
ABSTRACT
The mosquito-borne chikungunya virus (CHIKV) causes an acute febrile illness with rash, joint and muscle pain.A realtime RT-PCR assay for CHIKV detecting non-structural protein (nsP2; CHIKV nsP2-RT-qPCR) was set up. All the serodiagnosed CHIKV cases detected during 2009-2019 in Finland were screened with the assay, followed by isolations attempts and sequencing using Sanger and next generation sequencing (NGS). To validate the assay external and in-house quality control samples were used and all were correctly identified. Specificity of the assay was 100%. Assay was sensitive to detect CHIKV RNA in dilution of 10-8.During years 2009-2019 34 patients were diagnosed for acute CHIKV infection. Twelve out of 34 cases were positive by CHIKV nsP2-RT-qPCR.Two CHIKV isolations succeeded from two individuals infected originally in Thailand, 2019. From 12 CHIKV nsP2-RT-qPCR positive samples, five (42%) CHIKVs were successfully sequenced. In this study, CHIKVs from year 2019 clustered with CHIKV ECSA-lineage forming sub-cluster with strains from ones detected in Bangladesh 2017, and the ones from Jamaica (2014) within Asian lineage showing highest similarity to strains detected in Caribbean outbreak 2013-15. Majority of the CHIKV infections detected in Finland originates from Asia and virus lineages reflect the global circulation of the pathogen.
KEYWORDS:
Introduction
Diagnosis of febrile illnesses in travelers and people living in areas endemic for mosquito-borne diseases is a global challenge. There is a highly diverse group of etiological agents and diseases to consider, such as malaria, dengue, and enteric fevers [Citation1]. Several of the mosquito-borne microbes are clinically important viruses, such as dengue (DENV), Zika (ZIKV), West Nile (WNV), and chikungunya viruses (CHIKV). Since tropically circulating diseases may cause life-threatening complications, the correct identification of the causative pathogen is of importance.
Globally one of the most significant mosquito-borne viruses is the CHIKV which belongs to the genus Alphavirus and causes millions of infections annually. CHIKV circulates in the tropical and subtropical regions of Africa, Asia, and the Americas and its main vector species are the Aedes albopictus and A. aegypti mosquitoes [Citation2]. At the moment, three major genetic lineages are found: West African, East/Central/South African (ECSA) which includes the Indian Ocean lineage (IOL) that emerged in 2004, and the Asian lineage [Citation3]. Large outbreaks have occurred in Asia, for example, in Thailand during the years 2008–2009, 2013, and 2018–2019, but also in the Americas during 2013–15, and more recently, in Brazil in 2017–18 [Citation4–Citation12]. An acute CHIKV infection has a mean incubation time of three days and the clinical symptoms include fever, rash, severe joint and muscle pain, and edema in the joints [Citation13]. Infection is confirmed by either serology or molecular methods, but in endemic regions, the debilitating arthralgia can also be used for a fairly reliable clinical diagnosis [Citation13].
In Finland, CHIKV infections occur only in travelers, reflecting the epidemiological situation in the endemic tropical areas. The continental subarctic climate of Finland is not suitable for the vector species of CHIKV [Citation14], and consequently, there are no autochthonous cases of CHIKV in Finland, unlike the Mediterranean more precisely Italy and France [Citation15–Citation17].
HUSLAB (HUS Diagnostic center, Helsinki, Finland) performs the laboratory diagnostics of CHIKV infections in Finland. Thirty-four CHIKV infections have been serodiagnosed in Finnish travelers during the years 2009–2019. All of the collected samples were examined using CHIKV IgM ELISA (Euroimmun) and CHIKV nsp2-RT-qPCR followed by virus isolation, sequencing, and molecular epidemiology analysis. In addition, data from the validation of a real-time reverse-transcription PCR assay detecting CHIKV the non-structural protein (CHIKV nsP2-RT-qPCR) are provided.
Materials and methods
We screened retrospectively all the samples sent to HUSLAB (Helsinki, Finland) and serodiagnosed for acute CHIKV infection using the CHIKV nsP2-RT-qPCR followed by Sanger sequencing and next-generation sequencing (NGS) for a whole-genome sequencing approach. Virus isolation was attempted from all CHIKV nsP2-RT-qPCR positive samples.
Virus strains and samples for validation
In order to test the specificity of the CHIKV nsP2-RT-qPCR, a panel of 15 CHIKV antibody-negative human serum samples from Finnish individuals [originally tested negative using CHIKV hemagglutination inhibition tests and immunofluorescence assays, HUSLAB] was used. In addition, we tested RNA extracted from cell culture isolations of DENV 1–4 (Robert Koch Institute (RKI), Germany, and Zeptometrix Corporation, Buffalo, New York, USA), ZIKV (strain MR766, Zeptometrix), yellow fever virus (YFV, strains 17D and Asibi, RKI), and WNV (strain Eg101, in-house).
For validation and quality control purposes, we also tested CHIKV Caribbean (European Virus Archive) and Ross (in-house) strains as well as 12 external quality assessment (EQA) samples distributed by the European Network for Diagnostics of Imported Viral Diseases (EVD-Labnet). The EQA samples consisted of CHIKV strains H20235 Saint Martin/2013 (Asian genotype), 236 origin Seychelles and 3162 origin India (both of the East-Central-South-Africa genotype) as well as O’nyong-nyong virus (ONNV) strain Ahero, Sindbis virus (SINV) strain Edgar 339, and DENV-2 strain VR-345 (EQA). The sample set is described thoroughly by [Citation18].
Acute CHIKV disease samples
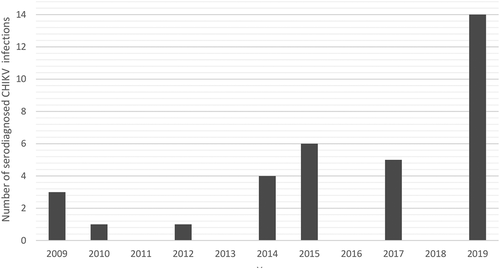
Altogether, 34 patients were diagnosed with acute CHIKV disease from 2009 until the end of December 2019 in Finland (). The annual variation of acute CHIKV cases in Finland is presented in . The primary diagnosis has been made using serology, i.e. rise in IgG using IFA or hemagglutination inhibition titers (detects total antibodies) and positive IgM test, either using immunofluorescence and/or ELISA (Euroimmun) (). All of the patients were tested to be negative for acute dengue fever (HUSLAB). All patient samples were handled anonymously and according to research permits (of projects TYH2016258, TYH2017257, and TYH2019263) of Helsinki University Hospital.
Table 1. Demographic data and laboratory findings from patients with acute CHIKV disease during years 2009–2019.
Figure 1. Annual variation of serodiagnosed acute CHIKV infections in Finland, years 2009–2019.
Extraction methods
Total nucleic acids were extracted from the serum and EDTA-blood samples using MagNA Pure LC instrument and Total Nucleic acid kit (Roche, Espoo, Finland). Total nucleic acid templates were used for nsP2-RT-qPCR and nested RT-PCR, both described below. For the processing of the nsP2-RT-qPCR reaction mixtures and total nucleic acid templates, QIAgility (Qiagen, Hilden, Germany) automated system was also used.
CHIKV RT-PCRs
We set up an in-house CHIKV nsP2-RT-qPCR based on previously published PCR primers (Centers for Disease Control and Prevention (CDC)) [Citation19] comprising a 101 bp long PCR product. Two detection probes were designed in this study, and different primers and probes are presented in more detail in .
Table 2. Primers and probes used in CHIKV RT-qPCR assay.
Real-time RT-PCR was performed using the SuperScript III Platinum One-Step qRT-PCR Kit (Invitrogen, Carlsbad, CA, USA) with 320 nM of the CHIKV-F primer, 240 nM of both CHIKV-R1 and CHIKV-R2 primers and 160 nM of each probe. Five µl of the extracted eluate was subjected to 25 µl PCR-reaction with cycling conditions of 1 cycle of 50°C for 30 min, 1 cycle of 95°C for 2 min, followed by 45 cycles of 95°C for 15 sec and 60°C for 50 sec.
For Sanger sequencing, nested RT-PCR primers described by [Citation20] were used. Nested RT-PCR was carried out using a final concentration of 40 pmol of each primer and Superscript® III Platinum® One-step RT-PCR System (Invitrogen) with the final template amount of 5 µl. The positive PCR products were purified using AMPure XP beads (Beckman-Coulter, CA, USA) and sequenced using Sanger method (Helsinki University sequencing services, Helsinki, Finland).
Virus isolation protocol
The CHIKV nsP2-RT-qPCR positive patient samples (serum/EDTA-blood) were inoculated to Vero cells in 2% FBS MEM with penicillin-streptomycin-nystatin. Seven days after the infection, RNA was extracted from the cultured cells using Viral RNA mini kit (Qiagen) and tested using CHIKV nsP2-RT-qPCR.
One sample (indicated in ; from year 2010) was not used for the virus isolation attempt since the CHIKV detected in this sample (taken in 2010) has been previously described by [Citation21].
Next-generation sequencing, NGS
For NGS, the original serum samples were filtered through 0.4 µm columns (Millipore, Burlington, Massachusetts, US) and treated with a mixture of micrococcal nuclease (New England BioLabs Ipswich, Massachusetts, US) and benzonase (Millipore) for 1 hour at 37°C, followed by RNA extraction using Trizol reagent (Thermo Fisher Scientific, Massachusetts, USA). Ribosomal RNA was removed using a NEBNext rRNA Depletion Kit (New England BioLabs) according to the manufacturer’s protocol. The sequencing library was prepared using a NEBNext Ultra II RNA Library Prep Kit (New England BioLabs). For cell culture isolates, RNA was extracted using Trizol reagent, followed by sequencing library preparation as described above. The library fragment sizes were measured using agarose gel electrophoresis and the concentrations using Qubit dsDNA BR Assay Kit (Life Technologies, Carlsbad, California, US) and NEBNext Library Quant Kit for Illumina (New England BioLabs). Sequencing was conducted using MiSeq Reagent Kit V2 with 150 bp reads.
Raw sequence reads were trimmed and low-quality, quality score <30, and short, <50 nt, sequences were removed using Trimmomatic [Citation22]. Thereafter, de novo assembly was conducted using MegaHit [Citation23]. Contigs were annotated using Blastn followed by re-assembly against the de-novo assembled CHIKV consensus sequences using BWA-MEM algorithm [Citation24] implemented in SAMTools version 1.8 [Citation25].
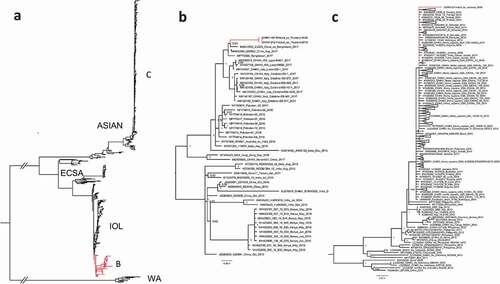
For phylogenetic analysis, all complete CHIKV coding sequences were downloaded from the GenBank (National Center for Biotechnology Information, NCBI). Identical sequences were removed using CD-HIT [Citation26] followed by alignment using ClustalW algorithm [Citation27] implemented in MEGA7 [Citation28]. The phylogenetic tree (-c)) was constructed using the Bayesian Markov chain Monte Carlo (MCMC) method, implemented in Mr Bayes version 3.2 [Citation29] using a GTR-G-I model of substitution with 2 independent runs and 4 chains per run. The analysis was run for 5 million states and sampled every 5000 steps.
Figure 2. A phylogenetic tree of complete CHIKV coding sequences detected during 2009–2019. (a) All complete CHIKV coding sequences acquired from GenBank. 2(b) CHIKV strains from Thailand clustered within ECSA-lineage (c) CHIKV strain from Jamaica clustered within the Asian lineage.
Results
CHIKV nsP2-RT-qPCR
The negative serum panel (n = 15) and controls representing other viruses (DENVs, ZIKV, YFVs, WNV, ONNV and SINV) all tested as negative by CHIKV nsP2-RT-qPCR, showing 100% specificity. In order to evaluate the analytical sensitivity of the assay, external EQA (EVD-Labnet) samples and CHIKV RNA dilutions were tested (). The CHIKV nsP2-RT-qPCR correctly identified all CHIKV positive EQA samples (n = 12; 100%), as well as the different ten-fold dilutions (10−5-10−8) of RNA extracted from cell culture supernatants of CHIKV strains Caribbean and Ross (100%). The median Ct values (five parallel reactions; standard deviation, SD; coefficient variation, CV%) for CHIKV Caribbean and Ross dilutions are presented in . The limit of detection was not calculated as there were no commercial or in-house purified and quantified CHIKV isolates or RNA available. However, the Ct value of 37 was considered the cut-off for positivity based on the parallel tests (). If the Ct value was over 37, the test was repeated and positivity was confirmed with other methods.
Table 3. Cycle threshold values of two different CHIKV dilution series using the nsP2 CHIKV RT-qPCR.
Clinical samples
Demographic data and laboratory findings are presented in . The most frequent source of infection for Finnish patients was Asia, accounting for ~60% percent of cases, and particularly Thailand (). This most likely simply reflects the amount of Finnish travelers to Asia compared to other CHIKV endemic areas. The median age of the patients was 35 years (range: 9–70 years) and 16 of the patients were female and 18 were male.
Twelve of the acute CHIKV disease patients (n = 34, , ), were positive using the CHIKV nsP2-RT-qPCR. From these CHIKV nsP2-RT-qPCR positive cases, five (42%) were successfully downstream processed using Sanger sequencing and/or NGS. Altogether three whole genomes were constructed using NGS (-c)). In this study, the whole-genome sequences obtained originated in the Caribbean (Jamaica 2014; 33-year-old male) and from Thailand (Phuket 2019; 52-year-old female and 51-year-old male). In addition, from two samples, CHIKV was sequenced using nested-RT-PCR (Sanger sequencing).
In the phylogenetic analysis, the strains from Thailand clustered within ECSA-lineage and formed a sub-cluster together with strains from Bangladesh and a Chinese traveler from Bangladesh 2017 ); [Citation30, Citation31]. These strains formed a sister clade for CHIKV strains sequenced during a CHIKV outbreak in Italy in 2017 [Citation32].
The strain from Jamaica clustered within the Asian lineage and showed the highest similarity with strains sequenced from a Caribbean outbreak during 2013–2015 (c); [Citation33].
The female patient from Thailand had reported fever and joint pain as clinical symptoms and the male patient had reported fever, rash, mild conjunctivitis, arthralgia, and the inability to walk. The male patient also had Legg-Calvé-Perthes disease as a pre-existing condition. No clinical symptoms or other information was available for the patients who contracted the infection in Jamaica.
We isolated CHIKVs from two CHIKV-seronegative patient samples (see serological data from ) in Vero cells and confirmed the isolation by CHIKV nsP2-RT-qPCR and NGS from the cell culture supernatant. Both of the isolates originated in Phuket, Thailand, in 2019 (CHIKV-197-Finland_ex_Thailand-2019, MN075149; CHIKV-212-Finland_ex_Thailand-2019, MN075150). The sequences were identical ()).
None of the other virus isolation attempts out of altogether 11 attempts were successful. Virus isolation did not succeed from an EDTA blood sample (2019; country visited: Thailand) which was positive for CHIKV RNA (Ct 24.5). As there is no validated protocol for NGS, an NGS run was not carried out in this sample.
Discussion
The CHIKV nsP2-RT-qPCR reaction and protocol were optimized for automated sample preparation and PCR pipetting robots [MagNA Pure LC instrument (Roche) and QIAgility, Qiagen], and was validated in diagnostic laboratory settings (HUSLAB). The validated CHIKV nsP2-RT-qPCR showed excellent performance with high specificity (100%) and good sensitivity and specificity with the EQA samples tested. The study was limited by not having a calculated limit of detection value due to the fact that commercial and quantified CHIKV RNA was not available at the time of the study. However, the CHIKV RNA was detected in a dilution of 10−8 and nsP2-RT-qPCR excellent intra-assay repeatability (). Overall, the data show that this CHIKV nsP2-RT-qPCR can be used as a diagnostic assay for the detection of acute CHIKV disease in the early phase of the infection.
Thirty-four travel-associated CHIKV cases have been diagnosed in Finland during 2009–19 (). 2019 had the highest number of CHIKV cases ( and ). In total, 14 (41.2%; 14/34) acute cases were diagnosed during that year. We collected all serum/blood samples available from all of the 34 patients and tested them with the CHIKV nsP2-RT-qPCR, and subsequently attempted isolation and Sanger/NGS sequencing from CHIKV nsP2-RT-qPCR-positive samples. Twelve CHIKV nsP2-RT-qPCR positive samples were detected, and out of these 12, two virus isolations succeeded and three whole-genome sequences were obtained (-c)).
In Finnish travelers, most of the CHIKV samples positive with CHIKV nsP2-RT-qPCR were taken less than one week after the onset of symptoms. Overall, the symptoms were typical for chikungunya and no fatal CHIKV disease cases have been reported in Finland ().
Nine Finnish patients returning from Thailand with severe symptoms have been described previously [Citation34]. In these cases, at 10–11 days after the onset of symptoms, a low level of CHIKV RNAemia was still detected in three individuals using CHIKV nsP2-Rt-qPCR.
In 2019, all the CHIKV strains detected from Finnish travelers originated in Thailand and clustered within ECSA-lineage, subclustering with strains identified in 2017 in Bangladesh [Citation30,Citation31]. In addition, these strains formed a sister clade with the ones reported by [Citation32] in Italy, 2017.
In 2014–2015, there was a peak of acute CHIKV cases () acquired from Asia and the Caribbean. During this time large CHIKV outbreaks were reported in the Americas as the CHIKV Asian lineage was introduced to and spread widely across the Caribbean, South and Central America, and the USA (Florida) [Citation33]. These epidemics were also reflected in Finnish travelers. The detected CHIKV strains from years 2014–15 belonged to Asian lineage and had the highest similarity with the strains reported to cause the outbreak in the Caribbean during 2013–15.
When using serological methods in the diagnosis of acute CHIKV infection, samples from acute and convalescent phase are needed. This can postpone the diagnosis of CHIKV infection. The molecular-based diagnostics, however, can be used particularly in the early phase and the diagnosis can be confirmed earlier than using serological methods. In addition, samples with CHIKV genome detected can be further used for sequencing and surveillance of CHIKV strains circulating. This is as important as vector-surveillance for monitoring CHIKV spreading to new areas or new strains evolving.
Author statements
The authors have declared no special author statements.
Author contributions
Jääskeläinen AJ (AJ), Kareinen L (LK), Smura T (TS), Kallio-Kokko H (HKK), Vapalahti O (OV).
AJ designed and set up the realtime RT-PCR system for CHIKV (CHIKV RT-qPCR), evaluated the CHIKV RT-qPCR and screened all the samples using the CHIKV RT-qPCR, collected and conducted the medical data and laboratory data, carried out the isolation attempts, and wrote and revised the manuscript.
LK carried out the Sanger sequencing by nested RT-PCRs and did part of isolation attempts, wrote, and revised the manuscript.
TS did the NGS and NGS data analysis and wrote and revised the manuscript.
HKK wrote and revised the manuscript.
OV wrote and revised the manuscript.
Key results
CHIKV strains were isolated and whole genomes were sequenced.
A realtime RT-qPCR (nsP2-RT-qPCR) was set up and validated with Finnish sample panels with excellent specificity and sensitivity.
Automated nucleic acid extraction followed by automated pipetting system was tested for CHIKV diagnostics.
Acknowledgments
We thank the Hanna Jarva (HUS Diagnostic center, HUSLAB and University of Helsinki, Finland) for language editing.
Disclosure statement
The authors have declared no conflicts of interest.
A phylogenetic tree was constructed using the Bayesian Markov chain Monte Carlo (MCMC) method, implemented in Mr Bayes version 3.2 using a GTR-G-I model of substitution with 2 independent runs and 4 chains per run.
Additional information
Funding
Notes on contributors
A. J. Jääskeläinen
A.J. Jääskeläinen is a clinical microbiologist, PhD and docent in virology, working at HUS Diagnostic Center, Helsinki University Hospital, Helsinki, Finland and University of Helsinki.
L. Kareinen
L. Kareinen is MSc and VDM, currently working at University of Helsinki.
T. Smura
T. Smura is a PhD and working at University of Helsinki.
H. Kallio-Kokko
H. Kallio-Kokko is a clinical microbiologist, PhD and docent in virology, working at HUS Diagnostic Center, Helsinki University Hospital, Helsinki, Finland.
O. Vapalahti
O.Vapalahti is a professor, MD, and PhD, working at University of Helsinki and HUS Diagnostic Center, Helsinki University Hospital, Helsinki, Finland.
References
- Scaggs Huang FA , Schlaudecker E. Fever in the returning traveler. Infect Dis Clin North Am. 2018;32(1):163–9.
- World Health Organization (WHO) . [cited 2020 Jul 6 ]. https://www.who.int/news-room/fact-sheets/detail/chikungunya
- Caglioti C , Lalle E , Castilletti C , et al. Chikungunya virus infection: an overview. New Microbiol. 2013;36(3):211–227.
- Aubry M , Teissier A , Roche C , et al. Chikungunya outbreak, French Polynesia, 2014. Emerg Infect Dis. 2015;21(4):724–726.
- Beau F , Lastère S , Mallet H-P , et al. Impact on blood safety of the last arboviruses outbreaks in French Polynesia (2012–2018). Transfus Clin Biol. 2020;27(1):4–9.
- Cao-Lormeau VM , Musso D . Emerging arboviruses in the Pacific. Lancet. 2014;384(9954):1571–1572.
- Chansaenroj J , Wanlapakorn N , Ngamsaithong C , et al. Genome sequences of chikungunya virus isolates from an outbreak in southwest Bangkok in 2018. Arch Virol. 2020;165(2):445–450.
- Lanciotti RS , Valadere AM . Transcontinental movement of Asian genotype chikungunya virus. Emerg Infect Dis. 2014;20(8):1400–1402.
- Leparc-Goffart I, Nougairede A, Cassadou S, et al. Chikungunya in the Americas. Lancet. 2014;383:514.
- Pereira Gusmão Maia Z , Mota Pereira F , Do Carmo Said RF , et al. Return of the founder Chikungunya virus to its place of introduction into Brazil is revealed by genomic characterization of exanthematic disease cases. Emerg Microbes Infect. 2020;9(1):53–57.
- Pongsiri P, V Auksornkitti, A Theamboonlers A, et al. Entire genome characterization of chikungunya virus from the 2008–2009 outbreaks in Thailand. Asian Pac J Tropical Biomedicine. 2010;27:167–176.
- Weaver SC , Forrester NL . Chikungunya: evolutionary history and recent epidemic spread. Antiviral Res. 2015;120:32–39.
- Weaver SC , Lecuit M , Campion EW . Chikungunya Virus and the Global Spread of a Mosquito-Borne Disease. N Engl J Med. 2015;372(13):1231–1239.
- European Centre for Disease Prevention and Control and European Food Safety Authority . Mosquito maps [internet]. Stockholm: ECDC; 2020. [cited Jun 29 ]. Available from https://ecdc.europa.eu/en/disease-vectors/surveillance-and-disease-data/mosquito-maps
- Grandadam M , Caro V , Plumet S , et al. Chikungunya virus, southeastern France. Emerg Infect Dis. 2011;17(5):910–913.
- Rezza G , Nicoletti L , Angelini R , et al. Infection with chikungunya virus in Italy: an outbreak in a temperate region. Lancet. 2007;370(9602):1840–1846. .
- Riccardo F , Venturi G , Di Luca M , et al. Secondary autochthonous outbreak of Chikungunya, Southern Italy, 2017. Emerg Infect Dis. 2019;25(11):2093–2095.
- Jacobsen S , Patel P , Schmidt-Chanasit J , et al. External quality assessment studies for laboratory performance of molecular and serological diagnosis of Chikungunya virus infection. J Clin Virol. 2016;76:55–65.
- Johnson BW , Russell BJ , Goodman CH . Laboratory diagnosis of Chikungunya virus infections and commercial sources for diagnostic assays. J Infect Dis. 2016;214(suppl 5):S471–S474.
- Sánchez-Seco MP , Rosario D , Quiroz E , et al. A generic nested-RT-PCR followed by sequencing for detection and identification of members of the alphavirus genus. J Virol Methods. 2001;95(1–2):153–161.
- Kurkela S , Sane J , Deren E , et al. Chikungunya virus as a causative agent of fever of unknown origin in Finnish travellers to tropics. J Clin Virol. 2012;54(3):289–290.
- Bolger AM , Lohse M , Usadel B . Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120.
- Li D , Liu C-M , Luo R , et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31(10):1674–1676.
- Li H . Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Cite as arXiv:1303.3997. 2013. https://www.researchgate.net/publication/236935760_Aligning_sequence_reads_clone_sequences_and_assembly_contigs_with_BWA-MEM/citation/download.
- Li H , Handsaker B , Wysoker A , et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079.
- Li W , Godzik A . Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659.
- Thompson JD , Higgins DG , Gibson TJ . CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680.
- Kumar S , Stecher G , Tamura K . MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–1874.
- Rönquist F , Teslenko M , van der Mark P , et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–4227.
- Pan J , Fang C , Yan J , et al. Chikungunya fever outbreak, Zhejiang Province, China, 2017. Emerg Infect Dis. 2019;25(8):1589–1591.
- Pyke AT , Moore PR , McMahon J . New insights into chikungunya virus emergence and spread from Southeast Asia. Emerg Microbes Infect. 2018;7(1):1–3.
- Lindh E, Argentini C, Remoli M.E., et al. The Italian 2017 Outbreak Chikungunya virus belongs to an emerging aedes albopictus-adapted virus cluster introduced from the Indian Subcontinent. Open Forum Infect Dis. 2018;12:6:ofy321.
- Tan Y , Pickett BE , Shrivastava S , et al. Differing epidemiological dynamics of Chikungunya virus in the Americas during the 2014-2015 epidemic. PLoS Negl Trop Dis. 2018;12(7):e0006670.
- Kantele A . Travellers as sentinels of chikungunya epidemics: a family cluster among Finnish travellers to Koh Lanta, Thailand, January 2019. Eurosurveillance. 2019;24(11). DOI:10.2807/1560-7917.ES.2019.24.11.1900162