
ABSTRACT
Neurofibromatosis-1 (NF-1) is a genetic neuro-cutaneous disorder that is associated with an increased prevalence of pheochromocytoma (PHEO). However, this association may not be commonly anticipated by physicians, as patients may be normotensive. In addition, NF-1 patients can be asymptomatic and/or normotensive. These factors can result in a delayed or missed diagnosis of pheochromocytoma leading to catastrophic complications. Currently, it is recommended to perform annual blood pressure monitoring in patients with NF-1 and to test for pheochromocytoma only if found to be hypertensive. However, recent studies show that this practice may lead to underdiagnosis of pheochromocytoma. Therefore, suggesting routine biochemical testing for pheochromocytoma in all patients with NF-1.
In this case report, we discuss the factors which can lead to a delayed diagnosis of pheochromocytoma in a patient with known NF-1 and hypertension.
1. Introduction
Neurofibromatosis-1 (NF-1), a syndrome with autosomal-dominant inheritance pattern, is caused by the inactivation of a tumor suppressor gene, resulting in the proliferation of various benign and malignant tumors. The disease has an incidence of 1 in 3,000. These patients have been noted to develop café au lait spots, neurofibromas, optic gliomas and hamartomas. Furthermore, increased rates of several vascular, endocrine and oncological conditions have been documented in NF-1 patients as compared to the general population [Citation1].
One of such conditions is pheochromocytoma, a catecholamine-secreting tumor that can lead to hypertension and increase the mortality rate in NF-1. However, this association between the two conditions is not well known and can be easily missed by the clinician. In addition, current guidelines on NF-1 recommend screening for pheochromocytoma only in the presence of hypertension or related symptoms [Citation2].
Here, we discuss a scenario where the diagnosis of pheochromocytoma was delayed in a patient with NF-1 and uncontrolled hypertension.
2. Case description
A 38-year-old female with history of NF-1 (diagnosed at the age of 13) presented to the emergency with right upper-quadrant abdominal pain, progressively worsening over the last 48 hours and associated with nausea and recurrent vomiting. Her vitals at the time of presentation included blood pressure of 224/133 and pulse of 98/min. Apart from typical features of NF-1 (café au lait spots and neurofibromas), the only significant examination findings were signs of dehydration and tenderness in the right upper quadrant of the abdomen. On questioning, she confirmed being diagnosed with hypertension 5 years back but was non-compliant to her medications as none helped improved her BP. She denied being investigated for secondary hypertension while the last anti-hypertensive prescribed to her was labetalol. It is possible if she had been compliant, she may have been diagnosed earlier. Due to concerns for high blood pressure and suspected cholecystitis, she was admitted to ICU for further management. Apart from leukocytosis of 19,000, the rest of her blood workup including bilirubin, liver enzymes and lipase was unremarkable while electrocardiogram had no acute changes. An urgent ultrasound of the abdomen showed cholelithiasis along with pericholecystic fluid suggestive of acute cholecystitis which was confirmed on CT abdomen/pelvis with contrast. She was immediately started on intravenous fluids and antibiotics while the general surgery team was consulted. Due to the patient being very sick, a cholecystostomy tube was placed to decompress the distended inflamed gall bladder.
Despite the use of multiple antihypertensive medications, the patient continued to have elevated blood pressure. Interestingly, a review of the CT abdomen/pelvis done on admission revealed the presence of bilateral adrenal masses, this finding was concerning for an undiagnosed PHEO. On further evaluation (), she was found to have elevated levels of free meta- and normetanephrine in plasma and urine samples which supported the initial suspected diagnosis.
Table 1. Workup of to evaluate secondary causes of hypertension in our patient.
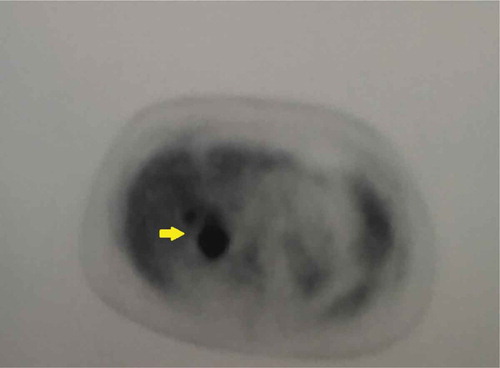
Presence of bilateral heterogenous adrenal masses on MRI abdomen/pelvis () did confirm the diagnosis but also indicated a more aggressive form of PHEO. Therefore, a PET scan was performed that demonstrated a highly metabolic right adrenal tumor with only mild activity in the left one () but no metastatic disease.
Figure 1. MRI abdomen/pelvis without contrast demonstrating bilateral solid slightly heterogeneous adrenal gland masses (yellow arrows).
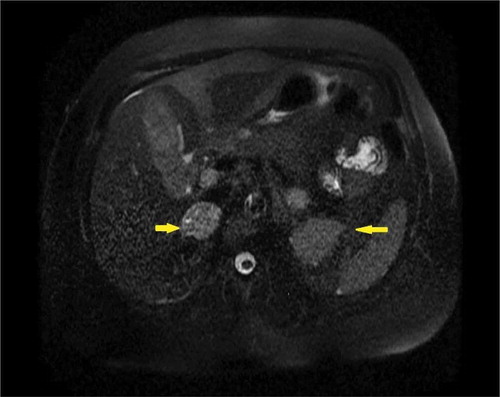
Figure 2. PET scan showing intense FDG (fluorodeoxyglucose) localization within the enlarged right adrenal gland (yellow arrow).
Treatment with prazosin (alpha-blocker) followed by labetalol (beta-blocker) and amlodipine led to a complete control of hypertension within days. After the appropriate treatment of acute cholecystitis and elevated blood pressure, the patient was discharged with a close follow-up in the outpatient endocrinology and general surgery clinics. A few weeks later, a laparoscopic cholecystectomy along with bilateral adrenalectomy was performed. On gross examination, both adrenal masses had features suggestive of pheochromocytoma which were confirmed on a histopathological analysis. Additional findings included vascular invasion and profound nuclear atypia in the right mass while the left adrenal mass only showed high cellularity.
Since her described hospital admission, the patient has remained normotensive and is being managed by an outpatient multidisciplinary team.
3. Discussion
Benign peripheral nerve tumors have been mentioned in the medical literature since the 13th century [Citation3]. But the definite clinical description was provided only in 1882 by von-Recklinghausen who named these tumors as ‘neurofibromas’ due to their origin from the peripheral nerve sheath and described the most well-known neurocutaneous syndrome in medical history-‘von-Recklinghausen disease’ or ‘Neurofibromatosis-1ʹ (NF-1) [Citation4].
NF-1 is an autosomal dominant disorder that occurs due to the inactivation of the NF1 gene resulting in loss of an important tumor suppressor protein called neurofibromin and subsequent development of various neural tumors [Citation5]. Although NF-1 is categorized as a ‘neurocutaneous’ disorder and is commonly associated with café au lait spots and neurofibromas, a significant number of the patients might lack these classical manifestations. Therefore, it is recommended to make the final diagnosis only if a patient fulfills the 1988-NIH criteria for NF-1 given in [Citation5].
Table 2. National institutes of health 1988 consensus development conference diagnostic criteria for Neurofibromatosis −1.
An interesting aspect of this syndrome is its association with various other conditions () [Citation1,Citation6] which have been found to reduce the average life expectancy of a NF-1 patient by 15 years [Citation7]. It is hypothesized that loss of neurofibromin leads to disrupted cellular function, uninhibited oncogenesis and vascular proliferation throughout the body and not just the nervous system. As a result, many ‘non-neurocutaneous’ manifestations with a high mortality rate can develop in NF-1 patients.
Table 3. Conditions associated with Neurofibromatosis-1.
One of such conditions is pheochromocytoma (PHEO), a rare catecholamines-secreting neoplasm arising from the adrenomedullary chromaffin cells. It usually presents as episodic or resistant hypertension in adults and increases the risk of labile pregnancy complications, myocardial infarction and cardiac arrest [Citation8]. Although mostly sporadic, approximately 30% of pheochromocytomas are associated with familial disorders such as MEN-2, von Hippel-Lindau syndrome (VHL) and NF-1 [Citation8].
Studies have shown that almost 5–7% of NF-1 patients develop pheochromocytomas or paragangliomas in their lifetimes [Citation7]. While the incidence rate of PHEO in NF-1 with hypertension has been reported to be as high as 20–50% [Citation9]. We have also described a similar scenario where a patient with NF-1 and hypertension was eventually diagnosed to have a PHEO.
Due to the increased incidence of PHEO, it is recommended to perform annual biochemical screening for PHEO in all patients with familial disorders (VHL and MEN syndromes). On the other hand, the current guidelines recommend screening the NF-1 patients with annual blood pressure measurement and checking for the presence of any unusual symptoms while biochemical testing for PHEO is advised only if a patient is found to be hypertensive and/or symptomatic [Citation1]. The rationale for this screening strategy is that although NF-1 patients have a higher risk of developing PHEO as compared to the normal population, the overall risk is still lower than that in patients with VHL and MEN syndromes [Citation10]. On the other hand, emerging medical evidence has demonstrated several pitfalls in the current conservative approach to screen NF-1 patients. For example, not all patients with an underlying PHEO may be hypertensive or symptomatic; therefore, screening only patients with suspected manifestations may result in under-diagnosis of PHEO. Gruber et al. [Citation2] demonstrated that even with elevated catecholamine production, 24% of NF1 patients with PHEO remain asymptomatic while 61%–80% do not have hypertension. In another study where NF-1 patients over the age of 18 years were imaged for PHEO, the majority of the patients diagnosed with PHEO-whether secreting or not lacked any associated symptoms [Citation11]. Another important fact is that normotensive NF-1 patients with pheochromocytoma have a similar post-operative mortality rate when compared to those with hypertension [Citation12]. Thus, failure to diagnose an underlying PHEO in NF-1 patients can have devastating consequences. Similarly, our patient developed acute cholecystitis and eventually underwent a cholecystectomy. Although she was found to have PHEO due to her hypertension in a timely manner, a missed diagnosis could have caused significant surgical complications. Based on these facts, it has been suggested that biochemical screening for PHEO in asymptomatic individuals with NF1 should be done routinely starting at the age of 18 years. However, the frequency of screening can be every 3 years instead of every year, as the prevalence of PHEO is lower in NF1 compared to VHL and MEN syndromes [Citation13]. In addition, some authorities have favored the routine screening of pheochromocytoma in all patients with NF-1 prior to any surgical procedures and/or pregnancy [Citation12].
Furthermore, it is currently recommended that after surgical removal of PHEO, all patients should get blood pressure checks and biochemical testing every 6 to 12 months for the first 3 years and then annually for the next 5 to 7 years. These guidelines include all patients diagnosed with PHEO regardless of their medical history [Citation14]. However, approximately 12% of NF-1 associated PHEOs are malignant having a high recurrence rate due to the underlying germline mutation with some cases reported to occur 50 years after the initial surgery [Citation15]. Thus, NF-1 patients with PHOE should be considered to get annual biochemical testing for a longer duration of time after the removal of the PHEO.
NF-1 associated PHEO usually presents with hypertension in the third or the fourth decade of life. These patients are generally recommended to be treated as the general population, unless found to have another cause. However, based on our experience with the discussed patient, we feel that an exception for NF-1 patients should be considered due to the associated high-risk. Our patient had been on various anti-hypertensive medications in the past but none of these medications could control her BP. In addition, her use of labetalol could have led to serious consequences due to unopposed alpha-stimulation. Therefore, we suggest using alpha-blockers as the first choice to control hypertension in all patients with NF-1 regardless of age and comorbidities until secondary causes have been ruled out.
4. Conclusion
Our case demonstrates many interesting learning points: First, a high index of suspicion should be maintained when managing patients with neurofibromatosis-1 as not all of them may have typical symptoms of pheochromocytoma. This is especially important when patients are going for a surgical procedure. Second, if found to be hypertensive then these patients while being worked-up for pheochromocytoma should be started on proper medical management which constitutes alpha-blockers followed by beta-blockers in order to avoid any complications. Third, the risk of malignancy in NF-1 associated pheochromocytoma is high as compared to sporadic cases. This warrants a close follow-up even after successful surgical removal of the tumor.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81–88.
- Gruber LM, Erickson D, Babovic-Vuksanovic D, et al. Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1. Clin Endocrinol (Oxf). 2017;86(1):141–149.
- Madigan P, Shaw RV. Neurofibromatosis in 13th century Austria? Neurofibromatosis. 1988;1(5–6):339–341.
- Antônio JR, Goloni-Bertollo EM, Trídico LA. Neurofibromatosis: chronological history and current issues. An Bras Dermatol. 2013;88(3):329–343.
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development conference. Arch Neurol. 1988;45(5):575–578.
- Walker L, Thompson D, Easton D, et al. A prospective study of neurofibromatosis type 1 cancer incidence in the UK. Br J Cancer. 2006;95(2):233–238.
- Rassmussen SA, Yang Q, Friedman JM. Mortality in Neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. 2001;68(5):1110–1118.
- Dluhy RG. Pheochromocytoma: the death of an axiom. N Engl J Med. 2002;346:1486–1488.
- Hari Kumar KVS, Shaikh A, Sandhu AS, et al. Neurofibromatosis 1 with pheochromocytoma. Indian J Endocrinol Metab. 2011;15(S4):S406–S408.
- King KS, Pacak K. Familial pheochromocytomas and paragangliomas. Mol Cell Endocrinol. 2014;386:92–100.
- Képénékian L, Mognetti T, Lifante JC, et al. Interest of systematic screening of pheochromocytoma in patients with neurofibromatosis type 1. Eur J Endocrinol. 2015;175(4):335–344.
- Petr EJ, Else T. Pheochromocytoma and paraganglioma in Neurofibromatosis type 1: frequent surgeries and cardiovascular crises indicate the need for screening. Clin Diabetes Endocrinol. 2018;4:1.
- Tate JM, Gyroffy JB, Colburn JA. The importance of pheochromocytoma case detection in patients with neurofibromatosis type 1: a case report and review of literature. SAGE Open Med Case Rep. 2017;5:2050313X17741016.
- Plouin P, Amar L, Dekkers OM, et al. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol. 2016;174:G1–10.
- Walther MM, Herring J, Enquist E, et al. von Recklinghausen′s disease and pheochromocytomAS. J Urol. 1999;162(5):1582–1586.