family level. These viral reads corresponded to 7 different viral families and 2 other viral groups, including unclassified dsDNA viruses and environmental samples ().
Table 1. Sample information, quality checks and host mapping of reads.
Table 2. Number of reads belonging to each viral family.
Figure 1. Taxonomic profiling of reads from Rhipicephalus spp. ticks.
Orthomyxoviridae family
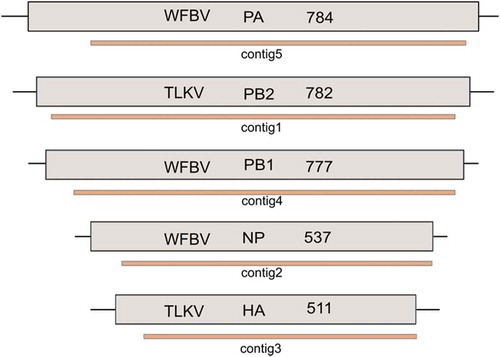
In total, 4008 reads (98% of all viral reads) were classified within the Orthomyxoviridae family (). At the amino acid level, the reads showed closest similarity to members of the Quaranjavirus genus, such as the Wellfleet Bay virus (WFBV), Tjuloc vius (TLV), Quaranfil virus (QRFV), Johnstol Atoll virus (JAV) and unclassified quaranjaviruses. No significant similarities were observed at the nucleotide level. Assembly of these reads generated 6 contigs with high sequencing depth and lengths ranging from 218 to 2353 nucleotides (nt) (). BLAST searches of these contigs revealed that they show similarities to the nearly full-length segments of Wellfleet Bay virus and Tjuloc virus ORFs: Hemagglutinin (HA), Nucleoprotein (NP), Polymerase basic 1 protein (PB1), Polymerase basic 2 protein (PB2), and Polymerase Acidic protein (PA) (). However, the identified ORFs exhibit high genetic diversity to known quaranjavirus genomes, with an amino acid identity of only 32–55%, indicating that these represent novel viral sequences belonging to the Quaranjavirus genus. One of the contigs (contig6, 218 nt long) showed an amino acid identity of 55% to the NP protein of WFBV; however, it did not assemble with the other NP-classified reads in the data set. The BLAST hit results for this contig included a hypothetical protein of the Tjuloc virus and Quaranfil virus with an identity of 32%. This contig could, therefore, be a matrix protein or another viral protein divergent from previously known quranjaviral segments. The 3′ UTR sequences of the HA and NP sequences were recovered using RACE analysis. The 3′ UTR could not be recovered from the other segments, nor could any 5′ UTR be recovered.
Table 3. Orthomyxoviridae family reads assembly, BLAST search and coverage.
Figure 2. Schematic representation of the contigs covering different segments of the quaranjavirus. The name of the reference virus, segment and length of the ORFs (in amino acids) are shown in boxes with black lines (top). Contigs that are aligned to each segment are represented as color-shaded thick lines (bottom).
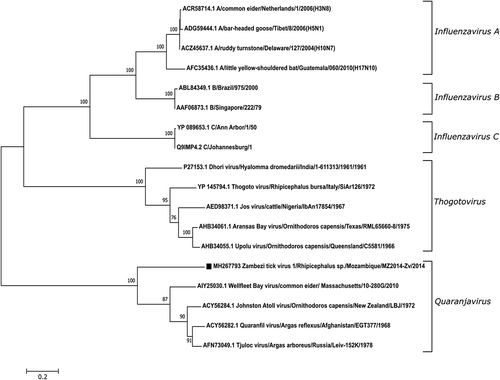
Evolutionary relationships was analyzed using PB1 as it is considered the most conserved of the orthomyxovirus genes. The phylogenetic analysis of the PB1 protein sequences, including Influenza A, B, C, Thogotovirus and Quaranjavirus, showed that PB1 from the novel quaranjavirus, identified in this study, formed a separate branch in the quaranjavirus group and was most closely related to WFBV (with 100% bootstrap support) ().
Figure 3. Phylogenetic analysis of novel quaranjavirus with other viruses in the Orthomyxoviridae family. The phylogeny consists of PB1 protein sequences from 19 different viruses including the novel quaranjavirus from the current study. A total of 664 amino acid positions were used to build the tree and bootstrap values >60% are displayed. The PB1 nucleotide sequence (contig4) from this study has been submitted to GenBank as Zambezi tick virus 1 (ZaTV-1) under the accession number MH267793. The GenBank accession number, virus name, host, location, strain and year are shown for each virus used in the analysis.
Other viral families
Sequences similar to Parvoviridae, Retroviridae, Coronaviridae, unclassified dsRNA viruses and environmental samples were also identified in the ticks (). Assembly of the sequences belonging to the Parvoviridae family generated 5 contigs ranging between 354–870 nt. These contigs showed an amino acid identity (26–62%) to the non-structural protein 1 of Cherax quadricarinatus densovirus (accession no. YP009134732.1), Lonestar tick densovirus 1 (ASU47551.1) and Ambidensovirus CaaDV1 (AR146481.1). A single contig (448 nt) was generated from the assembly of the coronavirus reads. This contig showed the closest identity to ORF1a of Duck coronavirus (AKF17723.1); however, the similarity at the amino acid level was only 24% (Supplementary Table S1). Sequences classified as Retroviridae family showed low sequence identity (25–35%) to known viral proteins (polymerase of Feline foamy virus, gag protein of human immune deficiency virus 1 and Simian immune deficiency virus) and it was not possible to assemble them into longer contigs, possibly due to the absence of overlaps or high sequence diversity.
Discussion
In this study, a metagenomic approach was used to identify viruses associated with Rhipicephalus ticks in the Zambezi Valley of Mozambique where the surveillance of tick-associated pathogens is limited. To the best of our knowledge, this is the first study where high-throughput sequencing have been used to explore the viruses present in ticks from Mozambique. The majority of the viral sequences belonged to the Orthomyxoviride family and was therefore studied in more detail; however, other viral families such as Parvoviridae and Coronaviridae were also identified. Many of these viral sequences showed very low sequence identity to known viruses, indicating that they most likely correspond to novel viruses.
The Orthomyxoviridae family consists of six recognized genera: Influenza A, B, and C, Isavirus, Thogotovirus, Quaranjavirus and the newly proposed Influenza D. Viruses in this family infect a broad range of hosts including humans, birds, swine, fish and arthropods, and transmission occurs by different routes such as fecal-oral, water, air, and direct contact [Citation20]. The genus Quaranjavirus was approved by ICTV in 2011 and included two new species, Quaranfil virus (QRFV) and Johnston Atoll virus (JAV), as well as a tentative member, Lake Chad virus (LKCV). These viruses were isolated in the 1950s and 1960s but were only recently recognized as quaranjaviruses. They appear to be mainly associated with ticks (QRFV and JAV) and birds (LKCV) [Citation21–Citation24]. In the present study, novel viral sequences showing sequence identity to quaranjaviruses were characterized from Rhipicephalus spp. ticks in Mozambique using high-throughput sequencing. These sequences are highly divergent from all known quaranjaviruses, with approximately 50% amino acid identity. BLASTx search analyses show that these sequences are most closely related to WFBV and TLV. In the phylogeny, the PB1 protein sequence from this study clustered with quaranjaviruses and showed closest relationship to WFBV. At this point, we are not sure if the sequences found in this study belong to one, two or more viruses. Future studies are needed to show this. The mechanism of transmission is uncertain, but it is speculated that soft and hard ticks are the primary vectors [Citation25,Citation26]. Several studies have identified quaranjaviruses in other arthropod vectors, such as spiders, mosquitoes and horseflies [Citation27]. Little is known about the molecular pathogenesis of quaranjaviruses. The viruses in this genus are globally distributed throughout the Middle East, Africa and Pacific regions.
Parvoviruses are associated with a variety of chronic diseases in humans and animals. However, these viral sequences have the ability to integrate into the chromosomal DNA of a wide range of hosts and may transmit to offspring. The parvovirus sequences identified in the current study had closest similarity to non-structural protein 1 of different densoviruses, which were shown previously to integrate into tick genomes such as in Ixodes, Amblyomma and Rhipicephalus genera [Citation28,Citation29]. We assume that these sequences are either endogenous viral elements of the tick genome or DNA from densoviruses remaining in the RNA due to incomplete DNase treatment during the sample preparation step.
Ticks can be co-infected with different bacteria and viruses and may thus be involved in co-transmission of different pathogens, which may enhance disease severity as reported previously [Citation30]. In contrast, endosymbionts may interact with arthropod-borne pathogens and play a crucial role in the fitness of the arthropod vector. For instance, Coxiella endosymbionts of Amblyomma ticks impair the transmission of Ehrlichia chaffiensis [Citation31]. Further, in another important arthropod vector, the mosquito, the Wolbachia symbiont can limit the replication of several pathogenic viruses such as Dengue virus, yellow fever virus, West Nile virus and different protozoans such as Plasmodium [Citation32–Citation34]. It has also been demonstrated that pathogens may influence the microbiome of arthropod vectors by modulating immune responses for its survival and infection [Citation35]. Thus, characterizing the microbiome of ticks and other arthropods is just the first step to understanding the complex interactions between the different microorganisms that reside within the vector.
In summary, we have identified a novel and highly divergent orthomyxovirus in Rhipicephalus spp. ticks in Mozambique. This study constitutes the initial step towards a more comprehensive understanding of the viruses circulating in ticks. Additional investigations are warranted on the functional and ecological aspects of the identified orthomyxovirus. This increased understanding of the tick-borne viruses highlights the role in emergence and transmission of disease-causing agents.
Supplemental Material
Download Zip (57 KB)Acknowledgments
The authors would like to acknowledge the support of the National Genomics Infrastructure (NGI)/Uppsala Genome Center and UPPMAX for providing assistance in massive parallel sequencing and computational infrastructure. Work performed at NGI/Uppsala Genome Center has been funded by RFI/VR and Science for Life Laboratory, Sweden. The authors also thank SLU Global Bioinformatics Centre, Swedish University of Agricultural Sciences, Uppsala for providing computational time for the data analysis. The authors would also like to thank the Swedish VR (SWE-2012-138) and FORMAS (2012-586) for financial support to conduct this study. The funder had no role in the study design, data collection and analysis, decision to publish, or reprinting of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data that support the findings will be available at NCBI Sequence Read Archive (SRA) with accession number: SRP109282.
Supplementary material
Supplementary data can be accessed here
Additional information
Funding
Notes on contributors
Harindranath Cholleti
Harindranath Cholleti he received bachelor degree in Biotechnology from Jawaharlal Nehru Technological University, India and MSc degree from Uppsala University. He is currently a PhD student at the Department of Biomedical Sciences and Veterinary Public Health, Swedish University of Agricultural Sciences, Sweden.
Juliette Hayer
Juliette Hayer she received master degree in Bioinformatics, Structural Biochemistry and Genomics and a PhD in Bioinformatics. She is currently a researcher at the Department of Animal Breeding and Genetics, Swedish University of Agricultural Sciences, Sweden.
Fernando Chanisso Mulandane
Fernando Chanisso Mulandane he is currently a PhD student at the Division of Molecular Diagnostics and Epidemiology, Biotechnology Center, Eduardo Mondlane University, Mozambique.
Kerstin Falk
Kerstin Falk she received her PhD and is currently an Associate Professor at the Karolinska Institute, Sweden.
Jose Fafetine
José Manuel Fafetine he received his bachelor degree in Veterinary Medicine from the Eduardo Mondlane University, Mozambique and his MSc degree in Tropical Veterinary Medicine from the Free University of Berlin, Germany. He obtained his PhD at the Faculty of Veterinary Science, University of Pretoria, South Africa. He is currently an Associate Professor at the Department of Para-Clinical, Veterinary Faculty and Biotechnology Center of the Eduardo Mondlane University, Mozambique.
Mikael Berg
Mikael Berg he received his bachelor degree in Microbiology at Uppsala University, Sweden. He is specialized in Veterinary Virology, and obtained his PhD from Swedish University of Agricultural Sciences. He is currently Professor in Veterinary Virology at the Department of Biomedical Sciences and Veterinary Public Health, Swedish University of Agricultural Science, Sweden.
Anne-Lie Blomström
Anne-Lie Blomström she received her master degree in Biology and PhD from the Swedish University of Agricultural Sciences. She is currently a researcher and Associate Professor in Molecular Virology at the Department of Biomedical Sciences and Veterinary Public Health, Swedish University of Agricultural Sciences, Sweden.
References
- Labuda M, Nuttall PA. Tick-borne viruses. Parasitology. 2004;129(Suppl): 1–8. PubMed PMID: 15938513.
- Ahantarig A, Trinachartvanit W, Baimai V, et al. Hard ticks and their bacterial endosymbionts (or would be pathogens). Folia Microbiol (Praha). 2013 Sep;58(5):419–8. PubMed PMID: 23334948.
- Clayton KA, Gall CA, Mason KL, et al. The characterization and manipulation of the bacterial microbiome of the rocky mountain wood tick, dermacentor andersoni. Parasit Vectors. 2015 Dec 10;(8):632. PubMed PMID: 26653035; PubMed Central PMCID: PMC4674957.
- Narasimhan S, Fikrig E. Tick microbiome: the force within. Trends Parasitol. 2015 Jul;31(7):315–323. PubMed PMID: 25936226; PubMed Central PMCID: PMC4492851.
- Machado-Ferreira E, Vizzoni VF, Balsemao-Pires E, et al. Coxiella symbionts are widespread into hard ticks. Parasitol Res. 2016 Dec;115(12):4691–4699. PubMed PMID: 27595990.
- Albanese M, Bruno-Smiraglia C, Di di Cuonzo G, et al. Isolation of thogoto virus from Rhipicephalus bursa ticks in western sicily. Acta Virol. 1972 May;16(3):267. PubMed PMID: 4680821.
- Charrel RN, Attoui H, Butenko AM, et al. Tick-borne virus diseases of human interest in Europe. Clin Microbiology Infection: Official Publication Eur Soc Clin Microbiol Infect Dis. 2004 Dec;10(12):1040–1055. PubMed PMID: 15606630.
- Hubalek Z, Rudolf I. Tick-borne viruses in Europe. Parasitol Res. 2012 Jul;111(1):9–36. PubMed PMID: 22526290.
- Wang YZ, Mu LM, Zhang K, et al. A broad-range survey of ticks from livestock in Northern Xinjiang: changes in tick distribution and the isolation of Borrelia burgdorferi sensu stricto. Parasit Vectors. 2015 Sep 04;(8):449. PubMed PMID: 26337627; PubMed Central PMCID: PMC4560164.
- Hartelt K, Oehme R, Frank H, et al. Pathogens and symbionts in ticks: prevalence of anaplasma phagocytophilum (Ehrlichia sp.), wolbachia sp., Rickettsia sp., and Babesia sp. in Southern Germany. Intl J Med Microbiol: IJMM. 2004 Apr;293(Suppl 37):86–92. PubMed PMID: 15146989.
- Tokarz R, Williams SH, Sameroff S, et al. Virome analysis of amblyomma americanum, dermacentor variabilis, and ixodes scapularis ticks reveals novel highly divergent vertebrate and invertebrate viruses. J Virol. 2014 Oct;88(19):11480–11492. PubMed PMID: 25056893; PubMed Central PMCID: PMC4178814.
- Sakamoto JM, Ng TFF, Suzuki Y, et al. Bunyaviruses are common in male and female Ixodes scapularis ticks in central Pennsylvania. PeerJ. 2016;4:e2324. PubMed PMID: 27602290; PubMed Central PMCID: PMC4991847
- Carpi G, Cagnacci F, Wittekindt NE, et al. Metagenomic profile of the bacterial communities associated with Ixodes ricinus ticks. PLoS One. 2011;6(10):e25604. PubMed PMID: 22022422; PubMed Central PMCID: PMC3192763
- Xia H, Hu C, Zhang D, et al. Metagenomic profile of the viral communities in Rhipicephalus spp. ticks from Yunnan, China. PLoS One. 2015;10(3):e0121609. PubMed PMID: 25799057; PubMed Central PMCID: PMC4370414
- Norval RA, Lawrence JA, Young AS, et al. Theileria parva: influence of vector, parasite and host relationships on the epidemiology of theileriosis in southern Africa. Parasitology. 1991 Jun;102(Pt 3):347–356. PubMed PMID: 1907728.
- Machado RZ, Teixeira MM, Rodrigues AC, et al. Molecular diagnosis and genetic diversity of tick-borne Anaplasmataceae agents infecting the African buffalo syncerus caffer from marromeu reserve in Mozambique. Parasit Vectors. 2016 Aug 17;(9):454. PubMed PMID: 27531003; PubMed Central PMCID: PMC4987998.
- Cholleti H, Hayer J, Abilio AP, et al. Discovery of novel viruses in mosquitoes from the Zambezi valley of Mozambique. PLoS One. 2016;11(9):e0162751. PubMed PMID: 27682810; PubMed Central PMCID: PMC5040392
- Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011 Mar 15;27(6):863–864. PubMed PMID: 21278185; PubMed Central PMCID: PMC3051327.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012 Mar 04;9(4):357–359. PubMed PMID: 22388286; PubMed Central PMCID: PMC3322381.
- Webster RG, Bean WJ, Gorman OT, et al. Evolution and ecology of influenza A viruses. Microbiol Rev. 1992 Mar;56(1):152–179. PubMed PMID: 1579108; PubMed Central PMCID: PMC372859.
- Presti RM, Zhao G, Beatty WL, et al. Quaranfil, Johnston atoll, and lake chad viruses are novel members of the family orthomyxoviridae. J Virol. 2009 Nov;83(22):11599–11606. PubMed PMID: 19726499; PubMed Central PMCID: PMC2772707.
- Taylor RM, Hurlbut HS, Work TH, et al. Arboviruses isolated from ARGAS TICKS IN Egypt: quaranfil, Chenuda, and Nyamanini. Am J Trop Med Hyg. 1966 Jan;15(1):76–86. PubMed PMID: 5901633.
- Kemp GE, Lee VH, Moore DL. Isolation of Nyamanini and Quaranfil viruses from Argas (Persicargas) arboreus ticks in Nigeria. J Med Entomol. 1975 Dec 30;12(5):535–537. PubMed PMID: 1223303.
- Clifford CM, Thomas LA, Hughes LE, et al. Identification and comparison of two viruses isolated from ticks of the genus Ornithodoros. Am J Trop Med Hyg. 1968 Nov;17(6):881–885. PubMed PMID: 4973055.
- Allison AB, Ballard JR, Tesh RB, et al. Cyclic avian mass mortality in the northeastern USA is associated with a novel orthomyxovirus. J Virol. 2015 Jan 15;89(2):1389–1403. PubMed PMID: 25392223; PubMed Central PMCID: PMC4300652.
- Kessell A, Hyatt A, Lehmann D, et al. Cygnet River virus, a novel orthomyxovirus from ducks, Australia. Emerg Infect Dis. 2012 Dec;18(12):2044–2046. PubMed PMID: 23171630; PubMed Central PMCID: PMC3557875.
- Li CX, Shi M, Tian JH, et al. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife. 2015 Jan 29;4. DOI:10.7554/eLife.05378 PubMed PMID: 25633976; PubMed Central PMCID: PMC4384744.
- Francois S, Filloux D, Roumagnac P, et al. Discovery of parvovirus-related sequences in an unexpected broad range of animals. Sci Rep. 2016 Sep 07;(6):30880. PubMed PMID: 27600734; PubMed Central PMCID: PMC5013282.
- Liu H, Fu Y, Xie J, et al. Widespread endogenization of densoviruses and parvoviruses in animal and human genomes. J Virol. 2011 Oct;85(19):9863–9876. PubMed PMID: 21795360; PubMed Central PMCID: PMC3196449.
- Krause PJ, Telford SR 3rd, Spielman A, et al. Concurrent lyme disease and babesiosis: evidence for increased severity and duration of illness. Jama. 1996;275(21):1657–1660.
- Klyachko O, Stein BD, Grindle N, et al. Localization and visualization of a coxiella-type symbiont within the lone star tick, Amblyomma americanum. Appl Environ Microbiol. 2007 Oct;73(20):6584–6594. PubMed PMID: 17720830; PubMed Central PMCID: PMC2075054.
- Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, et al. A wolbachia symbiont in aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell. 2009 Dec 24;139(7):1268–1278. PubMed PMID: 20064373.
- van den Hurk AF, Hall-Mendelin S, Pyke AT, et al. Impact of Wolbachia on infection with chikungunya and yellow fever viruses in the mosquito vector Aedes aegypti. PLoS Negl Trop Dis. 2012;6(11):e1892. PubMed PMID: 23133693; PubMed Central PMCID: PMC3486898
- Paradkar PN, Trinidad L, Voysey R, et al. Secreted vago restricts West Nile virus infection in culex mosquito cells by activating the Jak-STAT pathway. Proc Natl Acad Sci U S A. 2012 Nov 13;109(46):18915–18920. PubMed PMID: 23027947; PubMed Central PMCID: PMC3503207.
- Abraham NM, Liu L, Jutras BL, et al. Pathogen-mediated manipulation of arthropod microbiota to promote infection. Proc Natl Acad Sci U S A. 2017 Jan 31;114(5):E781–E790. PubMed PMID: 28096373; PubMed Central PMCID: PMC5293115.