
ABSTRACT
Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) is a relatively under-recognized hereditary cardiomyopathy. It is characterized pathologically by fibro-fatty infiltration of right ventricular (RV) myocardium and clinically by consequences of RV electrical instability. Timely intervention with device therapy and pharmacotherapy may help reduce the risk of arrhythmic events or sudden cardiac death. Here, we describe a classic case of a young adult with ARVC and a brief literature review. The patient presented with exertional palpitations and ARVC was suspected after his routine electrocardiogram (EKG) revealed symmetric T wave inversions and possible epsilon waves in right precordial leads. Subsequent work up showed fatty infiltration of RV myocardium on cardiac magnetic resonance imaging and inducible ventricular tachycardia from the right ventricle during electrophysiologic study. Those findings confirmed the diagnosis of ARVC and warranted treatment with implantable cardioverter defibrillator. It is always exciting to encounter rare pathological entities with classic clinical findings, especially when they present as a diagnostic challenge.We were able to provide correct diagnosis and management, thereby preventing the potentially lethal consequences. Therefore, it is important to recognize the possible EKG findings of ARVC and to know when to pursue further investigations and to implement therapies.
1. Case report
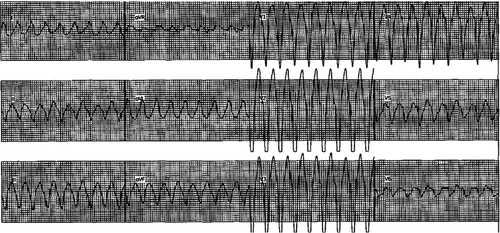
A healthy 33-year-old athletic male was evaluated in cardiology clinic for worsening exertional palpitations and non-specific chest discomfort. About five years ago, he had similar complaints and prior electrocardiogram (EKG) from the initial encounter showed diffuse symmetric T wave inversions in right precordial leads. Subsequently, he had extensive work up consisting of cardiac monitor, echocardiogram (ECHO), exercise stress test and coronary angiogram, which were all unremarkable. There was no personal or family history of unexplained syncope or cardiac arrest. At this visit, the patient’s EKG showed symmetrically inverted T waves and possible epsilon waves in the right precordial leads ( and ). ECHO was grossly unremarkable. Holter monitor showed frequent premature ventricular complexes of right ventricular (RV) origin and episodes of non-sustained ventricular tachycardia (NSVT) with left bundle branch block pattern (LBBB) (). Cardiac magnetic resonance imaging (CMRI) showed dyskinetic right ventricle with RV ejection fraction (EF) of 35% and foci of fat in RV side of interventricular septum (). The patient was informed of the possible diagnosis of ARVC. Electrophysiologic study (EPS) was done and showed easily inducible ventricular tachycardia (VT) from the right ventricle with LBBB pattern (). Dual-chamber implantable cardioverter defibrillator (ICD) was subsequently placed and patient was discharged on sotalol. After counseling, genetic testing was done and was positive for mutation in plakophilin-2 gene (PKP-2). Genetic testing was also recommended for his family. Follow up device interrogation has showed no therapies.
Figure 1. Resting 12-lead EKG showing symmetric T wave inversion in right precordial leads (V1, V2 and V3) (see black arrows).
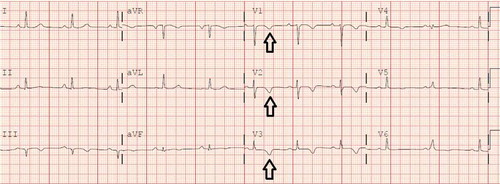
Figure 2. Possible epsilon waves in right precordial lead V3 (see black arrow).
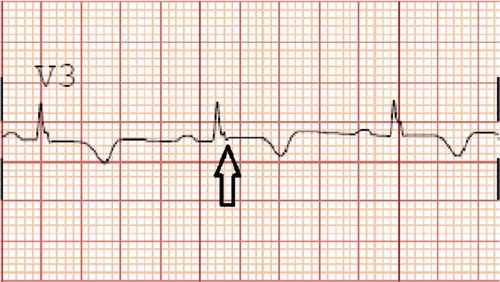
Figure 3. Holter monitor showing non-sustained ventricular tachycardia with left bundle branch block pattern.
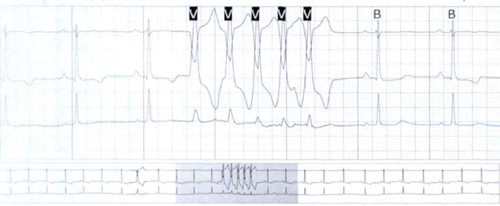
Figure 4. Cardiac MRI showing foci of fat in right ventricular side of interventricular septum (see white arrow).
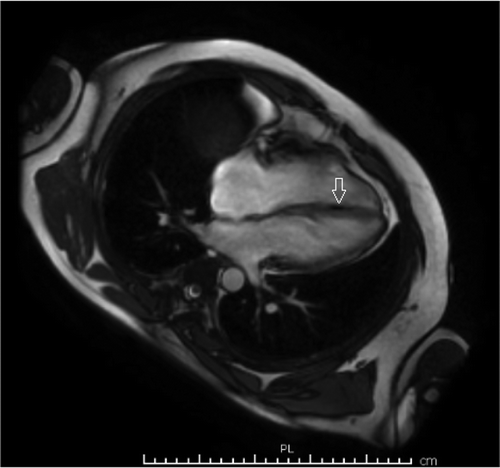
Figure 5. Twelve-lead EKG showing inducible ventricular tachycardia with left bundle branch block pattern and superior axis during electrophysiologic study.
2. Discussion
First reported by Dr Frank Marcus in 1982, ARVC is a progressive hereditary cardiomyopathy with a higher risk of ventricular arrhythmias and sudden cardiac death (SCD). It is characterized histologically by fibro-fatty replacement of RV myocardium with subsequent RV dilation and systolic dysfunction.[Citation1] Despite the name, ARVC may also involve the left ventricle and cause left-sided heart failure. The estimated prevalence is 1:2000–1:5000. Underlying pathogenesis is thought to be the mutation of several genes encoding desmosomes, which are responsible for cell to cell adhesion. Malfunctioning desmosomes account for myocyte detachment and cell death along with perpetual mechanical stress or myocardial contraction. This process leads to the initial phase of inflammation in affected myocardium with subsequent apoptosis and fibro-fatty replacement of the myocardium. Ventricular fibrillation (VF) may occur during the process of cell death whereas macro-reentrant ventricular tachycardia (VT) is related to fibrofatty scars in ARVC. Mutation of non-desmosomal genes have also been reported in autosomal dominant ARVC.[Citation2,Citation3]
Many patients may remain asymptomatic, but some patients may present with palpitations, dizziness, cardiogenic syncope, heart failure, ventricular arrhythmias or cardiac arrest. EKG findings, reported in approximately 30% of ARVC patients, include symmetrically inverted T waves and epsilon waves in the right precordial leads (V1, V2 and V3). Thought to be due to slowed depolarization of affected part of RV myocardium, epsilon waves are described as low-amplitude positive signals at the end of QRS complexes. Of ARVC associated ventricular arrhythmias, the most common one is monomorphic VT with LBBB pattern.[Citation4,Citation5] Supraventricular arrhythmias have also been reported. Cardiovascular mortality among ARVC patients is attributable to progressive heart failure and SCD in the order of common occurrence. The SCD risk is higher among those with history of ventricular tachycardia.[Citation6] The phenotypic penetrance is variable and is strongly related to the amount of exercise. James et al. [Citation7] reported endurance exercise level has an impact on phenotypic penetrance of the disease and onset of VT and SCD among athletes. This finding has also been described in an animal study.[Citation8]
Holter monitor may show NSVT with variable morphologies. ECHO can show regional RV wall motion abnormality, increased RV dimension (especially at the RV outflow tract) and reduced RV EF. With emerging technology, CMRI has played a major role in diagnosing ARVC with fairly high sensitivity and specificity, however inter-observer variability has been reported.[Citation9] EPS can be done diagnostically to look for inducible VT. The extent of the disease can also be identified by electroanatomic mapping.[Citation10] EPS can also help guide biopsy the endomyocardium. However, the use of endomyocardial biopsy (EMB) is controversial due to patchy areas of myocardial involvement and limited sensitivity from sampling errors.[Citation11] On the other hand, genetic testing may be helpful when the diagnosis of ARVC is borderline and when targeted genetic screening of first-degree relatives is planned. Neither EMB nor genetic testing is recommended as an initial step to establish the diagnosis.
Literature suggests that approximately 50% of ARVC patients are familial, which perhaps may be an underestimate. The disease can be inherited in autosomal dominant form or autosomal recessive form, with the former being the most common. Several genes have been reported in the autosomal dominant disease. One of them is plakophilin-2 gene (PKP-2), which encodes desmosomal protein plakophilin-2 and is the most common mutation identified in North America. Patients with PKP-2 mutation tend to have earlier onset of symptoms and arrhythmias with the mean age of onset being 28 (±11 years).[Citation2,Citation12] In our patient, genetic testing was positive for pathogenic variant in PKP-2 gene. Additionally, a recessive counterpart is less common and is associated with cutaneous manifestations. Both Naxos disease and Carvajal syndrome are inherited in autosomal recessive pattern and are associated with woolly hair, palmoplantar keratosis and cardiomyopathy.[Citation13,Citation14]
Making a correct diagnosis is critically important and can be challenging. Marcus et al. [Citation15] have proposed the 2010 revised Task Force Criteria, which include six categories as below:
Global or regional dysfunction and structural alterations in CMRI or ECHO
Tissue characterization of wall (fibrous replacement and percentage of residual myocytes in right ventricle)
Repolarization abnormalities on EKG (T wave inversion in V1, V2 and V3)
Depolarization/conduction abnormalities on EKG (epsilon wave in V1, V2 and V3)
Arrhythmias (VT with LBBB morphology and superior axis)
Family history (AVRC in a first degree relative confirmed with Task Force criteria or at autopsy)
Among the six categories, each of them is subdivided into major and minor criteria. Diagnosis is fulfilled as below:
Definite diagnosis with two major or one major and two minor criteria or four minor from different categories
Borderline diagnosis with one major and one minor or three minor criteria from different categories
Possible diagnosis with one major or two minor criteria from different categories
(Please refer to , adapted from Marcus et al. [Citation15] with permission).
Table 1. Revised task force criteria (adapted with permission from Marcus et al. [Citation15]).
The revised diagnostic criteria have provided an improved sensitivity with a preserved specificity compared to the original 1994 International Task Force Criteria. In our patient, the definite diagnosis of ARVC can be made since he has more than two major criteria which are CMRI finding of RV dyskinesia with foci of fat in RV myocardium, RV EF of 35% and RV end diastolic volume of 161 milliliters, T wave inversions and epsilon waves in right precordial leads and VT of LBBB pattern with superior axis during EPS.
Lifestyle modification is recommended for all patients with ARVC given the strong association between endurance exercise and incidence of ventricular arrhythmias. Not only do patients with ARVC need to abstain from all competitive sports, but they should also avoid any physical activity that causes palpitations or pre-syncope.[Citation7,Citation16] The fact that exercise increases the disease progression compels the use of β-blockers for sympathetic suppression and potentially improved clinical outcome. However, strong evidence to support the empiric use of β-blockers is lacking. Angiotensin-converting enzyme inhibitors may also limit structural progression of the disease and prevent ventricular arrhythmias.[Citation1] Patients with ARVC who progress to have heart failure should be treated with standard medical therapy.
Furthermore, ICD therapy may help treat ventricular arrhythmias and prevent SCD. The expert consensus is that ICD therapy is indicated for primary prevention for those at high risk and for secondary prevention for those with prior history of sustained VT or aborted cardiac arrest.[Citation17] However, the precise indications for ICD implantation among ARVC patients have not been well established, nor is there a consensus on stratifying the patients at high risk. Yet, the efficacy of ICD therapy in preventing SCD among those at high risk has been proven by randomized control trials. Successful termination of ventricular tachyarrhythmias in ARVC patients by anti-tachycardia pacing (ATP) is reported in a prospective study.[Citation18] As in all settings with appropriate implantation, ICD therapies carry the risks of pericardial effusion, perforation, device infection and inappropriate shocks. Of them, myocardial perforation and under-sensing of arrhythmias are of particular concern in ARVC due to thin areas of RV myocardium and progressive fibro-fatty infiltration respectively. Anti-arrhythmic drugs, often sotalol, may help reduce the incidence of ventricular arrhythmias. However, anti-arrhythmic medications are used as an adjunct therapy in ARVC patients with ICD devices and are no replacement for ICD therapy. Furthermore, experts recommend the use of sotalol or amiodarone in ARVC patients who cannot tolerate ICD therapy.[Citation17,Citation19] Due to patchy involvement of the myocardium, radiofrequency ablation cannot be considered as a definitive therapy. Surgery and heart transplantation are considered for refractory ventricular arrhythmias and end stage heart failure. In our patient, given the clinical symptom of palpitation, fibrofatty changes in RV myocardium and inducible VT in EPS, an ICD was implanted as a primary preventive therapy. This case was initially a diagnostic challenge, but also highlights the proper use of medical resources, such as a cardiac MRI.
Many patients tend to do well albeit all patients with ARVC are at a relatively increased risk of developing ventricular tachyarrhythmias and heart failure. Asymptomatic family members of an affected patient may develop ARVC later in life due to age-related phenotypic penetrance. EPS may play a role in risk-stratification, as patients with inducible sustained monomorphic VT are at a higher risk of SCD. Literature suggests that the overall prognosis of ARVC is better than that of other structural heart diseases which cause arrhythmias of ventricular origin.[Citation5,Citation6,Citation20]
3. Conclusion
ARVC is a hereditary cardiomyopathy and conveys a higher risk of ventricular arrhythmias and SCD. In our patient, incidental EKG findings after episodes of exertional palpitations led to the discovery of a potentially life-threatening disease and subsequent ICD implantation. Thus, it is critically important to recognize the possible EKG findings of ARVC and to know when to pursue further investigations and to implement therapies. Since its maiden description in 1982, ARVC has largely been explored in both pathophysiology and management. Nonetheless, there remain several unanswered questions regarding risk stratification, effective pharmacotherapy, optimal device strategy and the prognosis of the disease.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- James CA, Calkins H. Update on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C). Curr Treat Options Cardiovasc Med. 2013;15:476–487.
- Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation. 2006;113:1634–1637.
- Que D, Yang P, Song X, et al. Traditional vs. genetic pathogenesis of arrhythmogenic right ventricular cardiomyopathy. Europace. 2015;17:1770–1776.
- Dalal D, Nasir K, Bomma C, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005;112:3823–3832.
- Nava A, Bauce B, Basso C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36:2226–2233.
- Hulot J-S, Jouven X, Empana JP, et al. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2004;110:1879–1884.
- James CA, Bhonsale A, Tichnell C, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1290–1297.
- Kirchhof P, Fabritz L, Zwiener M, et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation. 2006;114:1799–1806.
- Tandri H, Castillo E, Ferrari VA, et al. Magnetic resonance imaging of arrhythmogenic right ventricular dysplasia: sensitivity, specificity, and observer variability of fat detection versus functional analysis of the right ventricle. J Am Coll Cardiol. 2006;48:2277–2284.
- Boulos M, Lashevsky I, Reisner S, et al. Electroanatomic mapping of arrhythmogenic right ventricular dysplasia. J Am Coll Cardiol. 2001;38:2020–2027.
- Basso C, Ronco F, Marcus F, et al. Quantitative assessment of endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy/dysplasia: an in vitro validation of diagnostic criteria. Eur Heart J. 2008;29:2760–2771.
- Dalal D, Molin LH, Piccini J, et al. Clinical features of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in plakophilin-2. Circulation. 2006;113:1641–1649.
- Protonotarios N, Tsatsopoulou A, Anastasakis A, et al. Genotype-phenotype assessment in autosomal recessive arrhythmogenic right ventricular cardiomyopathy (Naxos disease) caused by a deletion in plakoglobin. J Am Coll Cardiol. 2001;38:1477–1484.
- Norgett EE, Hatsell SJ, Carvajal-Huerta L, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761–2766.
- Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541.
- Ruwald A-C, Marcus F, Estes NA 3rd, et al. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2015;36:1735–1743.
- European Heart Rhythm Association; Heart Rhythm Society; Zipes DP, et al.. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death). J Am Coll Cardiol. 2006;48:e247–e346.
- Link MS, Laidlaw D, Polonsky B, et al. Ventricular arrhythmias in the North American multidisciplinary study of ARVC: predictors, characteristics, and treatment. J Am Coll Cardiol. 2014;64:119.
- Wichter T, Borggrefe M, Haverkamp W, et al. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation. 1992;86:29.
- Saguner AM, Medeiros-Domingo A, Schwyzer MA, et al. Usefulness of inducible ventricular tachycardia to predict long-term adverse outcomes in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2013;111:250–257.