
ABSTRACT
Mutations in β-catenin, especially at the residues critical for its degradation, render it constitutively active. Here, we show that mutant β-catenin can be transported via extracellular vesicles (EVs) and activate Wnt signalling pathway in the recipient cells. An integrative proteogenomic analysis identified the presence of mutated β-catenin in EVs secreted by colorectal cancer (CRC) cells. Follow-up experiments established that EVs released from LIM1215 CRC cells stimulated Wnt signalling pathway in the recipient cells with wild-type β-catenin. SILAC-based quantitative proteomics analysis confirmed the transfer of mutant β-catenin to the nucleus of the recipient cells. In vivo tracking of DiR-labelled EVs in mouse implanted with RKO CRC cells revealed its bio-distribution, confirmed the activation of Wnt signalling pathway in tumour cells and increased the tumour burden. Overall, for the first time, this study reveals that EVs can transfer mutant β-catenin to the recipient cells and promote cancer progression.
Introduction
Large-scale colorectal cancer (CRC) sequencing studies have shown that 93% of all tumours had at least one mutation in proteins implicated in the Wnt signalling pathway [Citation1]. Altogether, 16 different Wnt signalling genes were identified to be mutated among which APC accounted for 81% while β-catenin accounted for 5% [Citation2]. Mutations in APC and/or β-catenin have often been associated with the constitutive activation of Wnt signalling pathway and has been established as a major driver of CRC. In the presence of mutated APC, the destruction complex is rendered non-functional, resulting in an elevated level of cytoplasmic β-catenin that later translocates to the nucleus. If β-catenin carries mutations in its phosphorylation domains [Citation3], its degradation is inhibited even in the presence of wild-type APC and a functional destruction complex. Consequently, β-catenin translocates into the nucleus and initiates the transcription of Wnt target genes [Citation4]. Hence, it has been known that the phosphorylation sites in β-catenin have been long conserved from drosophila to humans (S33, S37, T41, S45).
Extracellular vesicles (EVs) are considered as mediators of intercellular communication both at local and distant sites [Citation5–Citation7]. EVs mediate cell-to-cell communication through the horizontal transfer of cargo molecules including proteins and nucleic acids [Citation8,Citation9]. In the context of cancer, EVs-mediated cell-to-cell communication has been shown to regulate several signalling pathways [Citation6,Citation10]. It is now well established that EVs regulate various pathophysiological processes in favour of cancer progression, including remodelling the tumour microenvironment, immune evasion, coagulation, vascular leakiness, establishing pre-metastatic niche, tropism for metastasis, transfer of chemoresistance and metastasis [Citation11,Citation12]. Importantly, many oncoproteins that are implicated in cancer progression (e.g., EGFRvIII and KRAS) are known to be secreted via EVs [Citation13,Citation14].
One of the proposed mechanisms of activating Wnt signalling involves EVs as cellular couriers to transfer Wnt ligands from one cell to another. It has been reported that EVs associated with Wnt ligands (Wnt3a and Wnt5a) and wild-type β-catenin can activate or antagonise Wnt signalling pathway in the recipient cells [Citation15–Citation18]. However, the association of oncogenic mutant β-catenin with EVs has not been studied. Subpopulations of cancer cells with different mutational loads and behavioural variations lead to intra-tumour heterogeneity [Citation19]. Although mutations in KRAS, APC and TP53 are known to be the key drivers in the development and progression of CRC [Citation20,Citation21], the different clonal subpopulations interact with each other and the surrounding stromal tissue via release of soluble factors and EVs.
Here, for the first time, we show that EVs carrying mutant β-catenin can stimulate Wnt signalling pathway in the recipient cells. EVs derived from LIM1215 human CRC cells altered the Wnt signalling activity in RKO CRC cells that bears wild-type β-catenin, both in vitro and in vivo. SILAC-based quantitative proteomics analysis confirmed the transfer of mutant β-catenin to the nucleus of the RKO cells. Intravenous administration of β-catenin containing EVs activated the Wnt signalling pathway and increased the tumour burden of mice implanted with RKO cells. These results suggest that EVs can transfer oncoproteins between cells and aid in cancer progression.
Materials and methods
List of materials (antibodies and primers) used in this study is provided in the Supplementary information.
Trypan blue
Conditioned medium with dead cells was collected and adherent cells were trypsinised using 0.25% (v/v) Trypsin-EDTA and complete medium. Subsequently, medium containing both dead and live cells was subjected to centrifugation at 500 g for 5 min. The pellet was collected and resuspended using 1 mL of 0.4% (v/v) Trypan Blue (Santa Cruz). Dead and live cells were counted using a Neubauer haemocytometer (La fontaine) and Countess® automated cell counter (Invitrogen™) to determine the cell viability.
Cell lines
RKO CRC cells were kindly donated by Prof. John Mariadason (Olivia Newton-John Cancer Research Institute, Melbourne). RKO CRC cells are wild type for APC and β-catenin. Human CRC cell lines LIM1215 were from the Ludwig Institute for Cancer Research in Melbourne. LIM1215 cells are wild type for APC but have a mutation in β-catenin (T41A). LIM1215 and RKO cells were cultured in RPMI and DMEM (with 1% Glutamax), respectively, supplemented with 10% (v/v) FCS and 100 units/mL of penicillin-streptomycin and incubated at 37°C with 5% CO2.
Isolation of EVs
Cells were seeded in 150 mm diameter culture dishes with 20 mL media and grown to 70–80% confluency. The cells were then washed with 1X PBS thrice and cultured with the respective media with EV-depleted FCS for 24 h. EV-depleted FCS was obtained by spinning FCS at 110,000 g for 18 h. Conditioned media (CM) was collected and centrifuged at 500 g for 10 mins to remove cell debris followed by 2000 g for 20 mins at 4°C. The supernatant collected after 2000 g was subjected to centrifugation at 10,000 g for 30 min at 4°C to remove large extracellular vesicles [Citation22]. The supernatant was then subjected to ultracentrifugation at 100,000 g (SW45Ti rotor, Beckman) for 1 h at 4°C. This step was repeated to wash the pellet with 1× PBS to collect EVs and the pellet was stored in -80°C for further analysis. In addition, EVs were isolated using OptiPrepTM density gradient separation as described [Citation23]. Briefly, a discontinued iodixanol gradient was set by diluting 60% w/v stock of OptiPrepTM aqueous solution (Sigma Life Sciences®) in 0.25 M sucrose/10 mM Tris, pH 7.5, to achieve a gradient consisting of 40%, 20%, 10% and 5% w/v solutions. The gradient was layered using 3 mL fractions each of 40%, 20%, 10% and 5% w/v iodixanol solution in a 12 mL polyallomer tube (Beckman Coulter). The EV pellet obtained after differential centrifugation was overlaid on the top of 5% w/v iodixanol solution and spun at 100,000 g at 4°C for 18 h. Fractions of 1 mL were collected from the top of the tube and diluted with 1.5 mL of 1× PBS and further subjected to centrifugation at 100,000 g at 4°C for 1 h. The pellet obtained was again washed with 1 mL 1× PBS and centrifuged at 100,000 g at 4°C for 1 h to collect EVs. The control OptiPrepTM gradient was run in parallel to determine the density of each fraction using 0.25 M sucrose/10 mM Tris, pH 7.5.
Western blotting
SDS-PAGE was used to separate proteins from samples and gels were transferred using the iBlotTM gel blotting system (Life Technologies) and XCell IITM Blot Module (Life Technologies). Skim milk was used to block membranes, which was later probed overnight with primary antibodies. Secondary antibodies used were anti-rabbit and anti-mouse which were conjugated to fluorophores. For visualisation of protein bands, the ODYSSEY CLx (LI-COR®) machine was used.
Luciferase assay
An equal number of cells were seeded in 12-well plates to reach 50–60% confluency. The cells were then transfected with 0.25 µg of TOPFlash or FOPFlash plasmids in the presence of 25 ng of renilla vector using turbofectin. After 24 h, cells were incubated with EVs (30 µg/mL) for another 24 h and finally harvested and lysed. Luciferase assays were conducted using a luciferase assay kit (Promega) according to the manufacturer’s protocol. Cell lysates were prepared using 200 μL of passive lysis buffer with vigorous shaking for 20 mins. Firefly and renilla luciferase activities were measured using a GloMax® 96 Microplate Luminometer (Promega).
SDS-PAGE and tryptic digestion
Equal amounts of protein samples were run in NuPAGE® Bis-tris gels (4–12%) using a MES SDS buffer. A constant voltage of 150 V was applied, and proteins were visualised using a Coomassie stain (Bio-Rad) for 1 h. Gels were destained using 20% methanol and 7.5% acetic acid in Milli-Q water overnight. Protein bands (20) were excised and subjected to in gel-digestion as described previously [Citation23,Citation24]. Briefly, the excised bands were reduced using 10 mM DTT (Bio-Rad) for 30 min, followed by 20 min alkylation using 25 mM iodoacetamide (Sigma®). The gel bands were digested overnight at 37°C with 150 ng of sequencing grade trypsin (Promega) and extracted using 100 µL solution of 50% (v/v) acetonitrile and 0.1% trifluoroacetic acid.
LC-MS/MS
LC MS/MS was carried out on a QExactive plus Orbitrap mass spectrometer (Thermo Scientific) with a nanoESI interface in conjunction with an Ultimate 3000 RSLC nanoHPLC (Dionex Ultimate 3000). The LC system was fitted with an Acclaim Pepmap nano-trap column (Dinoex-C18, 100 Å, 75 µm x 2 cm) and an Acclaim Pepmap RSLC analytical column (Dinoex-C18, 100 Å, 75 µm x 50 cm). The tryptic peptides were passed to the enrichment column through an injector at an isocratic flow of 5 µL/min of 2% v/v CH3CN containing 0.1% v/v formic acid for 5 min applied before the enrichment column was switched in-line with the analytical column. The eluents were 0.1% v/v formic acid (solvent A) and 100% v/v CH3CN in 0.1% v/v formic acid (solvent B). The flow gradient was (i) 0–5min at 3% B, (ii) 5–28 min, 3–25% B (iii) 28–30 min, 25–40% B (iv) 30–32 min, 40–85% B (v) 32–34 min, 85–85% B (vi) 34–34.1 min 85–3% and equilibrated at 3% B for 10 minutes before the next sample injection. The QExactive plus mass spectrometer was run in the data-dependent mode, whereby full MS1 spectra were operated in positive mode, 70 000 resolution, AGC target of 3e6 and maximum IT time of 50ms. Fifteen of the most intense peptide ions with charge states ≥2 and intensity threshold of 1.7e4 were isolated for MS/MS. The isolation window was set at 1.2 m/z and precursors fragmented using normalised collision energy of 30, 17 500 resolution, AGC target of 1e5 and maximum IT time of 100ms. Dynamic exclusion was programmed to be 30 sec. For SILAC-based mass spectrometry, LC-MS/MS was carried out on a LTQ Orbitrap Elite (Thermo Scientific) with a nanoelectrospray interface coupled to an Ultimate 300 RSLC nanosystem (Dionex) as described previously [Citation25].
Database searching and protein identification
Extract-MSn as part of Bioworks 3.3.1 (Thermo Scientific) was used to generate peak list files with the following parameters: minimum mass 300; maximum mass 5,000; grouping tolerance 0 Da; intermediate scans 200; minimum group count 1; 10 peaks minimum and total ion current of 100. Peak lists for LC-MS/MS runs were merged into a single mascot generic file format. Automatic charge state recognition was used due to the high-resolution survey scan (30,000). LC-MS/MS spectra were searched against the NCBI RefSeq human protein database in a target decoy fashion using X!Tandem Sledgehammer (2013.09.01.1). Searching parameters used were: fixed modification (carboamidomethylation of cysteine; +57 Da), variable modifications (oxidation of methionine; +16 Da) and N-terminal acetylation; +42 Da), three missed tryptic cleavages, 20 ppm peptide mass tolerance and 0.6 Da fragment ion mass tolerance. Protein identifications with at least 2 unique peptides were shortlisted and false discovery rate was less than 0.5% [Citation22].
Circos plot
Circos (v 0.67) was downloaded to generate the exome sequencing derived genomic data of LIM1215 cells and MS-derived proteomics data of LIM1215 whole cell lysates (WCL) and EVs. In-house Python scripts were used to generate files for SNVs and INDEL in Circos [Citation22].
RNA isolation
For RNA isolation, 1 mL of TRI® RT reagent was used in a 6-well plate for cells and 50 mg tissue. The cells were lysed and mixed with 50 µL of 4-bromoanisole (BAN) solution with vigorous mixing. The cell suspension was then centrifuged at 12,000 g for 5 min at 4°C to obtain phase separation. The aqueous layer (top part) was separated out carefully and then mixed with an equal volume of isopropanol. The suspension mixture was incubated at room temperature (RT) for 10–15 min and centrifuged at 12,000 g for 5 min at 4°C to pellet out RNA. Washing of the RNA pellet was carried out with 1 mL of 75% (v/v) ethanol and further pelleted by centrifugation. The resulting pellet was resuspended in DEPC-treated water and further stored at −20°C.
cDNA synthesis and qPCR
DNA analysis was carried out using iScriptTM cDNA synthesis kit according to manufacturer’s protocol (Bio-Rad). qPCR was carried out using SensiMixTM SYBR Low-ROX kit (Bioline) according to manufacturer’s instructions using Agilent LC140 qPCR machine. qPCR reaction mixture was heated at 95°C for 10 min for activating polymerase enzyme. The temperature settings used were 95°C for 15 sec, followed by 52°C for 15 sec and15 sec for 40 cycles.
Immunofluorescence
Cells (5 x 105) were overlaid on the sterilised coverslips in 6-well plates with 2 mL media. After 24–48 h, cells were washed thrice with 1× PBS followed by fixation with 4% (v/v) paraformaldehyde for 10 min in dark. Cells were then permeabilised with 0.1% (v/v) Triton X-100 for 10 min and subsequently blocked using 2% (w/v) bovine serum albumin for 1 h. Cells were stained with β-catenin primary antibody at 1:200 dilution (Santa Cruz Biotechnology). Cells were washed thrice with 1× PBS and then probed with Alexa Fluor® 488 conjugated goat anti-mouse IgG (Life Technologies) at 1:200 dilution for 45 min. TO-PRO®-3 iodide (Invitrogen™) at 1:1000 was used as the nuclear stain. Zeisis LSM510 confocal microscope was used to visualise the cells under 40×/1.3 oil DIC M27 objective. ImageJ and Zen software were used for image analysis.
Wound healing
Cells were allowed to reach 100% confluence (monolayer) in 6-well plates. Pipette tips were used to scratch the wound. Medium was replaced to fresh medium with or without LIM1215 cell-derived EVs (30 µg/mL). The plates were then incubated for 18 h at 37°C in 5% CO2. The width of the wound gap was captured under the microscope.
Co-culture assay
In this assay, polycarbonate cell culture inserts (0.4 µm pore size) in 24-well plates (Thermo Scientific™) were used. RKO cells (1000 cells) were seeded in the bottom chambers. The plates were incubated for 24 h at 37°C in 5% CO2. RKO cells were then transfected with TOPFlash/FlopFlash and renilla plasmids and 24 h later the top chambers were seeded with RKO cells (1000 cells) or with LIM1215 cells (1000 cells). Then, the plates were incubated at 37°C in 5% CO2 before performing Luciferase assay on RKO cells in the bottom chamber.
SILAC labelling
RKO cells were cultured in SILAC media containing lysine (Lys8) and arginine (Arg10). Cells underwent at least 9 cell doublings to incorporate the labelled amino acids within the proteome. RKO cells were treated with EVs derived from LIM1215 cells cultured in RPMI media and subcellular fractionation was performed. WCL, cytoplasm and nuclear fraction were subjected to mass spectrometry. Proteome Discoverer was used to quantitate the MS data.
Subcellular fractionation
Subcellular fractionation was performed using a NE-PERTM nuclear and cytoplasmic extraction kit as per manufacturer’s instructions (Thermo Scientific). Cells were harvested and centrifuged at 300 g for 5 min. The cells were washed using 1× PBS and again centrifuged at 300 g for 5 min. Ice cold CER1 was added to the pelleted cells and mixture was vortexed vigorously for 15 sec followed by incubation on ice for 10 min. Ice cold CER II was then added as per manufacturer’s instructions. The mixture was then vortexed and incubated on ice for a min and later centrifuged at 16,000 g for 5 min at 4°C. The resulting supernatant was separated and labelled as the cytoplasmic fraction. The pellet was washed with 1× PBS and treated with ice cold NER reagent, followed by incubation on ice for 40 min. The mixture was vortexed every 10 mins within 40 min incubation and centrifuged at 16,000 g for 10 min to collect the supernatant (nuclear fraction).
Transmission electron microscopy and nanoparticle tracking analysis
EVs were characterised using transmission electron microscopy (TEM) as previously described [Citation23]. Briefly, samples (0.2 µg/µL) were examined using a Jeol JEM-2100 TEM operated at 200 kV. Samples were applied to copper grids for 2 min and excess material was drained by blotting. Samples were then air dried and stained using 2% w/v uranyl acetate solution (Electron Microscopy Services). NanoSight NS300 machine (Malvern Instruments, Malvern, UK) equipped with a sample chamber and 640 nm laser was used to analyse the size distribution of EVs. EVs samples (1–2 μg/mL) were disaggregated using a needle and syringe, before injection into the NanoSight sample cubicle. The frame rate was 30 frame per second. Data analysis was done using the NanoSight NTA 3.2 software.
PKH67 labelling of EVs for in vitro experiments
EV samples were incubated with PKH67 dye (Sigma®) according to manufacturer’s instructions with sight modifications. EV samples (100 µg) were mixed with 230 µL of Diluent C and 2 µL of PKH67 dye. This mixture was then gently suspended using a pipette for 5 min and FCS depleted of EVs were added to the EV mixture to stop the staining reaction. Labelled EVs were then ultracentrifuged at 100,000 x g and resuspended in 1× PBS. As a negative control, FCS depleted of EVs was mixed with PKH67 dye. Once RKO cells attached to the coverslips, they were incubated with EVs for the following time points (1, 2 and 4 h) at 37°C. The cells were then washed thrice with 1× PBS and fixed using 4% formaldehyde for 15 min. The uptake assay was assessed using the LSM 510 Zesis confocal microscope.
Establishing tumour xenografts in athymic nude
RKO cells (2.5 x106) were subcutaneously injected in 6–8 week-old athymic nude mice. After tumour formation, 10 µg of LIM1215 cell-derived EVs labelled with 2.5 µM DiR dye was injected twice a week intravenously. Control mice were injected with PBS. Later, mice were sacrificed before the tumours grew to 1500 mm3. Tumour size was measured daily using digital calipers and tumour volume was calculated according to the formula ½(W2 x L). Tumour bearing mice were anesthetised (isoflurane/O2) and imaged using IVIS software. Mice were euthanised and tissues were dissected. Tissues were snap frozen in liquid nitrogen and later stored at -80°C for RNA/protein extraction. Tissues were embedded in Tissue-Tek O.C.T (Sakura Finetek) and blocks were frozen in dry ice for observation by microscopy.
Statistical analysis
Statistical analysis was performed using a two-tailed t-test and p-values of <0.05 were considered significant. In vitro experiments were conducted in triplicates. In vivo experiments were performed using 4–8 mice per group. Error bars in graphical data represents ± SEM.
Results
Proteogenomic analysis revealed the secretion of mutant β-catenin via EVs
To identify mutant proteins that are secreted by a cell via EVs, we adopted a global proteogenomics approach (Figure S1A). Using exome sequencing, a total of 31,304 INDELs and 48,575 SNVs were identified in the LIM1215 CRC cells (Supplementary Table 1; data deposited to colorectal cancer atlas [Citation26]: (http://www.colonatlas.org/). A customised mutant protein database with 10,111 sequence variations (nonsynonmous SNVs – Supplementary Table 2) was constructed using the exome sequencing data (human RefSeq protein sequences as reference). Next, EVs were isolated from LIM1215 CRC cells by differential centrifugation coupled with ultracentrifugation and was subjected to high-resolution mass spectrometry (MS)-based proteomics analysis. The MS/MS spectra files obtained from the LIM1215 CRC-cell-derived EVs samples were searched against the customised mutant protein database using X!Tandem search engine. Using this integrated genomics and proteomics approach, a total of 89 mutant proteins with an arbitrary cut-off of 2 unique peptides or more than 4 MS/MS spectra (Supplementary Table 3) were identified in EVs (Figure S1B). The exome sequencing data, mutant proteins and abundance of all identified proteins are depicted in the circos plot ()). Interestingly, mutant β-catenin (T41 > A) that can constitutively activate Wnt signalling pathway was secreted via EVs from LIM1215 CRC cells. The MS/MS spectra of a peptide mapped to mutant β-catenin secreted via EVs is represented in ). Though the secretion of wild-type β-catenin via EVs has been shown previously, this is the first report to our knowledge describing the EV-mediated secretion of β-catenin that is mutated in the critical phosphorylation residues (T41A).
Figure 1. Characterisation of EVs derived from LIM1215 CRC cells.
(A) The circos plot represents the genomic and proteomics data obtained from exome sequencing of cells and MS analysis of EVs, respectively. (B) MS/MS spectra of mutant β-catenin (T41 > A) peptide identified in EVs is depicted. (C) Western blot analysis of LIM1215 whole-cell lysate (WCL) for β-catenin and enrichment of β-catenin and EV-enriched markers in EV pellet compared to secretome and 10K. (D) NTA depicting the size distribution of EVs isolated from LIM1215 CRC cells. (E) Western blot analysis (10 µg) of fractions collected from OptiPrepTM density gradient centrifugation (LIM1215 cells) showed enrichment of β-catenin, Alix and TSG101 in fraction 7 and 8. (F) A TEM image of EVs (fraction 7) isolated by OptiPrepTM gradient corresponding to density 1.10 g/mL. (G and H) Western blotting of a panel of CRC cells (20 μg) with mutated and wild-type β-catenin for Alix, TSG101and β-catenin. Western blot analysis indicated the presence of Alix, TSG101 and β-catenin in EV samples (20 μg) isolated from CRC cells carrying both mutated and wild-type β-catenin.
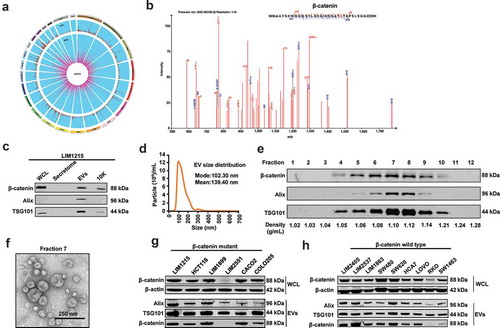
Validation of β-catenin secretion via EVs
To validate the secretion of β-catenin by orthogonal methods, EVs were isolated from LIM1215 cells. Additionally, the pellet obtained after 10,000 g (10K) and LIM1215 secretome (soluble secreted proteins) were also examined for the presence of β-catenin. Western blot analysis revealed that EVs were enriched with β-catenin and the EV markers Alix and TSG101 when compared to the 10K pellet that contains larger EVs such as ectosomes ()). However, β-catenin could not be detected in the LIM1215 cell secretome indicating that β-catenin secretion indeed follows the non-classical secretory pathway via EVs. Next, isolated pellets were characterised by nanoparticle tracking analysis (NTA) to evaluate the particle size distribution of EVs in liquid suspension ()). NTA revealed the presence of nanovesicles with a mean size of 139.4 nm consistent with the size range of EVs. Calnexin which is considered as a negative control for EV preparations, could not be seen in Western blotting (Figure S1C) indicating the purity of the isolated EVs. To prove that β-catenin is contained within EVs and is not associated, the isolated EVs were subjected to iodixanol density gradient separation (OptiPrepTM) centrifugation. Fractions of increasing density were collected, and Western blot analysis was performed using EV enriched proteins Alix and TSG101 to identify EV enriched samples. As shown in ), LIM1215 CRC cell-derived EVs were enriched in fractions 7 and 8 corresponding to density 1.10 and 1.12 g/mL, respectively. This density is consistent with previously reported studies conducted on different cell types and biological fluids [Citation23,Citation27]. Furthermore, β-catenin was detected in fractions 4–9 similar to the EV markers. Next, transmission electron microscopy of EVs obtained from fraction 7 corresponding to density 1.10 g/mL revealed vesicles that were consistent with the size and morphology of EVs ()). Overall, these results suggest that mutant β-catenin is secreted via EVs from LIM1215 CRC cells and are enriched in fractions 7 and 8 corresponding to density 1.10 and 1.12 g/mL.
Packaging of mutated β-catenin in EVs is not selective
It has been reported that β-catenin is mutated at least in 5% of CRC patients. To check whether the secretion of mutant β-catenin via EVs is conserved across multiple cell lines, we performed Western blot analysis on WCL from seven CRC cell lines that are known to have a mutation in β-catenin. Except for LIM2551, the other CRC cell lines expressed high levels of mutant β-catenin ()). Western blot analysis of EVs isolated from the six CRC cells confirmed the secretion of mutant β-catenin via EVs. Mutant β-catenin was not detected in LIM2551 CRC cell-derived EVs as the β-catenin antibody used here could not detect the truncated version of β-catenin (data not shown). Regardless, these observations confirm that mutant β-catenin is indeed being targeted for secretion via EVs. To confirm whether EV packaging of β-catenin is selective between mutant and wild type, CRC cells bearing wild-type β-catenin were selected and subjected to Western blotting. Among the CRC cells that contain wild-type β-catenin, RKO cells showed low levels of endogenous β-catenin (). EVs isolated from the CRC cells with wild-type β-catenin also secreted β-catenin via EVs, hence, confirming that there is no mechanism of selective packaging of mutant β-catenin in the EVs.
LIM1215 cells exhibit high levels of endogenous mutant β-catenin and high Wnt activity
Next, endogenous levels of nuclear and cytosolic β-catenin were assessed in LIM1215 (mutant) and RKO (wild-type) CRC cells using confocal microscopy. Consistent with the mutations, β-catenin (T41 > A) was localised predominantly to the nucleus and to a lesser extent in the plasma membrane of LIM1215 cells ()). However, RKO cells bearing wild-type β-catenin showed cytosolic distribution of β-catenin. Additionally, RKO cells exhibited lower levels of β-catenin compared to LIM1215 cells consistent with the Western blotting results (). These data suggest that LIM1215 cells may have constitutive activation of the Wnt signalling pathway due to the high levels of nuclear β-catenin. To test whether nuclear localisation of β-catenin correlated with high Wnt signalling activity, a luciferase assay was performed after transfection with TOPFlash/FOPFlash and renilla plasmids for 24 h. TOPflash is a luciferase reporter that consists of wild-type TCF binding region whereas FOPflash is used as a negative control containing the mutated TCF binding region upstream of the luciferase gene. Consistent with the confocal results, the luciferase assay revealed high Wnt signalling activity in LIM1215 cells compared to the RKO cells ()). Approximately, a 2676-fold (luciferase activity of LIM1215 compared to RKO cells) increase in Wnt signalling activity was observed in LIM1215 cells that had a mutation in β-catenin ()).
Figure 2. Endogenous localisation of β-catenin in LIM1215 and RKO cells.
(A) Confocal microscopy was performed to determine the localisation of β-catenin (green fluorescence) in LIM1215 and RKO cells. Cells were fixed on the coverslip and stained with anti-β-catenin. β-catenin was mainly localised to the nucleus and to an extent in plasma membranes in LIM1215 cells (scale bar, 20 μm). In RKO cells, wild-type β-catenin was mainly localised to the cytoplasm (scale bar, 40 μm). (B) LIM1215 and RKO cells were transfected with TOPFlash/FOPFlash and renilla plasmids. After 24 h, cells were lysed and analysed for luciferase activity. LIM1215 cells exhibited higher level of Wnt signalling activity compared to RKO cells. Error bar represents ±SEM; **denotes P ≤ 0.01, n = 3. (C) Schematic diagram of co-culture of LIM1215 and RKO cells. (D) RKO cells cultured with LIM1215 cells showed a 2.5-fold increase in Wnt signalling activity. (E) Western blotting for Wnt target gene expression in RKO cells. C: Control; T-Treated (co-culture with LIM1215 cells). (F) LIM1215 cell-derived EVs (100 µg) were incubated with RKO cells for 1, 2 and 4 h at 37°C. PKH67-labelled EVs were taken up by RKO cells after 4 h. Fluorescence was captured using a confocal microscope, Zeiss LSM 510 (magnification, × 100, scale bar 10 μm). (G) Upon incubation of EVs (30 µg/mL), Wnt signalling activity was 2-fold higher in LIM1215 and 5-fold higher in RKO cells compared to their respective controls. Error bar represents ±SEM; **denotes P ≤ 0.01, n = 3. (H) Wound healing assay of RKO cells in the presence (T) and absence of EVs (C – 30 µg/mL). Error bar represents ±SEM; **denotes P ≤ 0.01, n = 3.
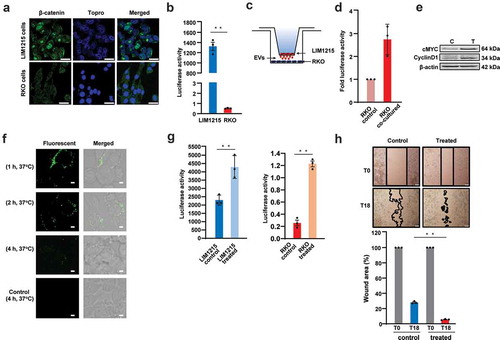
EVs activate Wnt signalling pathway in the recipient cells
To examine whether constitutively active β-catenin could activate Wnt signalling in the recipient cells, a co-culture model was used to mimic the transfer of EVs from LIM1215 to RKO cells ()). RKO cells were seeded in the bottom chamber and after 24 h, RKO cells were transfected with TOPFlash/FOPFlash and renilla plasmids for 24 h. RKO cells were co-cultured with LIM1215 cells in the top chamber for next 24 h. As a control, RKO cells were seeded in the bottom chamber in the absence of LIM1215 cells. The results of the luciferase assay on RKO cell lysates suggested that there was more than a 2-fold increase in Wnt signalling when RKO cells were co-cultured with LIM1215 cells ()). Western blot analysis confirmed the upregulation of Wnt target genes including cMYC and CyclinD1 ()). These results confirm the activation of Wnt signalling in RKO cells when co-cultured with LIM1215 cells.
In co-culture experiments, the functional effects seen in the recipient cells can be due to the soluble secreted fraction and EVs. Hence, to examine whether EVs alone can activate Wnt signalling pathway, EVs were isolated from LIM1215 cells and incubated with RKO cells. Prior to this, the uptake of EVs by RKO cells was studied using confocal microscopy. EVs were labelled with the green fluorescent dye PKH67 and incubated with recipient cells and analysed at three time points at 37°C. At 1 and 2 h, EVs were not taken up by the RKO cells but were attached to the plasma membrane, as confirmed by confocal microscopy ()). However, after 4 h incubation, the EVs were taken up by the RKO cells. As a control, LIM1215 cells were incubated with PKH67 dye premixed with FCS which quenches any excess dye.
Next, LIM1215 cell-derived EVs (30 µg/mL) were incubated with LIM1215 and RKO CRC cells for 24 h to study the alteration in Wnt signalling pathway. It must be noted that RKO is wild type for APC and β-catenin. Upon incubation of EVs with LIM1215 cells, there was a 2-fold increase in Wnt signalling activity in comparison to the untreated cells. Similarly, in RKO cells, there was a 5-fold increase in luciferase activity upon incubation with EVs ()). These results suggest that LIM1215 cell-derived EVs can be taken up by the recipient cells and can induce the Wnt signalling pathway. To investigate the influence of LIM1215 cell-derived EVs in migration of RKO cells, a wound healing assay was performed. LIM1215 cell-derived EVs significantly accelerated wound healing in RKO cells by 18 h ()). The percentage of wound area was 4.5% in RKO cells treated with EVs compared with 37% wound area in untreated RKO cells ()). These results suggest that LIM1215 cell-derived EVs can accelerate migration of RKO CRC cells, though this functional effect should be attributed to the total cargo of EVs and not mutant β-catenin alone. Conversely, RKO cells incubated with EVs derived from SW620 cells (wild-type β-catenin) showed no activation of Wnt signalling pathway (Figure S2A). Moreover, there was no observable change in the expression levels of Wnt signalling target genes (Figure S2B) in RKO cells confirming the importance of mutant β-catenin in activating Wnt signalling pathway.
LIM1215 cell-derived EVs increase the expression of Wnt target genes
To test whether the incubation of EVs with RKO cells resulted in the significant increase of Wnt target genes, Western blotting and qPCR analysis were performed. As shown in ), the expression of the Wnt target gene cMYC was increased in a dose dependent manner. Although β-catenin expression did not change with different concentration of EVs, a prominent increase was observed when the cells were incubated with 150 µg/mL of EVs. It has been reported that an increase in β-catenin expression is induced by pAKT levels in endometrial cancer cell lines [Citation28]. It is known that pAKT mediates phosphorylation of β-catenin and promotes increased stability of β-catenin in the cytoplasm [Citation29,Citation30]. Therefore, pAKT expression levels were examined in RKO cells incubated with different concentrations of EVs. However, no changes in the pAKT levels were observed in RKO cells upon treatment with different concentrations of EVs ()). qPCR analysis confirmed the significant increase in the mRNA levels of Wnt target genes cMYC and Cyclin D1 when RKO cells were treated with EVs ()). Though a high increase in cMYC protein expression was observed only when RKO cells were treated with 150 µg/mL of LIM1215 cell-derived EVs, the mRNA expression was 2-fold upregulated upon incubation with 20 µg/mL LIM1215 cell-derived EVs. This discrepancy could be attributed to the lack of positive correlation between the mRNA and protein expression. In addition, it is also possible that high Wnt activity may be required to stabilise cMYC expression in RKO cells. To confirm the increase in β-catenin in recipient cells, RKO cells were incubated with 150 µg/mL of EVs for 24 h and subjected to confocal analysis. As shown in ), RKO cells showed marked increase in the levels of β-catenin when incubated with EVs. However, nuclear localisation of β-catenin could not be confirmed by confocal microscopy.
Figure 3. EVs activate Wnt signalling pathway in recipient RKO cells.
(A) Western blotting of RKO cells with increasing concentration of EVs showed a dose-dependent increase in cMYC protein levels. (B) qPCR analysis revealed increased mRNA expression of cMYC and Cyclin D1 upon treatment of RKO cells with 2 different concentrations of EVs (* denotes P ≤ 0.05, n = 3, Error bar represents ±SEM). (C) Confocal microscopy revealed increased expression levels of β-catenin in RKO cells incubated for 24 h with 150 µg/mL of LIM1215-derived EVs, compared to the control RKO cells. Fluorescence was captured using a confocal microscope, Zeiss LSM 510 (magnification, × 100, scale bar 10 μm). (D) Venn diagram depicts the number of proteins transferred from EVs to the cytoplasm and nucleus of the RKO cells. (E) MS/MS spectrum of a lightly labelled peptide from mutant β-catenin transferred to the nucleus of the RKO cell.
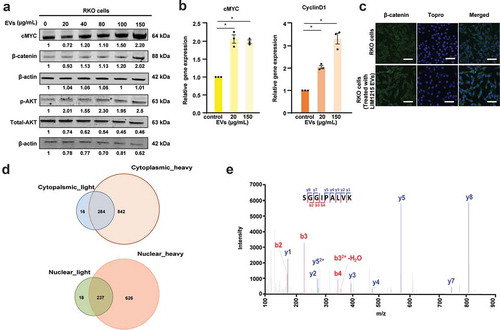
SILAC confirms the transfer of mutant β-catenin from EVs to the nucleus of RKO cells
To confirm whether mutant β-catenin from EVs could be transferred to the nucleus of the RKO cells, a Stable Isotope Labelling with Amino acids in Cell culture (SILAC) based proteomics approach was performed. Using SILAC, proteins were labelled with either light or heavy amino acids. LIM1215 cells were grown in normal media and EVs were isolated. EVs containing proteins with light amino acids were incubated with RKO cells that were grown in media containing heavy amino acids for 9 passages. A schematic view of this workflow is depicted in Figure S3A. Following treatment of RKO cells with EVs (150 µg/mL), subcellular fractionation and Western blotting was performed on the fractions to confirm the purity of the preparations. As shown in Figure S3B, the nuclear fraction was enriched with Topo II β indicating the enrichment of nuclear proteins. Conversely, cytoplasmic fraction was enriched with cytoplasmic marker β-tubulin which was undetected in the nuclear fraction. Additionally, β-actin which is considered as a loading control for WCL, was enriched in the cytoplasmic fraction when compared to the nuclear fraction. These results confirm the enrichment of nuclear and cytoplasmic fractions. Next, the WCL, cytoplasmic and nuclear extracts of RKO cells treated with EVs were subjected to MS analysis. In this way, light-labelled EV proteins could easily be separated from the heavy lysine and arginine-labelled proteins in RKO cells. A total of 144 and 113 proteins from EVs were transferred to the cytoplasm and nucleus of RKO cells, respectively ()). The proteins transferred include histones and methyl transferases (Supplementary Table 4). Importantly, SILAC-based MS results confirmed the presence of two light peptides corresponding to β-catenin in the nuclear fraction of the RKO cells ()). This confirms that EV-derived β-catenin was indeed transferred to the nucleus of recipient cells and activates the Wnt signalling pathway.
LIM1215 cell-derived EVs activate Wnt signalling and increase tumour burden in vivo
Next, EVs were injected into mice bearing RKO cells and its impact on Wnt signalling was monitored. Prior to that, the biodistribution of LIM1215 cell-derived EVs was examined in vivo. EVs were labelled with the lipophilic near infrared fluorescent dye, DiR, and the fluorescence could only be detected in EV fractions and not in control (DiR-PBS) using the in vivo imaging system (IVIS) () and Figure S4). Next, mouse xenografts were generated by subcutaneous implantation of RKO cells. Two-weeks after tumours were palpable, mice were intravenously administered with either DiR-labelled EVs (10 µg) or PBS. Mice were then imaged using IVIS 24 and 96 h post injection ()). Analysis of the organs excised 96 h post injection showed accumulation of DiR-EVs mainly in the liver, lungs, spleen and the GI-tract ()). Importantly, the DiR-EVs administered intravenously were also able to reach the RKO xenograft tumour ()). To investigate whether Wnt signalling activity was affected upon the uptake of EVs, Western blot and qPCR analysis for Wnt target genes were performed on the tumour tissue lysates from control and EV treated mice. As shown in ), an increase in the Wnt target gene cMYC was observed at the protein level in treated mice (T) compared to the control mice (C). In addition, qPCR analysis showed an increase in cMYC, and Axin2 mRNA levels in the tumour tissue of mice treated with LIM1215 cell-derived EVs ()). This confirms the ability of EVs in inducing the Wnt signalling pathway in the tumour tissue. Next, mice bearing RKO CRC cells were administered with LIM1215 cell-derived EVs (10 µg) intravenously twice a week and the tumour volume was monitored until it reached 1500 mm3 ()). A significant increase in the tumour volume was observed when LIM1215 cell-derived EVs were administered to mice bearing RKO CRC cells. Although the Wnt signalling activation can be attributed to the mutant β-catenin, the increase in tumour burden cannot be attributed solely to mutant β-catenin alone and hence the entire EV cargo could play a critical role in this process. Taken together, these results suggest that LIM1215 cell-derived EVs can induce the Wnt signalling pathway in recipient RKO cells and increase the tumour burden.
Figure 4. EVs activate Wnt signalling and increase tumour burden in vivo.
(A) DiR-labelled EVs and PBS control were subjected to fluorescent imaging using IVIS in a 96-well plate. DiR-labelled EVs showed a strong fluorescent signal as opposed to no signal in the control treated with PBS. (B) In vivo fluorescence images of CRC RKO mouse xenograft model injected with DiR-labelled LIM1215 cell-derived EVs at 24 and 96 h. Values correspond to total radiant efficiency [(p/sec/cm2/sr)/(µW/cm2)], n = 5. (C) IVIS images from representative organs from mice injected with labelled EVs (10 µg) and PBS. Values correspond to total radiant efficiency [(p/sec/cm2/sr)/(µW/cm2)], n = 5. (D) Western blot analysis was performed on tumour tissue lysates. Protein levels of the Wnt target gene cMYC was evaluated (n = 4). (E) qPCR analysis was performed on tumour tissue lysates from control and EV-treated mice. The mRNA expression of Wnt target genes (cMYC, Cyclin D1 and Axin2) was found to be elevated in EV-treated mice compared to control mice. Error bar represents ±SEM; *denotes P ≤ 0.05, n = 4. (F) Schematic diagram representing the mouse experiment to examine the relative tumour burden. (G) Mice administered with EVs (10 µg) showed a significant increase in tumour volume (* denotes P ≤ 0.05, n = 4, Error bar represents ±SEM).
![Figure 4. EVs activate Wnt signalling and increase tumour burden in vivo.(A) DiR-labelled EVs and PBS control were subjected to fluorescent imaging using IVIS in a 96-well plate. DiR-labelled EVs showed a strong fluorescent signal as opposed to no signal in the control treated with PBS. (B) In vivo fluorescence images of CRC RKO mouse xenograft model injected with DiR-labelled LIM1215 cell-derived EVs at 24 and 96 h. Values correspond to total radiant efficiency [(p/sec/cm2/sr)/(µW/cm2)], n = 5. (C) IVIS images from representative organs from mice injected with labelled EVs (10 µg) and PBS. Values correspond to total radiant efficiency [(p/sec/cm2/sr)/(µW/cm2)], n = 5. (D) Western blot analysis was performed on tumour tissue lysates. Protein levels of the Wnt target gene cMYC was evaluated (n = 4). (E) qPCR analysis was performed on tumour tissue lysates from control and EV-treated mice. The mRNA expression of Wnt target genes (cMYC, Cyclin D1 and Axin2) was found to be elevated in EV-treated mice compared to control mice. Error bar represents ±SEM; *denotes P ≤ 0.05, n = 4. (F) Schematic diagram representing the mouse experiment to examine the relative tumour burden. (G) Mice administered with EVs (10 µg) showed a significant increase in tumour volume (* denotes P ≤ 0.05, n = 4, Error bar represents ±SEM).](/cms/asset/479e37d6-a7a3-45d7-b066-5edb4f9e1768/zjev_a_1690217_f0004_oc.jpg)
Discussion
It is well known that tumours are comprised of different subpopulations of cancer cells that exhibit salient genetic and behavioural variations leading to intra-tumour heterogeneity [Citation19,Citation31,Citation32]. This diversity within a single tumour is known to be attributed to the different subpopulations of cells displaying genetic and epigenetic factors and acquiring random mutations. In context of CRC, mutations in KRAS, APC and TP53 are known to be the key drivers in the development and progression of the disease [Citation20,Citation21]. Mutations in APC are most common in CRC and are crucial activators of the Wnt signalling pathway. Apart from APC mutations, β-catenin mutations are also responsible for aberrated Wnt signalling activity in CRC [Citation33]. The frequency of APC, KRAS, TP53 and β-catenin mutations occurring alone or in combination may vary. Therefore, different clonal subpopulations bearing different mutational loads could potentially interact with each other and with normal colon cells. It is also speculated that minor clones within the other subclones interact via release of soluble factors (growth factors and cytokines) [Citation34]. We postulated that EVs could potentially play a role in intra-tumour heterogeneity by the intracellular transfer of genetic information within the tumour cells [Citation25].
Several studies have shown that EVs could switch the phenotype of normal cells to a malignant phenotype upon internalisation [Citation35–Citation37]. Recently, two studies demonstrated that EVs from cancer cells phenotypically convert mesenchymal cells to fibroblasts [Citation38,Citation39]. Furthermore, it has been shown that EVs released by cancer associated fibroblasts promotes metastasis of breast cancer cells by upregulation of Wnt-PCP signalling [Citation40]. These above-mentioned studies strongly indicate that EVs regulate several different pathways and could alter the phenotype of the target cells.
Though wild-type β-catenin was detected in EVs [Citation15,Citation17], this is the first report on the presence of mutant β-catenin that is constitutively active in EVs. Though the secretion of mutant proteins via EVs has already been established [Citation13,Citation14], this study examined the functional role of EVs bearing mutant β-catenin on recipient cells in the context of the Wnt signalling pathway. The results suggest that EVs could stimulate Wnt signalling activity in the target cells by the transfer of molecular cargo in vitro and in vivo. Activation of canonical Wnt/β-catenin signalling is regulated by Wnt ligands or via mutations in APC and/or β-catenin. In our study, we show that the transfer of mutant β-catenin can activate the Wnt signalling pathway. However, the role of Wnt ligands in the activation of Wnt signalling cannot be ruled out completely as Wnt ligands have been previously shown to be transferred via EVs [Citation16,Citation18]. We would also like to emphasise that the increase in migration of RKO cells or the tumour burden upon treating with EVs should be attributed to the entire EV cargo and not to mutant β-catenin alone. Nevertheless, the results suggest that circulating EVs can be rapidly taken up by the target cells in the tumour microenvironment and allows for the amplification of the signal in favour of tumour progression.
Author contributions
S.M. conceived and directed the entire project; H.K., P.F., S.A.C., I.A., M.L., L.G., S.B., M.S., H.A.S., C.C., C.G.A. and S.M. performed the experiments; S.K., and S.M. performed the bioinformatics analysis; C.S.A. performed the mass spectrometry; H.K., P.F. and S.M. drafted and finalised the manuscript with inputs from other authors; P.F., H.K. and S.M. prepared the figures; all authors read and approved the manuscript.
Supplemental Material
Download Zip (7.4 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Kim DK, Kang B, Kim OY, et al. EVpedia: an integrated database of high-throughput data for systemic analyses of extracellular vesicles. J Extracell Vesicles. 2013;2.
- The Cancer Genome Atlas, N. Muzny DM, Bainbridge MN, Chang K, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330.
- Ilyas M, Tomlinson IPM, Rowan A, et al. β-Catenin mutations in cell lines established from human colorectal cancers. Proc Nat Acad Sci. 1997;94:10330–13.
- Løvig T, Meling G, Diep C, et al. APC and CTNNB1 mutations in a large series of sporadic colorectal carcinomas stratified by the microsatellite instability status. Scand J Gastroenterol. 2002;37:1184–1193.
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383.
- Cossetti C, Iraci N, Mercer TR, et al. Extracellular vesicles from neural stem cells transfer IFN-gamma via Ifngr1 to activate Stat1 signaling in target cells. Mol Cell. 2014;56:193–204.
- Zhao K, Bleackley M, Chisanga D, et al. Extracellular vesicles secreted by Saccharomyces cerevisiae are involved in cell wall remodelling. Commun Biol. 2019;2:305.
- Anand S, Samuel M, Kumar S, et al. Ticket to a bubble ride: cargo sorting into exosomes and extracellular vesicles. Biochim Biophys Acta Proteins Proteom. 2019.
- Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750.
- Peinado H, Aleckovic M, Lavotshkin S, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–891.
- Wortzel I, Dror S, Kenific CM, et al. Exosome-Mediated Metastasis: communication from a Distance. Dev Cell. 2019;49:347–360.
- Chitti SV, Fonseka P, Mathivanan S. Emerging role of extracellular vesicles in mediating cancer cachexia. Biochem Soc Trans. 2018;46:1129–1136.
- Al-Nedawi K, Meehan B, Micallef J, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619–624.
- Demory Beckler M, Higginbotham JN, Franklin JL, et al. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol Cell Proteomics. 2013;12:343–355.
- Dovrat S, Caspi M, Zilberberg A, et al. 14-3-3 and beta-catenin are secreted on extracellular vesicles to activate the oncogenic Wnt pathway. Mol Oncol. 2014;8:894–911.
- Gross JC, Chaudhary V, Bartscherer K, et al. Active Wnt proteins are secreted on exosomes. Nat Cell Biol. 2012;14:1036–1045.
- Chairoungdua A, Smith DL, Pochard P, et al. Exosome release of beta-catenin: a novel mechanism that antagonizes Wnt signaling. J Cell Biol. 2010;190:1079–1091.
- Koch R, Demant M, Aung T, et al. Populational equilibrium through exosome-mediated Wnt signaling in tumor progression of diffuse large B-cell lymphoma. Blood. 2014;123:2189–2198.
- Hardiman KM, Ulintz PJ, Kuick RD, et al. Intra-tumor genetic heterogeneity in rectal cancer. Lab Investig. 2016;96:4.
- Hsieh J-S, Lin S-R, Chang M-Y, et al. APC, K-ras, and p53 gene mutations in colorectal cancer patients: correlation to clinicopathologic features and postoperative surveillance. Am Surg. 2005;71:336–343.
- Armaghany T, Wilson JD, Chu Q, et al. Genetic alterations in colorectal cancer. Gastrointestinal Cancer Research: GCR. 2012;5:19–27.
- Keerthikumar S, Gangoda L, Liem M, et al. Proteogenomic analysis reveals exosomes are more oncogenic than ectosomes. Oncotarget. 2015;6:15375–15396.
- Kalra H, Adda CG, Liem M, et al. Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics. 2013;13:3354–3364.
- Anand S, Foot N, Ang CS, et al. Arrestin-domain containing protein 1 (Arrdc1) regulates the protein cargo and release of extracellular vesicles. Proteomics. 2018.
- Fonseka P, Liem M, Ozcitti C, et al. Exosomes from N-Myc amplified neuroblastoma cells induce migration and confer chemoresistance to non-N-Myc amplified cells: implications of intra-tumor heterogeneity. J Extracell Vesicles. 2019;8:1597614.
- Chisanga D, Keerthikumar S, Pathan M, et al. Colorectal cancer atlas: an integrative resource for genomic and proteomic annotations from colorectal cancer cell lines and tissues. Nucleic Acids Res. 2016;44:D969–D974.
- Tauro BJ, Greening DW, Mathias RA, et al. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods. 2012;56:293–304.
- Saegusa M, Hashimura M, Kuwata T, et al. Requirement of the Akt/beta-catenin pathway for uterine carcinosarcoma genesis, modulating E-cadherin expression through the transactivation of slug. Am J Pathol. 2009;174:2107–2115.
- Nava P, Kamekura R, Quiros M, et al. IFNγ-induced suppression of beta-catenin signaling: evidence for roles of Akt and 14.3. 3ζ. Mol Biol Cell. 2014;25:2894–2904.
- Fang D, Hawke D, Zheng Y, et al. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282:11221–11229.
- Barranha R, Costa JL, Carneiro F, et al. Genetic heterogeneity in colorectal cancer and its clinical implications. Acta Med Port. 2015;28:370–375.
- Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892.
- Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7537.
- Inda -M-D-M, Bonavia R, Mukasa A, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010;24:1731–1745.
- Suetsugu A, Honma K, Saji S, et al. Imaging exosome transfer from breast cancer cells to stroma at metastatic sites in orthotopic nude-mouse models. Adv Drug Deliv Rev. 2013;65:383–390.
- Roma-Rodrigues C, Fernandes AR, Baptista PV. Exosome in tumour microenvironment: overview of the crosstalk between normal and cancer cells. Biomed Res Int. 2014;2014:10.
- Taylor DD, Gercel-Taylor C. The origin, function, and diagnostic potential of RNA within extracellular vesicles present in human biological fluids. Front Genet. 2013;4:142.
- Cho JA, Park H, Lim EH, et al. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int J Oncol. 2012;40:130–138.
- Cho JA, Park H, Lim EH, et al. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol Oncol. 2011;123:379–386.
- Luga V, Wrana JL. Tumor-stroma interaction: revealing fibroblast-secreted exosomes as potent regulators of Wnt-planar cell polarity signaling in cancer metastasis. Cancer Res. 2013;73:6843–6847.