
ABSTRACT
Tumour metastasis suppressor KAI1/CD82 inhibits tumour cell movement. As a transmembrane protein, tetraspanin CD82 bridges the interactions between membrane microdomains of lipid rafts and tetraspanin-enriched microdomains (TEMs). In this study, we found that CD82 and other tetraspanins contain cholesterol recognition/interaction amino-acid consensus (CRAC) sequences in their transmembrane domains and revealed that cholesterol binding of CD82 determines its interaction with lipid rafts but not with TEMs. Functionally, CD82 needs cholesterol binding to inhibit solitary migration, collective migration, invasion and infiltrative outgrowth of tumour cells. Importantly, CD82–cholesterol/–lipid raft interaction not only promotes extracellular release of lipid raft components such as cholesterol and gangliosides but also facilitates extracellular vesicle (EV)–mediated release of ezrin–radixin–moesin (ERM) protein Ezrin. Since ERM proteins link actin cytoskeleton to the plasma membrane, we show for the first time that cell movement can be regulated by EV-mediated releases, which disengage the plasma membrane from cytoskeleton and then impair cell movement. Our findings also conceptualize that interactions between membrane domains, in this case converge of lipid rafts and TEMs by CD82, can change cell movement. Moreover, CD82 coalescences with both lipid rafts and TEMs are essential for its inhibition of tumour cell movement and for its enhancement of EV release. Finally, our study underpins that tetraspanins as a superfamily of functionally versatile molecules are cholesterol-binding proteins.
Abbreviations: Ab: antibody; CBM: cholesterol-binding motif; CCM: cholesterol consensus motif; CRAC/CARC: cholesterol recognition or interaction amino-acid consensus; CTxB: cholera toxin B subunit; ECM: extracellular matrix; ERM: ezrin, radixin and moesin; EV: extracellular vesicles; FBS: foetal bovine serum; mAb: monoclonal antibody; MST: microscale thermophoresis; pAb: polyclonal antibody; and TEM: tetraspanin-enriched microdomain
Introduction
Tetraspanins are linked to clinical outcomes of cancer patients, regulate cancer progression in animal models and alter cancer cell behaviours such as cell movement in vitro [Citation1]. For instance, expression of tetraspanin KAI1/CD82 is correlated with favourable prognosis in patients with solid malignant tumours [Citation2], and decreased or lost expression of CD82 is frequently observed in invasive and metastatic solid malignant tumours [Citation2]. Although it is well recognized that CD82 inhibits tumour cell movement [Citation2], the mechanism for this inhibition remains unclear at the molecular level.
Like other tetraspanins, CD82 is physically associated with cell adhesion proteins, growth factor receptors and gangliosides to form multimolecular membrane complexes or tetraspanin-enriched microdomains (TEMs) [Citation3–Citation5]. Also, CD82 is present in lipid rafts; and the distribution of CD82 to lipid rafts is cholesterol dependent [Citation6–Citation9]. Moreover, CD82 modulates the interactions between TEMs and lipid rafts [Citation8]. But the mechanism for CD82 coalescence with lipid rafts remains unclear at the molecular level.
CD82 contains several structural elements important for its functions. CD82 can be palmitoylated at five intracellular cysteine residues, and the palmitoylation is needed for CD82 inhibition of tumour cell movement [Citation10]. Transmembrane helix–helix interactions mediated by three polar residues in CD82 transmembrane segments [Citation11], and N-glycosylation at three asparagine residues in CD82 large extracellular loop [Citation12,Citation13] also contribute to cell movement-regulatory activity of CD82. CD82 attenuates the plasma membrane–initiated organization of actin cytoskeleton [Citation14], and such attenuation correlates with reduced movement of CD82-overexpressed tumour cells. But the mechanism for this attenuation remains unclear at the molecular level.
Earlier studies demonstrated that tetraspanins such as CD9, CD81 and CD82 physically bind to cholesterol [Citation7,Citation15]. A recent study on the crystal structure of CD81 revealed that a cholesterol molecule situates within the pocket formed by CD81 transmembrane domains [Citation16]. We found that CD82 links TEMs to lipid rafts and CD82 endocytosis is cholesterol dependent [Citation8]. Thus, identifying the structural element(s) in CD82 molecule critical for its interaction with cholesterol or lipid rafts is essential for understanding the role of CD82–cholesterol interaction in CD82 functions.
In this study, we identified CRAC motifs in CD82 and other tetraspanins, based on the conservative sequence of -L/V-(X)(1–5)-Y-(X)(1–5)-R/K-, in which X represents one to five residues of any amino acid [Citation17]. By analysing the mutant of one of these motifs, we found that this motif is needed for CD82–cholesterol and CD82-lipid raft interactions but not for CD82–TEM interaction. We also found that CD82–cholesterol/–lipid raft interaction is essential for TEM constituent CD82-mediated inhibition of tumour cell movement and invasive outgrowth by releasing (1) plasma membrane–cytoskeleton linker Ezrin and (2) lipid raft components from the cells via extracellular vesicles (EVs), to disengage the plasma membrane–cytoskeleton connection and subsequently impair tumour cell movement.
Materials and methods
Reagents
Primary antibodies (Abs) used in this study were CD82 monoclonal Abs (mAbs) TS82b, 6D7, and 8E4 [Citation18], CD9 mAbs MAB7 and C9BB [Citation19], Annexin-A2 mAb (Santa Cruz Biotechnology, CA), Ezrin polyclonal Ab (pAb) (Santa Cruz Biotechnology), TIMP1 pAb (Santa Cruz Biotechnology), ezrin–radixin–moesin (ERM) mAb and p-ERM pAb (Cell Signaling Technology, MA), and normal goat IgG (Santa Cruz Biotechnology). Secondary Abs were Alexa488- or Alexa594-conjugated goat anti-mouse IgG, goat anti-rabbit IgG, and rabbit anti-goat IgG Abs (ThermoFisher, MA), Cy3-conjugated donkey anti-goat IgG Ab (Jackson ImmunoResearch Laboratories, PA), Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG Ab (Sigma-Aldrich, MO), and Horseradish Peroxidase (HRP)-conjugated goat anti-mouse and anti-rabbit IgG Abs (Sigma-Aldrich).
Extracellular matrices (ECMs) laminin 111 and fibronectin were obtained from ThermoFisher and collagen type-I of rat tails from Corning (NY). Fluorescent probes include Alexa488-conjugated phalloidin and cholera toxin B subunit (CTxB) (ThermoFisher), Filipin (Sigma-Aldrich), DAPI (Sigma-Aldrich) and Alexa488-conjugated Annexin-V recombinant protein (ThermoFisher). Other reagents were bovine serum albumin (BSA) (AMRESCO, TX), FluorSave (EMD Millipore, MA), TAK-475 squalene synthase inhibitor (Sigma-Aldrich) and competent Escherichia coli, lipofectamine 2000, and G418 (ThermoFisher).
Du145 and PC3 human metastatic prostate cancer cell lines, LnCap human prostate cancer cell line and HT1080 human fibroblastoma cell line were obtained from ATCC (VA) and cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Corning) supplemented with 10% foetal bovine serum (FBS) (Biowest, TX), penicillin and streptomycin. Exosome-depleted FBS (Exo-FBS) was obtained from System Biosciences (CA) and used for cell culture subject to exosome isolation.
Other reagents include site-directed mutagenesis kit (Agilent, CA), BCA protein assay kit (ThermoFisher) and Amplex red cholesterol quantification kit (ThermoFisher).
Identification of CRAC, CARC, CRAC-like and CARC-like motifs
Sequences of CD82 and other tetraspanins from different species were aligned. CRAC motifs were identified by analysing amino sequences within transmembrane domains of CD82 and other tetraspanins. The consensus sequences of CRAC, CARC, CRAC-like and CARC-like motifs were described as -[L/V]-[X](1–5)-[Y]-[X](1–5)-[R/K]-, [R/K]-[X](1–5)-[Y]-[X](1–5)-[L/V], -[L/V]-[X](1–5)-[F]-[X](1–5)-[R/K]- and [R/K]-[X](1–5)-[F]-[X](1–5)-[L/V], respectively, where (X)(1–5) represents one to five residues of any amino acid [Citation20].
Site-directed mutagenesis, overexpression and knockdown of CD82
The CRAC motif in the exoplasmic leaflet of the third transmembrane domain of CD82 was mutated based on CD82-containing pCDNA3.1 construct as a template. Amino acid residues leucine105, tyrosine107 and lysine112 were mutated to alanine residues. The mutant was named after the three mutated amino acid residues of the cholesterol-binding motif (CBM): CD82-LYK. The list of primers is as follows:
Table 1. Primer sequences for site directed mutagenesis.
The pCDNA3.1 vector and pCDNA3.1-CD82 WT, -CD82 LYK mutant and -CD82 FM [Citation10] mutant constructs were separately transfected into Du145 cells. CD82-positive clones were established by G418 selection, collected by flow cytometric cell sorting and maintained in culture as stable transfectants. Of note, CD82 FM mutant is palmitoylation-deficient mutant of CD82 [Citation10]. CD82 WT and LYK mutant were also constructed into pEGFP-C1 vector, and the resulting constructs were expressed in Du145 cells for the production of eGFP-CD82 WT and eGFP-CD82 LYK mutant fusion proteins, respectively.
LnCap cells were transfected with CD82 shRNA plasmids (sc-35734-SH) (Santa Cruz Biotechnology, CA) or non-silencing control plasmid to generate LnCap-CD82 KD or -control KD cells, respectively. CD82 shRNA is a mixture of three different plasmids that target distinctive regions of CD82 [Citation21]. The hairpin sequences are GATCCGGGCCCTCTTCTACTTCAATTCAAGAGATTGAAGTAGAAGAGGGCCCTTTTT, GATCCGCCCTCTTCTACTTCAACATTCAAGAGATGTTGAAGTAGAAGAGGGCTTTTT and GATCCCAAGGGTGTGTATATTGTATTCAAGAGATACAATATACACACCCTTGTTTTT. After transfection and puromycin selection, hundreds of resistant clones were pooled, sorted by flow cytometry and then used as CD82 KD or control KD stable transfected cells. LnCap cells were also transfected with siRNA to establish transient transfected cells. The siRNAs used in this study include non-targeting control siRNA pool and human CD82 siRNA-1 (sense: 5′-GGGCCCUCUUCUACUUCAAtt-3′ and antisense: 5′-UUGAAGUAGAAGAGGGCCCtt-3′) (Dharmacon, CO) and human CD82 siRNA-2 (sense: 5′-CCCAU CCUGACUGAAAGUATT-3′ and antisense: 5′-UACUUUCAGUCAG GAUGGGTT −3′) (Sigma-Aldrich) [Citation22].
Cholesterol-peptide binding assay
Cholesterol/chloroform solution at 10 mg/ml was transiently coated on plastic film and promptly air-dried to generate a thin smooth layer of cholesterol. Then, 20 µl of biotin-conjugated peptides (synthesized by Biomatik, DE) dissolved in 6 M urea at a concentration of 0.25 mM was blotted on the cholesterol-coated film for 1 h. The film was washed and biotin was detected with HRP-conjugated streptavidin. Peptides used in the binding assay are (1) VTAGALFYFNMGKLK-biotin, which covers the region containing CD82 WT fourth CBM; (2) VTAGAAFAFNMGALK-biotin, which covers the region containing CD82 LYK mutation and (3) FTTFTVTKYWFYRLK-biotin, corresponding to the transmembrane region of caveolin-1 that contains a cholesterol-binding sequence.
Microscale thermophoresis
Microscale thermophoresis (MST) was used to determine cholesterol-binding affinity to human CD82 as it allows for rapid assessment of binding using small amounts of protein directly from cell lysate [Citation23]. Du145 cells expressing eGFP fused full-length human CD82, or the CD82 LYK mutant, were grown in T75 flasks to 70% confluence and detached using ice cold Phosphate-buffered saline (PBS) and trypsin. Twenty million cells were harvested by centrifugation and washed with repeated 30 ml volumes of cold PBS. Cells were then resuspended in 500 μl Radioimmunoprecipitation assay (RIPA) lysis buffer with added 1× DHALT protease inhibitor cocktail and 2 mM Phenylmethylsulfonyl Fluoride (PMSF). Cells were gently agitated using a tissue homogeniser in a 2-ml conical bottom tube and immediately transferred to a high-speed microfuge tube to clear lysates at 34,000 × g at 4°C for 15 min to reduce background fluorescence. Concentration of eGFP-CD82 was determined using a GFP dosage kit (Abnova KA0911) with optimal fluorescence yield (λ = 470 nm, LED power = 40%) titrated to 700–900 units for binding experiments. All experiments were performed at 22°C using premium-coated capillaries on a NanoTemper Monolith NT.115 (NanoTemper Technologies, CA) with data collected and analysed using the NTControl v2.2.1 and MO. Affinity Analysis v2.1.2 software is previously described [Citation24].
Flow cytometry
Cells were detached with Ethylenediaminetetraacetic Acid (EDTA) (2 mM)/PBS at 90% confluence, then incubated with primary Ab for 1 h on ice. Then, cells were washed three times with ice cold PBS and incubated with FITC-conjugated secondary Ab for 1 h on ice, followed by washing three more times and analysis with FACSCalibur (BD biosciences, NJ).
Cell movement assays
Collective cell migration was examined with wound healing assay. Briefly, cells were cultured to confluence in a six-well plate and pre-incubated with mitomycin (5 µg/ml) at 37°C for 1 h. Then scratches were generated with 200-µl pipette tips and photographed at 0 and 24 h time points. Wound healing was measured with ImageJ.
Solitary cell migration was examined with Transwell migration assay. Briefly, inserts with 8-µm pore size were coated with laminin (10 µg/ml) or fibronectin (10 µg/ml) at 4°C overnight and then blocked with heat-inactivated BSA at 37°C for 1 h. Cells suspended in 0.1% BSA/DMEM were added to the inserts that were placed in 24-well plates loaded with 1% FBS-containing DMEM. The cells were then incubated at 37°C for 3–6 h and fixed with 4% paraformaldehyde. The cells that didn’t migrated through the pores were removed with cotton swabs from the inserts, while the cells that migrated onto the bottom surface of the inserts were further stained with 0.1% (w/v) crystal violet for counting.
Directional cell invasiveness was measured by the ability of cells moving through three-dimensional matrix environment towards a gradient of growth factors. Briefly, 2 × 105 cells were suspended in FBS-free DMEM and loaded to upper chamber of Transwell inserts that were pre-coated with 30 µl of type-I collagen gel (1 mg/ml). DMEM containing 5% FBS was added to lower chamber, and the cells were incubated in 37°C, 5% CO2 incubator for 48 h, fixed, and stained with 0.1% crystal violet. The cells that were remained in upper chamber were removed with cotton tips, and the number of the cells that invaded through collagen gel per field was quantified.
Random cell invasiveness was measured by the ability of cells moving within three-dimensional matrix environment. Briefly, 2 × 105 cells were suspended in 20 µl of type-I collagen gel (1 mg/ml). After gelation, the cell-containing gel was encapsulated in 1 ml of type-I collagen gel (1 mg/ml). Light microscopic images were captured at 0 and 72 h after the second gelation was completed.
Cell-matrix adhesion and gel contraction assays
For cell adhesion assay, 48-well plates were coated with laminin-111 (10 µg/ml) or fibronectin (10 µg/ml) at 4°C overnight and blocked with heat-inactivated BSA (1 mg/ml) at 37°C for 60 min. Cells (1 × 103/well) were loaded to the matrix-coated wells and incubated at 37°C for 30 min. Non-adherent and loosely attached cells were removed with PBS washes. The remaining attached cells were fixed, stained and counted.
For gel contraction assay, cells were mixed with collagen-I solution (1 mg/ml, pH = 7.4). The cell suspensions underwent gelation at 37°C in 5% CO2, followed by addition of complete media. Diameters of the gels were measured and compared at 0 and 48 h for determination of contraction.
Sucrose density gradient floatation analysis
Cells at 90% confluence were rinsed with ice-cold PBS and incubated with 1% Brij 98 or 0.05% Triton X-100 in TNE buffer (20 mM Tris–HCl, pH 7.4; 150 mM NaCl; and 1 mM EDTA) [Citation25]. Cells were scraped off culture dishes and homogenised in an ice-cold, loose-fitting Teflon-glass homogeniser for 20 strokes. Then, for each sample, 2 ml of the homogenate was mixed with 2 ml of 80% sucrose in centrifugation tubes and sequentially overlaid with 5 ml of 30% and 1 ml of 5% sucrose. The samples were centrifuged at 15,000–20,000 × g at 4°C for 21 h in a SW41Ti rotor (Beckman Coulter, Fullerton, CA). For each tube, fractions of 1 ml were collected sequentially from top to bottom of the tube. The fractions were further examined in Western blot.
Western blot
Cells at 80% confluence were washed twice with ice-cold PBS, and then lysed on ice with RIPA lysis buffer (1% NP-40, 0.2% SDS, 50 mM Tris–HCl at pH 7.4, 150 mM NaCl and 2 mM EDTA) supplemented with protease inhibitors (1 mM PMSF, 10 µg/ml aprotinin, and 10 µg/ml leupeptin). After centrifugation at 13,000 × g at 4°C for 20 min, the supernatants were collected, mixed with 2× Laemmli sample buffer and boiled, followed by SDS–PAGE separation and electric transferring to nitrocellulose membrane. The membrane was blocked with 5% fat-free milk in PBS/0.05% Tween-20, blotted sequentially with primary Ab at RT for 1 h and HRP-conjugated secondary Ab at RT for 1 h and washed for chemiluminescence analysis.
Co-immunoprecipitation
Cells at the confluence stage were lysed at 4°C for 20 min with lysis buffer containing 1% Brij 98, 50 mM HEPES (pH 7.4), 150 mM NaCl and 2 mM CaCl2 supplemented with protease inhibitors. Supernatants were collected after 13,000 × g centrifugation at 4°C for 20 min. Cell lysate was precleared with protein A/G-conjugated sepharose beads and incubated with primary Ab (1 μg Ab/500 μg total protein of cell lysate) and the beads at 4°C for 8 h. The immunoprecipitates were then washed with ice-cold lysis buffer and processed for Western blot.
Immunofluorescence and confocal microscopy
Immunofluorescence and confocal microscopic analysis were performed as described previously [Citation19]. Cells cultured on glass coverslips for 2 days were washed with PBS and fixed with 4% paraformaldehyde, followed by cell membrane permeabilisation with 0.1% Brij 98 and blockade with 20% goat serum. The cells were incubated sequentially with primary Abs and fluorochrome-conjugated secondary Abs. Then, the cells were washed, mounted and imaged with a Leica SP2 or SP8 confocal microscope.
Cell apoptosis was assessed with TUNEL assay with the in situ cell death detection kit (Sigma-Aldrich), by following the protocol provided by the manufacturer, and then analysed with fluorescent microscopy.
EV purification and analysis
Cells at 80% confluence were cultured in DMEM supplemented with 1% Exo-FBS for 2 days. The cell culture supernatants were collected and centrifuged at 300 × g, 2000 × g, 10,000 × g and 100,000 × g. EVs were recovered from 100,000 × g pellet and washed once with PBS [Citation26].
After centrifugation at 2000 × g for 10 min to remove cell debris, the cell culture supernatants were also examined for EVs with NanoSight NS300 instrument (Malvern Instruments, Malvern, UK), by following the instruction provided by the manufacturer. The number and size of EVs were analysed with the nanoparticle tracking analysis system (NTA3.2, Build 3.2.16) of the instrument.
Statistical analysis
Quantifications were based typically on three to four individual experiments. Data are presented as mean ± SD or Standard Error of the Mean (SEM). Student’s t-test was used to determine significance of difference, *, ** and *** represent p values less than 0.05, 0.01 and 0.001, respectively.
Results
Tetraspanins contain CRAC motifs in their transmembrane domains
Based on the consensus sequence of CRAC (-L/V-(X)(1–5)-Y-(X)(1–5)-R/K-) [Citation17], CRAC-like, CARC (−R/K−(X)(1−5)−Y−(X)(1−5)−L/V−) [Citation20] and CARC-like motifs, we identified four CBMs in or near the first, second and third transmembrane regions of human CD82 ()). Two motifs are located by the membrane inner leaflet, and the other two motifs are located by the outer leaflet. Motifs 1, 3 and 4, but not motif 2, are well conserved in CD82 across species ()). Thirty out of 33 human tetraspanins contain at least one CRAC, CRAC-like, CARC and/or CARC-like motifs in the transmembrane domains () and Figure S1(a)), underlining cholesterol binding as a general property of tetraspanins. The CBMs in tetraspanins CD9 and CD81 are also well conserved ()), indicating that cholesterol-binding is a common structural property of tetraspanins. Eighteen out 33 tetraspanins contain CBMs that pass through the outer leaflet of the plasma membrane.
Figure 1. Cholesterol-binding motifs in tetraspanins. (a) Schematic representation of cholesterol-binding motifs (CBMs) in human CD82. (b) Sequence alignment of the transmembrane segments of CD82 from different species to identify CRAC, CARC and CARC-like motifs. (c) Summary of CRAC, CRAC-like,CARC and CARC-like motifs in human tetraspanins that have been frequently studied experimentally. Locations of the motifs, relative to outer and inner leaflets, are presented. (d) Sequence alignment of CARC-like motifs in CD9 and CD81.
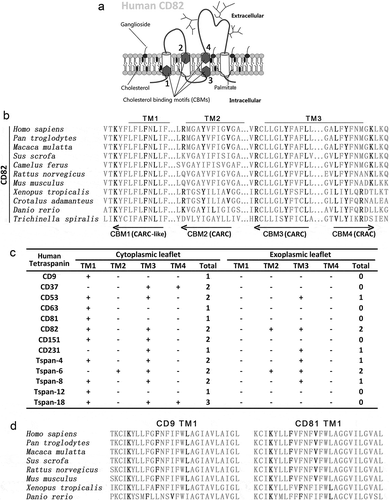
Another type of CBM, the threonine–leucine pair [Citation27], which mediates secreted protein–cell membrane interaction, was found in transmembrane domains and extracellular regions of several tetraspanins (Figure S1(b)). Functions of these threonine–leucine pairs in tetraspanins remain to be studied experimentally. We also searched for cholesterol consensus motif (CCM) [Citation28], which consists of (W/Y)(I/V/L)(K/R) sequence on one helix and (F/Y/R) residue on an adjacent helix, in tetraspanins. We identified a CCM in human CD81 molecule, with WLR sequence in the first and Y residue in the second transmembrane domain (Figure S1(c)). Of note, CD81 is the only tetraspanin with available structure of whole molecule, which is the premise for CCM prediction.
CBM in the third transmembrane domain of CD82 interacts with the cholesterol and lipid raft in the outer leaflet
To examine whether the putative CBMs in CD82 indeed interact with cholesterol, we selected the fourth CBM for experimental analyses, for the reasons described in the Discussion section. First, we used dot blot assay to address if this motif binds cholesterol in vitro. The peptides corresponding to the regions of (1) this CBM, (2) the LYK mutant and (3) caveolin-1 cholesterol-binding motif were blotted on cholesterol-coated film ()). Both CD82 WT and caveolin-1 peptides bound cholesterol, while the LYK mutation led to strong reduction in cholesterol binding. Next, we used MST, which detects the binding affinity of two biomolecules in solution by the changes in fluorescence of one biomolecule in a temperature gradient, to confirm the observation. Using GFP–CD82 WT and GFP–CD82 LYK mutant proteins that were expressed in Du145 cells at a similar level (Figure S2), we demonstrated that cholesterol binds to CD82 WT protein (KD = 5.89 µM) but not CD82 LYK mutant ()).
Figure 2. CD82–cholesterol binding and its molecular functions. (a) Cholesterol-overlaid film was blotted with the peptides of the fourth cholesterol-binding motif of CD82, corresponding region of CD82 LYK mutant, and cholesterol-binding motif of caveolin-1. See materials and methods for details. (b) The cell lysates of Du145-eGFP-CD82 WT and -eGFP-CD82 LYK mutant transfectants were used for cholesterol binding in MST assay as described in materials and methods. N = 4 individual experiments. (c) CD82 expression levels in Du145 transfectants were assessed by flow cytometry for the cell surface expression of CD82 and by Western blot for total cellular CD82 proteins. (d) Sucrose density gradient flotation analysis on the fractional distributions of CD82 proteins in Du145-CD82 WT and -CD82 LYK mutant transfectants under cell lysis conditions of either 0.05% Triton X-100 or 1% Brij 98 detergent. (e) Immunofluorescence staining of CD82 and lipid raft marker GM1 in Du145-CD82 WT and -CD82 LYK mutant cells. Scale bar: 10 µm. And Mander's correlation co-efficients M1, M2 between GM1 and CD82 stainings were quantified (n=3). (f) Co-immunoprecipitation of CD82 and CD9 in Du145 transfectant cells. Cells were lysed with 1% Brij 98. See materials and methods for details.
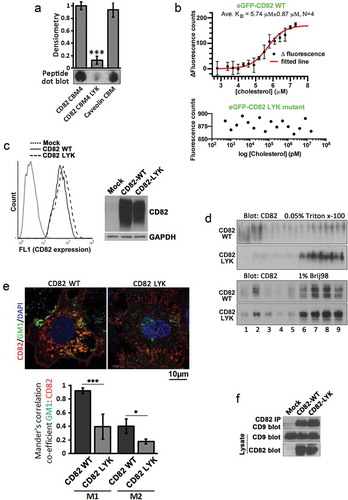
To determine the biological role of CD82–cholesterol interaction, we established Du145 stable transfectants of CD82 WT and CD82 LYK mutant, which were expressed at an equivalent level ()). Sucrose density gradient floatation was used for analysis on the distributions of CD82 WT and LYK mutant proteins in different densities after cell lysis with Triton X-100 and Brij98. Partitions of CD82 proteins to lipid rafts were substantially reduced in CD82 LYK mutant cells, compared with CD82 WT cells, under Triton X-100 lysis conditions, but remained similar between the two transfectants with 1% Brij98 ()). When the cells were lysed with Triton X-100, protein content across different fractions remained the same between Du145 transfectants (Figure S3(a)). However, cholesterol content in Du145-CD82 WT cells was markedly reduced in fraction 2, compared to those in Mock and LYK mutant cells (Figure S3(b)). As quality controls for the floatation experiments, non-raft protein transferrin and raft protein flotillin-1 were indeed distributed in non-raft fractions and raft fractions, respectively (Figures S3(c,d)).
Membrane domains resistant to Triton X-100 tend to contain exoplasmic leaflet raft lipids, such as gangliosides, while the domains resistant to Brij98 have both inner and outer leaflet lipids [Citation29]. Therefore, the fourth CBM in CD82 coalesces CD82 specifically to exoplasmic leaflet rafts, as predicted. By co-staining CD82 and a membrane raft marker ganglioside GM1, we found that CD82-WT, but not CD82-LYK mutant, was co-localized with GM1-enriched membrane regions ()). The CBM does not influence CD82 interaction with TEM, as demonstrated by CD82 and CD9 co-immunoprecipitation ()).
The LYK mutation unlikely disrupts global structure of CD82, given that (1) this CBM is positioned in CD82 transmembrane region; (2) CD82 mAbs 6D7, 8E4 and TS82b are able to recognize their antigen epitopes in CD82 in multiple assays and (3) CD82 LYK mutant associates with TEMs equally well as CD82 WT does.
CD82–cholesterol binding is important for CD82-mediated inhibition of tumour cell movement
Since inhibition of cell movement is an eminent effect that CD82 exerts on tumour cells, we examined the roles of CD82 binding to cholesterol in a variety of modes of tumour cell movement. As shown in , respectively, unlike CD82 WT, CD82 LYK mutant no longer inhibited solitary cell migration onto laminin-111 or fibronectin in Transwell migration assay and collective cell migration in wound healing assay. This mutant was unable to inhibit cell invasion through type-I collagen gel either ()). When Du145 transfectants were encapsulated as cell masses in collagen gel, random cell invasion into surrounding matrix was inhibited by CD82 WT but not by CD82 LYK mutant ()). When Du145-Mock, -CD82 WT, -CD82 LYK mutant and -CD82 FM mutant transfectants were cultured individually in collagen gel, Mock, CD82 LYK mutant and CD82 FM mutant transfectants exhibited robust protrusive outgrowth, while CD82 WT completely lost protrusive outgrowth but produced vesicle type of structures around cells ()), suggesting that (1) both cholesterol binding and palmitoylation are required for CD82 inhibition of invasive growth of tumour cells and (2) CD82 WT probably converted protrusive membrane structures into EVs.
Figure 3. CD82–cholesterol interaction and tumour cell movement. (a) Transwell migration of Du145 transfectant cells was examined towards ECMs and 1% FBS. Numbers of the cells that migrated through the inserts were counted and presented as mean ± SEM (n = 3 individual experiments). *: p < 0.05. (b) Wound healing abilities of Du145 transfectant cells were assessed in scratch assay with the areas that the cells recovered at 24 h after the wounds were created (mean ± SD, n = 3 individual experiments, *: p < 0.05). (c) Invasion of Du145 transfectant cells through three-dimensional ECM towards 1% FBS. Numbers of the cells that invaded through the gels and inserts were counted and presented as mean ± SEM (n = 3 individual experiments). **: p < 0.01. (d) Invasion of Du145 transfectant cells, after dispatching from the cell masses, into three-dimensional ECMs or collagen-I gels. (e) Invasive outgrowth in 3D culture. Individual cells of Du145 transfectant were cultured in collagen-I gel for 2 days, and stained with membrane dye Dio C16 for fluorescence imaging. Scale bar: 10 µm. (f) and (g) Effects of TAK-475 on Transwell cell migration. Du145 (f) and LnCap (g) transfectant cells were treated with DMSO (0.1%) or TAK-475 (20 µM) in serum-free media for 48 h. The cells (10,000 per insert) were loaded to the inserts that were pre-coated with collagen-I (10 µg/ml) and allowed to migrate for 24 h. The numbers of the cells that migrated through the inserts were counted and presented as mean ± SEM (n = 3 individual experiments). *, p < 0.05 and **, p < 0.01.
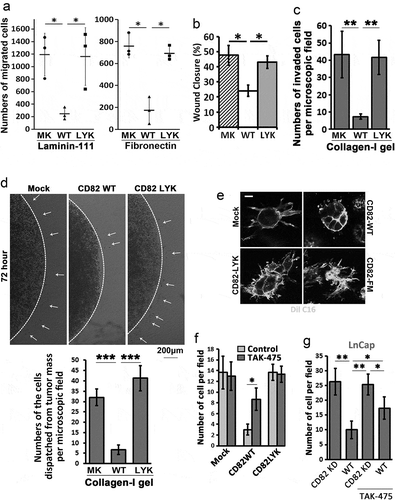
To substantiate the observations, we treated the serum-starved Du145 transfectant cells with TAK-475 (Lapaquistat), a squalene synthase inhibitor [Citation30], to inhibit cellular synthesis of cholesterol. TAK-475 reduced total cellular cholesterol by 30% (Figure S4). With TAK-475 treatment, CD82 WT partially but markedly lost inhibitory activity on cell migration ()). Surprisingly, TAK-475 didn’t alter migration of Du145-Mock and CD82 LYK mutant cells. These observations suggest reciprocal importance of cholesterol and CD82 in their migration-regulatory functions. To confirm these findings, we silenced CD82 in LnCap prostate cancer cell, which express endogenous CD82, by shRNA (Figure S5(a)). As expected, CD82 knockdown cells exhibited elevated migration ()). Such elevation partially lost upon TAK-475 treatment, suggesting that cholesterol contributes at least to migration-inhibitory activity of CD82 to some extends.
Roles of CD82–cholesterol binding in cellular functions relevant to cell movement
To understand how CD82–cholesterol interaction is needed for CD82 inhibition of movement, we evaluated cellular functions relevant to cell movement and also associated with cell adhesion proteins and/or actin cytoskeleton.
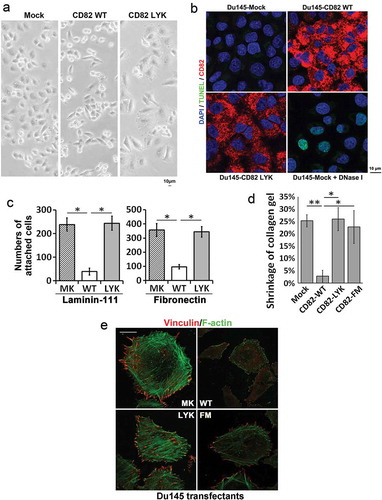
Compared to Du145-Mock cells, Du145-CD82 WT cells spread poorly and exhibited spindle-like shapes ()), as reported in an earlier study [Citation14]. Du145-CD82 LYK mutant cells displayed morphology similar to the Mock cells ()). Overexpression of CD82 WT and LYK mutant did not induce apoptosis in Du145 cells ()). For cell-ECM adhesion, CD82 WT inhibited cell adhesion onto laminin-111 and fibronectin, as we reported earlier [Citation31], but LYK mutant failed to do so ()). The ability to contract collagen gel was lost in CD82 WT, but not Mock, CD82 LYK mutant, or CD82 FM mutant transfectants ()). CD82 WT substantially inhibited stress fibre and focal adhesion formations, as reported by an earlier study [Citation14], while CD82 LYK or CD82 FM mutants did not ()).
Figure 4. Effects of CD82–cholesterol interaction on cell movement-related functions. (a) Morphology and spreading of Du145 transfectant cells in cell culture. Scale bar: 10 µm. (b). Apoptosis of Du145 transfectant cells. The cells cultured on glass coverslips for 2 days were fixed, permeabilised and then examined with TUNEL assay, followed by staining for CD82 and nuclei and by imaging with fluorescent microscopy. Scale bar: 10 µm. Du145-Mock cells that were treated with DNase-I (10 units/ml) at RT for 10 min for the generation of DNA breaks served as positive control. (c). Cell-matrix adhesion of Du145 transfectant cells onto laminin 111- or fibronectin-coated substratum. (d). Cell retraction. Du145 transfectant cells were cultured in collagen-I gel (1 mg/ml) for 2 days. Diameters of the gels were measured at day 0 and 2, and the gel contraction is presented as ratio of the reduction in gel diameter to initial gel diameter (mean ± SEM, n = 3 individual experiments, *: p < 0.05, **: p < 0.01). (e) Fluorescence staining of stress fibres and focal adhesions in Du145 transfectant cells. The cells were fixed, permeabilised and labelled with phalloidin or vinculin mAb and imaged by confocal microscopy. Scale bar: 10 µm.
Together, these observations underline that CD82–cholesterol interaction, along with CD82 palmitoylation, play key roles in CD82-mediated inhibition of cell adhesion proteins and/or actin cytoskeleton.
CD82–cholesterol interaction and CD82 palmitoylation are crucial for CD82-mediated disconnection of the plasma membrane from actin cytoskeleton and inhibition of ERM activity
To explore how CD82 reorganizes actin cytoskeleton to inhibit cell movement and cell adhesion, we examined ERM proteins, which link actin cytoskeleton to the anchorage proteins in the plasma membrane such as CD44 and tetraspanins [Citation32,Citation33].
Phosphorylation or activity of ERM proteins was markedly reduced in CD82 WT cells, but not in CD82 LYK and CD82 FM transfected cells, although total ERM proteins remain equivalent among the four transfectants ()) when cells were lysed directly from cell culture dishes (cell lysis in situ), however, detached cells (cell lysis in suspension) showed a reduction of total ERM proteins ()), indicating some of the ERM proteins were separated with the cells during detachment. The observation that CD82 expression leads to reduced ERM phosphorylation was reproduced in PC3 and CD82 positive LnCap adenocarcinoma cells, and HT1080 fibrosarcoma cells, where overexpression of CD82 in PC3 and HT1080 cells led to reduced ERM phosphorylation, while CD82 knocking down in LnCap cells increased ERM phosphorylation, though the total ERM protein level was not obviously changed ()). These results suggest that the connection between actin cytoskeleton and the plasma membrane becomes weakened with CD82 expression likely through inhibition of ERM phosphorylation, and that the cholesterol interaction and palmitoylation of CD82 are both needed for CD82 to inhibit this connection.
Figure 5. Effects of CD82 and its cholesterol binding on ERM proteins, the plasma membrane–actin cytoskeleton connectors. (a) Western blot analysis of total and phosphorylated ERM (Ezrin (Thr567)/Radixin (Thr564)/Moesin (Thr558)) proteins in Du 145 transfectant cells. For cell lysis in suspension, the cells were detached and spun down, and cell pellets were lysed with RIPA buffer. For cell lysis in situ, the cells attached to culture dishes were directly lysed with RIPA buffer. (b) Total and phosphorylated ERM (Ezrin (Thr567)/Radixin (Thr564)/Moesin (Thr558)) proteins in PC3, HT1080, and LnCap transfectants were examined with Western blot. The cells were lysed directly in culture dishes. (c) Regulation of Ezrin distribution by CD82 or its cholesterol binding. Cells cultured on glass coverslips for 4–5 days were fixed, permeabilised and incubated with Ezrin Ab and the secondary Ab, phalloidin, and DAPI, and examined with confocal microscopy. Scale bar: 10 µm. (d) Releases of ERM proteins via EVs. Exosomes isolated from culture supernatants of the cells were lysed with RIPA buffer, equal amounts of EV proteins (5 μg/lane) were loaded to SDS–PAGE, and ERM and CD9 proteins were examined in Western blot. (e)–(f) The analyses were performed as described in (c). (g) Du145-CD82 WT transfectant cells were cultured on glass coverslips in the serum-free DMEM containing either DMSO (0.1% v/v), TAK-475 (20 µM) or cholesterol (15 µg/ml)/BSA(2%) for 48 h, fixed, stained for Ezrin and F-actin, and photographed with confocal microscopy. Scale bar: 10 µm. Extracellular Ezrin proteins were quantified with ImageJ and presented as relative fluorescence intensity (mean ± SEM, n = 3 individual experiments). *: p < 0.05, **: p < 0.01 and ***: p < 0.001. (h) Cells were treated with DMSO (0.1% v/v) or TAK-475 (20 µM) in serum-free DMEM for 48 h, detached with 2 mM EDTA/PBS and lysed with RIPA buffer. Cell lysates were examined for Ezrin and actin in Western blot.
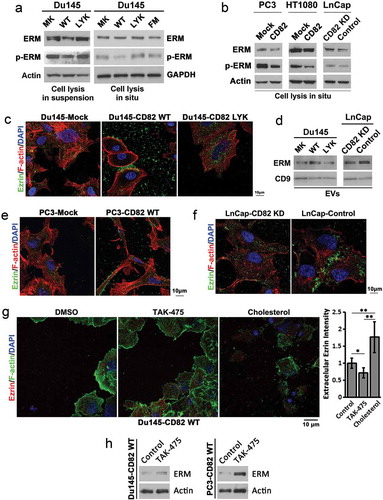
The localization of ERM proteins was determined by immunostaining of Ezrin in Du145, PC3 and LnCap transfectant cells. In Du145 cells, CD82 WT expression (1) mainly promoted Ezrin release to extracellular environment and (2) also translocated Ezrin to membrane bleb-like structures at cell periphery and intercellular space ()), while Ezrin in Du145-Mock and -CD82 LYK mutant cells displayed minimal but similar levels of extracellular release and similar pattern of intracellular distribution. We confirmed that the exosomes from Du145-CD82 WT cells contained more ERM proteins, compared to those from Du145-Mock and -LYK mutant cells ()). In PC3 cells, CD82 WT expression also promoted Ezrin localization to cell periphery and extracellular environment ()). In LnCap cells, which express CD82 endogenously, Ezrin tended to localize to intercellular and membrane bleb-like structures ()); while upon CD82 knockdown by shRNA (Figure S5(a)), Ezrin distribution became similar to those in Du145- and PC3-Mock cells. The same observations were made in Lncap cells upon CD82 knockdown by siRNAs (Figure S5(b–d)). Consistently, silencing CD82 in LnCap cells resulted in less exosomal release of ERM proteins ()). These observations suggest that (1) CD82 regulates extracelluar release and intracellular distribution of ERM proteins, (2) such regulation depends on its cholesterol binding and (3) EVs mediate the extracellular release of ERM proteins.
To further the findings, we found that lowering cellular cholesterol by TAK-475 treatment reduced extracelluar release of Ezrin while adding cholesterol to the cells promoted the release ()). Concomitantly, TAK-475 treatment increased intracellular level of Ezrin ()). As ERM proteins play crucial roles in cell adhesion and migration [Citation34], cholesterol-regulated discharge of Ezrin likely contributes directly to CD82 restraining of cell adhesion and migration.
CD82 induces extracellular release of ezrin by microvesicles and such induction requires CD82–cholesterol interaction
Du145-CD82 WT cells, not Du145-CD82 LYK or -CD82 FM mutant cells, exhibited substantially increased deposition of cholesterol and ganglioside GM1 in extracellular environment ()). Phosphatidylserine, a marker of microvesicles [Citation35] and labelled by Annexin V recombinant protein, and Annexin A2, a marker of EVs [Citation36,Citation37] and labelled with Annexin A2 Ab, were substantially increased in the extracellular environment in the CD82 WT cells, compared to other transfectant cells ()). A large majority of EVs, labelled by Annexin A2, contain GM1 (Figure S6, arrowheads and arrows). These observations indicate that (1) CD82 overexpression increases EV production, (2) CD82-promoted extracellular releases of lipid raft components are mediated, at least, partially by EVs and (3) CD82–cholesterol binding and CD82 palmitoylation are both needed for the CD82-enhanced releases of EVs and lipid rafts.
Figure 6. CD82 and its cholesterol-binding differentially regulate cellular release of EVs. (a) Extracellular staining by filipin and Alexa488-conjugated CTxb in Du145 transfectants. Equal number of the cells were cultured on glass coverslips for 2 days, then fixed and labelled with filipin or Alexa488-conjugated CTxb. For filipin staining, intercellular regions were imaged. For CTxb staining, pericellular regions were imaged. Scale bar: 10 µm. (b) Distributions of Annexin V and Annexin A2 in Du145 transfectant cells. Alexa488-conjugated recombinant Annexin V was used for phosphatidylserine labelling, while Annexin-A2 Ab was used for Annexin-A2 staining. Scale bar: 10 μm. (c) Colocalization of Ezrin with GM1 or Annexin A2 in EVs. For Ezrin and GM1 co-staining, the cells were labelled with the Abs, Alexa488-conjugated CTxB and DAPI. For Ezrin and Annexin A2 co-staining, the cells were incubated sequentially with the primary Abs, Cy3-conjugated donkey anti-goat IgG, normal goat IgG and Alexa594-conjugated goat anti-mouse IgG. Images were obtained by confocal microscopy. Scale bar: 10 µm. (d) The cells were seeded in six-well plate at 50% confluence and cultured in DMEM containing 1% exosome-depleted FBS for 2 – 3 days. The culture supernatants were collected, spun at 2000 × g for 10 min to remove cell debris, and then analysed with NanoSight instrument for EV number and size. Data are presented as mean ± SD (n = 3 individual experiments). *: p < 0.05 and **: p < 0.01.
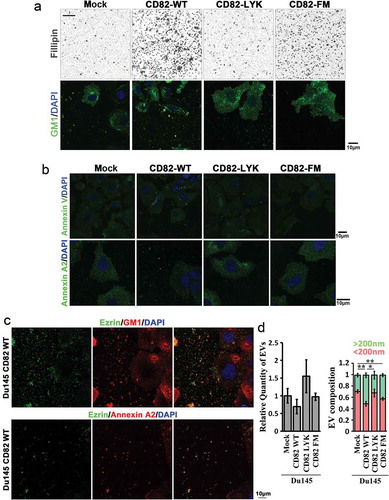
The property of Ezrin-containing EVs was further studied by co-staining Ezrin with GM1 and Annexin A2. In the EVs released from Du145-CD82 WT cells, Ezrin can be co-stained with GM1 or Annexin A2 ()), suggesting a close relationship between Ezrin and lipid raft releases, in the form of EVs.
We further analysed the numbers and sizes of EVs released by Du145 transfectant cells. The numbers of EVs released into culture media were not significantly altered among the four Du145 transfectants (), left panel). The portion of microvesicles (>200 nm) in EVs was increased in Du145-CD82 WT cells, compared to those of Du145-Mock and -CD82 LYK mutant cells, while the portion of exosomes (<200 nm) in EVs was decreased in Du145-CD82 WT cells (), right panel). Together with the increased Ezrin release upon CD82 expression, these observations suggest that (1) Ezrin is released preferentially via microvesicles, (2) Ezrin becomes highly concentrated in EVs in the presence of CD82 and/or (3) Ezrin-containing EVs adhered onto substratum are different from the EVs floating in the culture supernatants.
Discussion
Cholesterol binding appears to be a common feature of tetraspanins
From the peptide sequence analysis of tetraspanins, CBMs are identified in 30 out of 33 human tetraspanins, indicating that cholesterol binding is a common property of almost all tetraspanins and suggesting that cholesterol binding serves as a physical link between TEM and lipid raft [Citation38,Citation39]. This finding also suggests that coalescence with lipid rafts could be a common feature for TEMs. Moreover, the numbers and positions (corresponding to either inner or outer leaflet of the plasma membrane) of CBMs vary among tetraspanins, indicating that tetraspanins likely exhibit differences in extent and pattern to interact with lipid rafts.
Together with earlier observations, we extrapolate that tetraspanin palmitoylation facilitates tetraspanins to form TEMs while tetraspanin cholesterol binding promotes tetraspanins to interact with lipid rafts. Importantly, both interactions are functionally critical, exemplified in this study by the necessity of CD82–cholesterol binding for CD82-mediated inhibition of tumour cell movement.
CD82 binding to cholesterol is required for CD82-mediated inhibition of tumour cell movement
Like CD82 palmitoylation [Citation10], CD82 binding to cholesterol is crucial for CD82-mediated inhibition of tumour cell movement. In this study, we have systematically analysed various modes of tumour cell movement, including solitary migration on two-dimensional ECMs, collective migration on two-dimensional substratum, invasion through three-dimensional ECM, invasive outgrowth within three-dimensional ECM and invasive dispatch from tumour cell mass. CD82 inhibits all types of tumour cell movement, while CD82–cholesterol or -lipid raft interaction is needed for the inhibition.
CD82 inhibits tumour cell movement likely by reducing the activities of cell adhesion proteins and/or actin cytoskeleton, because cell spreading, cell-ECM adhesion, cell traction and focal adhesions become all compromised upon CD82 expression. Notably, CD82 inhibitions of these activities all entail CD82–cholesterol or -lipid raft interaction, underpinning the functional importance of such an interaction.
CD82 drives cellular discharge of ERM proteins by its cholesterol binding
Our earlier study revealed that CD82 overexpression attenuates the plasma membrane–initiated organization of actin cytoskeleton [Citation14]. But the in-depth mechanism still remains unknown. Indeed, tumour cell adhesion, spreading and movement, all of which CD82 inhibits, require the engagement of actin cytoskeleton with the plasma membrane. In this study, we demonstrated that CD82 downregulates total and phosphorylated ERM proteins, which link actin cytoskeleton to the plasma membrane. Equally important, CD82 binding to cholesterol or coalescence with lipid rafts is required for this downregulation.
In addition, we found that CD82 promotes cell periphery distribution and extracellular release of Ezrin. Likewise, the changes in Ezrin localization are lost in CD82 LYK mutant, supporting a key role of CD82–cholesterol binding in regulating Ezrin or the plasma membrane–cytoskeleton connection. Interestingly, Ezrin protein displays bleb- or vesicle-like structures at the cell peripheries. It is unclear whether Ezrin-containing EVs are generated directly from Ezrin-positive membrane blebs as microvesicles or from late endosome as exosomes.
The ERM protein-bound membrane molecules such as EWI2, CD44 and PIP2 also interact with CD82 physically and/or functionally. Specifically, CD82 is physically associated with EWI2 [Citation4], prevents CD44 coalescence with lipid rafts and TEMs [Citation21] and alters PIP2 presence at the plasma membrane [Citation14]. These observations, together with our new findings, underline a close relationship between CD82 and ERM proteins and imply the importance of ERM proteins in CD82 functions such as inhibition of cell movement.
CD82-enhanced release of EVs depends on CD82–cholesterol interaction and correlates with CD82-mediated inhibition of tumour cell movement
Besides Ezrin, CD82 also promotes the discharge of lipid raft components including cholesterol and GM1 ganglioside to extracellular environment. Although the mechanism for this release/shedding remains unknown, the process is dependent on the cholesterol-binding property of CD82 but largely independent of CD82 palmitoylation. As tetraspanins are connected to EVs [Citation40], the release/shedding is likely mediated by EVs, evidenced by Ezrin colocalization of EV marker Annexin-A2 and Ezrin presence in isolated EVs. In fact, CD82 promotes exosome-mediated release of β-catenin to diminish Wnt signalling [Citation41]. Reciprocally, crosslinking of GM1 promotes release of CD82 to extracellular environment (our unpublished data), underlining mutual regulation between lipid rafts and CD82.
As lipid rafts contain important molecules such as Src, NOS and CD44, cellular discharge of lipid rafts could impact cell functions such as movement and adhesion. Thus, CD82-driven and its cholesterol binding-mediated discharges of lipid rafts and ERM proteins likely contribute to CD82 inhibition of tumour cell movement.
Given the observations that (1) CD82 promotes cellular discharges of both ERM proteins and lipid raft components, (2) CD82 promotes EV production and (3) these discharged molecules colocalize with EV markers, it is plausible to extrapolate that CD82 facilitates extracellular release of ERM proteins at, near and/or with lipid rafts through EVs.
To this end, our study reveals that extracellular shedding/release of the components crucial for cell movement serves as a novel mechanism for CD82 to inhibit tumour cell movement. This mechanism is probably equally, if not more, important as other mechanism that CD82 employs to inhibit cell movement such as endocytosis [Citation8,Citation21].
CD82 modulates membrane microdomains interaction through cholesterol binding
Among four CBMs of CD82, two are localized by the inner or cytoplasmic leaflet, suggesting CD82 association with the inner leaflet lipid rafts such as caveolae [Citation42], which contain relatively more cholesterols. In fact, the CBMs by the inner leaflet are present in all CBM-containing tetraspanins such as tumour regulator CD151 and exosome marker CD63. Since exosomes typically carry CD63, the CBM in CD63 could facilitate (1) trafficking of cholesterol and lipid rafts to multivesicular bodies and then (2) release of them by exosomes. This release can be augmented, as CD63 is physically associated with other tetraspanins. Hence, tetraspanin–cholesterol interactions could serve as a mechanism for cellular efference of lipid rafts in which cholesterol and glycosphingolipids are enriched.
The other two motifs of CD82 are by the outer or exoplasmic leaflet and may contribute to CD82 association with the outer leaflet lipid rafts, which contain relatively more glycosphingolipids such as gangliosides [Citation43] as glycosphingolipids reside only in the outer leaflet. Cholesterol modulates the ganglioside conformation [Citation44] and is required for forming and stabilizing lipid rafts, while glycosphingolipids cluster with cholesterol and render lipid rafts more resistant to cholesterol depletion [Citation45]. Because absolute content of cholesterol in outer leaflet is approximately 10 times higher than the one in inner leaflet [Citation46], we have focused on the CBMs of CD82 passing through the outer leaflet.
In addition, CD82 inhibition of tumour cell movement depends on glycosphingolipids [Citation39,Citation47–Citation49]. But CD82 unlikely binds glycosphingolipids directly, given that sphingolipid recognition motif [Citation50] cannot be found in CD82. As glycosphingolipids are clustered with cholesterol in the outer leaflet [Citation51], CD82 may interact glycosphingolipids indirectly through cholesterol. Thus, for the functional relevance, we pay more attention to the CBMs by the outer leaflet.
As the motif at CD82 third TM domain shares higher homology with canonical CRAC motif, we analysed this one extensively in this study. Since (1) three of the four residues between tyrosine and lysine in this motif (LFYFNMGK) are hydrophobic and (2) tyrosine can position at the membrane–water interface [Citation52], we predict that this motif could be fully and/or partially embedded in outer leaflet. As a lysine residue immediately out of the transmembrane domain of murine insulin receptor is responsible for interacting ganglioside GM3 [Citation53], this motif may also mediate CD82 interaction with the ganglioside.
Notably, cholesterol binding of CD82 is needed for CD82-lipid raft interaction but not for CD82–TEM interaction. CD82 LYK mutant no longer partitions to light membrane fractions or interacts with lipid rafts under the Triton X-100 cell lysis condition, which disrupts most of TEMs. CD82 LYK mutant still partitions to light membrane fractions under the Brij98 cell lysis condition, which keeps TEMs largely intact, because other cholesterol-binding tetraspanins such as CD9 and CD81 likely bridge the indirect interaction between CD82 LYK mutant and lipid rafts. In contrast, palmitoylation of tetraspanins optimizes TEM formation but is not essential for teraspanin–lipid raft interaction [Citation54]. These observations underscore labour division but function commonality between different structural elements of tetraspanins.
From this and our earlier studies, we conclude that the function of CD82 as inhibitor of tumour cell invasiveness needs its interaction with both of TEMs and lipid rafts. For the first time, our study conceptualizes that tumour cell movement can be regulated by extracellular shedding/release of the components crucial for cell movement, such as the plasma membrane–actin cytoskeleton linker Ezrin (Figure S7). Based on the studies from us and elsewhere, we also predict that some tetraspanins such as CD82 could be binding proteins and/or transporters for sterols and glycosphingolipids such as cholesterol and gangliosides to transfer these lipids into, out of and/or between cells, as several tetraspanins actively undergo (1) endocytosis through endosomes and (2) exocytosis by EVs.
Supplemental Material
Download Zip (7.7 MB)Acknowledgments
XAZ is an Oklahoma TSET Cancer Research Scholar. We thank Dr. Rodney Tweten for helpful discussion and Ms. Kathy Kyler for English editing. We thank the Peggy and Charles Stephenson Cancer Center for the use of the Molecular Imaging Core, which provided instruments for Nanoparticle Tracking Analysis.
Supplementary Material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Hemler ME. Tetraspanin proteins promote multiple cancer stages. Nat Rev Cancer. 2014;14:49–17.
- Feng J, Huang C, Wren JD, et al. Tetraspanin CD82: a suppressor of solid tumors and a modulator of membrane heterogeneity. Cancer Metastasis Rev. 2015;34:619–633.
- Rubinstein E, Le Naour F, Lagaudrière-Gesbert C, et al. CD9, CD63, CD81, and CD82 are components of a surface tetraspan network connected to HLA-DR and VLA integrins. Eur J Immunol. 1996;26:2657–2665.
- Zhang XA, Lane WS, Charrin S, et al. EWI2/PGRL associates with the metastasis suppressor KAI1/CD82 and inhibits the migration of prostate cancer cells. Cancer Res. 2003;63:2665–2674.
- Odintsova E, Sugiura T, Berditchevski F. Attenuation of EGF receptor signaling by a metastasis suppressor, the tetraspanin CD82/KAI-1. Curr Biol. 2000;10:1009–1012.
- Delaguillaumie A, Harriague J, Kohanna S, et al. Tetraspanin CD82 controls the association of cholesterol-dependent microdomains with the actin cytoskeleton in T lymphocytes: relevance to co-stimulation. J Cell Sci. 2004;117:5269–5282.
- Charrin S, Manié S, Thiele C, et al. A physical and functional link between cholesterol and tetraspanins. Eur J Immunol. 2003;33:2479–2489.
- Xu C, Zhang YH, Thangavel M, et al. CD82 endocytosis and cholesterol-dependent reorganization of tetraspanin webs and lipid rafts. Faseb J. 2009;23:3273–3288.
- Claas C, Stipp CS, Hemler ME. Evaluation of prototype transmembrane 4 superfamily protein complexes and their relation to lipid rafts. J Biol Chem. 2001;276:7974–7984.
- Zhou B, Liu L, Reddivari M, et al. The palmitoylation of metastasis suppressor KAI1/CD82 is important for its motility- and invasiveness-inhibitory activity. Cancer Res. 2004;64:7455–7463.
- Bari R, Zhang YH, Zhang F, et al. Transmembrane interactions are needed for KAI1/CD82-mediated suppression of cancer invasion and metastasis. Am J Pathol. 2009;174:647–660.
- Wang H, Zhang W, Zhao J, et al. N-glycosylation pattern of recombinant human CD82 (KAI1), a tumor-associated membrane protein. J Proteomics. 2012;75:1375–1385.
- Ono M, Handa K, Withers DA, et al. Glycosylation effect on membrane domain (GEM) involved in cell adhesion and motility: a preliminary note on functional alpha3, alpha5-CD82 glycosylation complex in ldlD 14 cells. Biochem Biophys Res Commun. 2000;279:744–750.
- Liu WM, Zhang F, Moshiach S, et al. Tetraspanin CD82 inhibits protrusion and retraction in cell movement by attenuating the plasma membrane-dependent actin organization. PLoS One. 2012;7:e51797.
- Silvie O, Charrin S, Billard M, et al. Cholesterol contributes to the organization of tetraspanin-enriched microdomains and to CD81-dependent infection by malaria sporozoites. J Cell Sci. 2006;119:1992–2002.
- Zimmerman B, Kelly B, McMillan BJ, et al. Crystal structure of a full-length human tetraspanin reveals a cholesterol-binding pocket. Cell. 2016;167:1041–1051 e1011.
- Epand RM. Cholesterol and the interaction of proteins with membrane domains. Prog Lipid Res. 2006;45:279–294.
- Charrin S, Le Naour F, Oualid M, et al. The major CD9 and CD81 molecular partner. Identification and characterization of the complexes. J Biol Chem. 2001;276:14329–14337.
- Zhang F, Michaelson JE, Moshiach S, et al. Tetraspanin CD151 maintains vascular stability by balancing the forces of cell adhesion and cytoskeletal tension. Blood. 2011;118:4274–4284.
- Baier CJ, Fantini J, Barrantes FJ. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci Rep. 2011;1:69.
- Wei Q, Zhang F, Richardson MM, et al. CD82 restrains pathological angiogenesis by altering lipid raft clustering and CD44 trafficking in endothelial cells. Circulation. 2014;130:1493–1504.
- Oncotarget. 2017 Aug 1; 8(31): 51559–51568. doi:10.18632/oncotarget.18086
- Khavrutskii L, Yeh J, Timofeeva O, et al. Protein purification-free method of binding affinity determination by microscale thermophoresis. J Vis Exp. 2013;78:e50541
- Jerabek-Willemsen M, Wienken CJ, Braun D, et al. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev Technol. 2011;9:342–353.
- Slimane TA, Trugnan G, Van ISC, et al. Raft-mediated trafficking of apical resident proteins occurs in both direct and transcytotic pathways in polarized hepatic cells: role of distinct lipid microdomains. Mol Biol Cell. 2003;14:611–624.
- Kowal J, Arras G, Colombo M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113:E968–977.
- Farrand AJ, LaChapelle S, Hotze EM, et al. Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc Natl Acad Sci U S A. 2010;107:4341–4346.
- Rosenhouse-Dantsker A, Noskov S, Durdagi S, et al. Identification of novel cholesterol-binding regions in Kir2 channels. J Biol Chem. 2013;288:31154–31164.
- Pike LJ, Han X, Gross RW. Epidermal growth factor receptors are localized to lipid rafts that contain a balance of inner and outer leaflet lipids: a shotgun lipidomics study. J Biol Chem. 2005;280:26796–26804.
- Nishimoto T, Amano Y, Tozawa R, et al. Lipid-lowering properties of TAK-475, a squalene synthase inhibitor, in vivo and in vitro. Br J Pharmacol. 2003;139:911–918.
- He B, Liu L, Cook GA, et al. Tetraspanin CD82 attenuates cellular morphogenesis through down-regulating integrin alpha6-mediated cell adhesion. J Biol Chem. 2005;280:3346–3354.
- Tsukita S, Oishi K, Sato N, et al. ERM family members as molecular linkers between the cell surface glycoprotein CD44 and actin-based cytoskeletons. J Cell Biol. 1994;126:391–401.
- Sala-Valdes M, Ursa Á, Charrin S, et al. EWI-2 and EWI-F link the tetraspanin web to the actin cytoskeleton through their direct association with ezrin-radixin-moesin proteins. J Biol Chem. 2006;281:19665–19675.
- Arpin M, Chirivino D, Naba A, et al. Emerging role for ERM proteins in cell adhesion and migration. Cell Adh Migr. 2011;5:199–206.
- Muralidharan-Chari V, Clancy JW, Sedgwick A, et al. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci. 2010;123:1603–1611.
- Grill D, Matos ALL, de Vries WC, et al. Bridging of membrane surfaces by annexin A2. Sci Rep. 2018;8:14662.
- Stewart S, Gessler F, Pluchino S, et al. Inside-out: unpredicted annexin A2 localisation on the surface of extracellular vesicles. Matters 2016. ISSN: 2297-8240.
- Di Scala C, Baier CJ, Evans LS, et al. Relevance of CARC and CRAC cholesterol-recognition motifs in the nicotinic acetylcholine receptor and other membrane-bound receptors. Curr Top Membr. 2017;80:3–23.
- Todeschini AR, Dos Santos JN, Handa K, et al. Ganglioside GM2/GM3 complex affixed on silica nanospheres strongly inhibits cell motility through CD82/cMet-mediated pathway. Proc Natl Acad Sci U S A. 2008;105:1925–1930.
- Andreu Z, Yanez-Mo M. Tetraspanins in extracellular vesicle formation and function. Front Immunol. 2014;5:442.
- Chairoungdua A, Smith DL, Pochard P, et al. Exosome release of beta-catenin: a novel mechanism that antagonizes Wnt signaling. J Cell Biol. 2010;190:1079–1091.
- Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50.
- Yu RK, Tsai YT, Ariga T, et al. Structures, biosynthesis, and functions of gangliosides–an overview. J Oleo Sci. 2011;60:537–544.
- Lingwood D, Binnington B, Róg T, et al. Cholesterol modulates glycolipid conformation and receptor activity. Nat Chem Biol. 2011;7:260–262.
- Ilangumaran S, Hoessli DC. Effects of cholesterol depletion by cyclodextrin on the sphingolipid microdomains of the plasma membrane. Biochem J. 1998;335(Pt 2):433–440.
- Liu S-L, Sheng R, Jung JH, et al. Orthogonal lipid sensors identify transbilayer asymmetry of plasma membrane cholesterol. Nat Chem Biol. 2017;13:268–274.
- Todeschini AR, Dos Santos JN, Handa K, et al. Ganglioside GM2-tetraspanin CD82 complex inhibits met and its cross-talk with integrins, providing a basis for control of cell motility through glycosynapse. J Biol Chem. 2007;282:8123–8133.
- Wang XQ, Yan Q, Sun P, et al. Suppression of epidermal growth factor receptor signaling by protein kinase C-alpha activation requires CD82, caveolin-1, and ganglioside. Cancer Res. 2007;67:9986–9995.
- Li Y, Huang X, Zhang J, et al. Synergistic inhibition of cell migration by tetraspanin CD82 and gangliosides occurs via the EGFR or cMet-activated Pl3K/Akt signalling pathway. Int J Biochem Cell Biol. 2013;45:2349–2358.
- Bjorkholm P, Ernst AM, Hacke M, et al. Identification of novel sphingolipid-binding motifs in mammalian membrane proteins. Biochim Biophys Acta. 2014;1838:2066–2070.
- Lingwood D, Kaiser HJ, Levental I, et al. Lipid rafts as functional heterogeneity in cell membranes. Biochem Soc Trans. 2009;37:955–960.
- Yau WM, Wimley WC, Gawrisch K, et al. The preference of tryptophan for membrane interfaces. Biochemistry. 1998;37:14713–14718.
- Kabayama K, Sato T, Saito K, et al. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc Natl Acad Sci U S A. 2007;104:13678–13683.
- Charrin S, le Naour F, Silvie O, et al. Lateral organization of membrane proteins: tetraspanins spin their web. Biochem J. 2009;420:133–154.