
ABSTRACT
Hepatitis A virus (HAV), a classic nonenveloped virus, has recently been found to be released mainly in the form of quasi-enveloped HAV (eHAV) by hijacking host endosomal sorting complexes required for transport (ESCRT) complexes. Unlike the nonenveloped virion, eHAV contains the viral protein pX on the surface of the HAV capsid as an extension of VP1. How HAV capsids acquire the host envelope and whether the pX protein is involved in this process were previously unknown. Here, we analyse the role of pX in foreign protein secretion in exosome-like extracellular vesicles (EVs) and the formation of eHAV. Fusion of pX to eGFP guided eGFP into exosome-like EVs through directing eGFP into multivesicular bodies (MVBs), and apoptosis-linked gene 2-interacting protein X (ALIX) release was significantly enhanced. Coimmunoprecipitation (co-IP) demonstrated the interaction between pX and the ALIX V domain. Removal of the C-terminal half of pX abolished eHAV release and reduced the interaction between the HAV virion and ALIX. Finally, the C-terminal half of pX alone was sufficient for loading eGFP into EVs by interacting with ALIX. In conclusion, the C-terminal part of pX is important for eHAV production and may have potential for large protein complex loading into exosome-like EVs for therapeutic purposes.
Introduction
Hepatitis A virus (HAV) is a faecal-orally transmitted picornavirus that often causes acute hepatitis and even death [Citation1]. The recent discovery of membrane-cloaked “quasi-enveloped” HAV (eHAV) has rekindled investigators’ interest in HAV transmission and supports the idea that nonenveloped viruses are not always naked [Citation2]. Host-derived membranes can help HAV avoid antibody neutralization in the patient’s serum [Citation2], while the nonenveloped HAV virion is highly resistant to heat and acid inactivation [Citation3,Citation4]. Three large structural proteins are responsible for capsid assembly: VP0, VP3 and VP1pX. VP1pX, the largest of these structural proteins, is cleaved by a cellular protease to remove pX from particles containing HAV [Citation5]. pX has been wrongly called “2A”, a nonstructural protein in other picornaviruses that acts as a viral protease or an autocatalytic peptide [Citation6]. The only known function of pX is carried out by its N-terminal 12 amino acids (aa), which are required for efficient structural protein processing and infectious virion production [Citation7,Citation8]. Intriguingly, several lines of evidence indicated that pX might be related to the biogenesis of eHAV virions. For example, the pX structural protein is found in only eHAV particles [Citation9,Citation10], and the pX portion of eHAV particles is located on the surface of the viral capsid as an extension of VP1 [Citation11]. However, the role of pX in eHAV formation awaits exploration.
The release of many enveloped RNA viruses is tightly associated with endosomal sorting complexes required for transport (ESCRT) complexes [Citation2,Citation12], which perform unique topological membrane bending and scission reactions away from the cytoplasm [Citation13]. The ESCRT complex is required for various biological events, such as the biogenesis of multivesicular bodies (MVBs), cytokinesis and retrovirus budding [Citation13]. A late domain in the Gag protein of retroviruses recruits the ESCRT complex components TSG101 and ALIX for viral budding from the plasma membrane [Citation12,Citation14]. Moreover, recent research has found that the nonenveloped RNA virus can also acquire cellular membranes for nonlytic release [Citation2]. Poliovirus and enterovirus, which belong to the Picornaviridae family, seem to egress infected hosts via autophagosome-derived vesicles containing LC3-II [Citation15,Citation16]. Hepatitis E virus, which belongs to the Hepeviridae family, appears to exploit a TSG101-dependent pathway for its nonlytic release [Citation17–Citation19]. HAV, a unique member of the Picornaviridae family, occupies a position between “modern” picornaviruses and more “primitive” precursor insect viruses [Citation11]. Unlike the above viruses, HAV can hijack an ALIX-related exosome-like pathway to release its membrane-cloaked eHAV form [Citation10]. Gradient density centrifugation indicated that eHAV has a density of ~1.05–1.10 g/cm3, which is similar to that of exosomes, whereas nonenveloped HAV has a density of ~1.27 g/cm3. Proteomic analysis revealed that eHAV particle-enriched fractions contain the exosome markers CD9, CD81 and CD63 [Citation20]. Exosomes are generally defined as secreted membrane vesicles corresponding to the intraluminal vesicles (ILVs) of late endosomal compartments secreted upon the fusion of MVBs with the plasma membrane [Citation21]. In addition, HAV was shown to be coimmunoprecipitated with ALIX, especially after detergent treatment [Citation10,Citation20]. The interaction between the ESCRT system and conventional late domains within virus structural proteins is important for virion envelopment. However, because of steric hindrance, YPXnL traditional late domains within HAV capsid proteins are not accessible and on the surface of the HAV virion [Citation11], suggesting the presence of a novel sorting signal that interacts with the ESCRT complex.
Here, we hypothesize that pX plays a key role in virion envelopment through interacting with ESCRT complexes. By combining biochemical and virological approaches, we demonstrate that the C-terminal part of pX is important for the production of eHAV. Since the C-terminal part of pX alone can guide eGFP secretion through exosome-like extracellular vesicles (EVs), this study might shed light on a new route for large protein complex loading into exosome-like EVs.
Results
Ectopic expression of eGFPpX in Huh7 cells results in exosome-mediated eGFP release
pX is present on only the surface of the virus capsid in eHAV; therefore, we hypothesized that pX is responsible for virion envelopment. To test this hypothesis, we established a simplified model by fusing pX to the C-terminus of eGFP and evaluated eGFPpX secretion ()). Western blot analysis showed that pX fusion did not affect the intracellular expression of eGFP or EV markers ALIX, CD81, CD63, VPS4B, TSG101, HSP70 and syntenin-1 (Figure S1(a)). However, extracellular ALIX levels were significantly higher in the supernatant of Huh7-eGFPpX cells than in that from control cells (Supplementary Figure 1(b)), suggesting a correlation between eGFPpX expression and ALIX secretion. Using differential centrifugation, we found that eGFPpX could be pelleted at 10,000 × g and 100,000 × g with the increased release of ALIX, while the release of other exosome markers, the autophagosome marker LC3-II and the ectosome marker ACTN4 was not affected by pX ()). These observations suggested that the fusion of eGFP with pX induced eGFP secretion in exosome-like EVs. To further corroborate these findings, we performed density gradient analysis. eGFPpX was located in density fractions containing exosome-like EVs (1.05–1.10 g/cm3), where the majority of ALIX was detected ()). Moreover, we applied nanoparticle tracking analysis (NTA) to analyse EVs in the cell culture supernatant. NTA in side scatter mode showed that the extracellular particle profiles of Huh7-eGFP and Huh7-eGFPpX were similar and exhibited a peak diameter of 100 nm () right), indicating that pX fusion did not alter the production of EVs. In contrast, EVs from only Huh7-eGFPpX were detected by NTA in fluorescence mode and exhibited a peak diameter of 100 nm () left), suggesting that eGFPpX is associated with EVs. To further confirm this finding, we performed immuno-electron microscopy (EM) using an anti-eGFP antibody. eGFPpX was specifically associated with EVs, while eGFP was not ()). Furthermore, eGFPpX was pulled down from the culture supernatant by CD63-specific or membrane-specific precipitation (), Supplementary Figure 1(b)). To better quantify the secreted eGFPpX+ particles, we performed high-sensitivity flow cytometry (HSCFM), which enables light scattering detection of EVs as small as 40 nm in diameter. The main peak size of EVs, including CD63+ particles, CD81+ particles and eGFPpX+ particles, was approximately 70 nm ()). The percentages of CD63+ particles, CD81+ particles and eGFPpX+ particles were approximately 67.3% (58.7% CD63+/eGFPpX− and 8.6% CD63+/eGFPpX+), 8.9% (6.4% CD81+/eGFPpX− and 2.5% CD81+/eGFPpX+) and 10.1% ()), respectively. Approximately 85.1% of eGFPpX-loaded EVs are CD63+ positive (8.6%/10.1%). In addition, EV-associated eGFPpX was resistant to protease treatment unless NP-40 was added (Supplementary Figure 1(c)), suggesting that eGFPpX is located inside exosome-like EVs. In agreement with previous finding from density gradient profiling ()) and NTA analysis ()), little to no EV (diameter smaller than 100 nm) from eGFP expressing cells and naïve Huh7 cells were found GFP positive (Supplementary Figure 1(d–g)). Overall, these results demonstrated that pX can direct eGFP into exosome-like EVs and that ALIX appears to be involved in this process.
Figure 1. Exogenous expression of recombinant eGFPpX protein stimulates the release of eGFPpX via EVs. (a) Schematic representation of the recombinant eGFPpX protein. (b) Differential centrifugation analysis of EVs secreted from indicated cells. The harvested cell culture supernatant was concentrated and centrifuged by differential centrifugation as described in the Methods section. (c) Density gradient centrifugation analysis of EVs secreted from indicated cells. Concentrated cell culture supernatant was loaded at the bottom of tube for density gradient centrifugation as described in methods. (d) EVs diameter distribution analysis by NTA. The concentrated cell culture supernatant from indicated cells was analysed by NTA. Left: NTA detection of green fluorescence with a 530/25 emission filter; right: NTA with side scatter mode. (e) Immuno-EM analysis of EVs in the concentrated cell culture supernatant. Anti-GFP was used as the primary antibody. The gold is 10 nm in diameter. The green arrow indicates diffuse eGFP. The yellow arrow indicates EV-associated eGFP. The scale bar represents 20 nm. (f) eGFPpX is coimmunoprecipitated by anti-CD63. Human anti-CD63 magnetic beads (Invitrogen, LT-02241) were incubated with the concentrated cell culture supernatant as described in the Methods section.
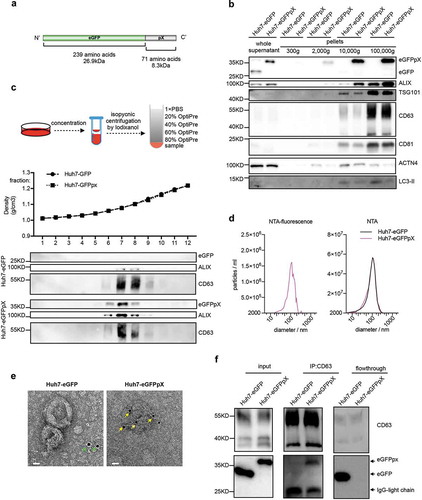
Figure 2. Sizing and molecular profiling of EV-associated eGFPpX via a Flow NanoAnalyzer. AF647-conjugated anti-CD63 or anti-CD81 antibody was used to label the EVs. (a–d) Size profiling of the purified EVs with side scattering mode (a), the FITC channel (b) and the APC channel (c–d). (e) Profiling of CD63+/eGFPpX+ EV particles. (f) Profiling of CD81+/eGFPpX+ EV particles.
pX directs eGFP loading into exosome-like vesicles in MVBs by interacting with ALIX
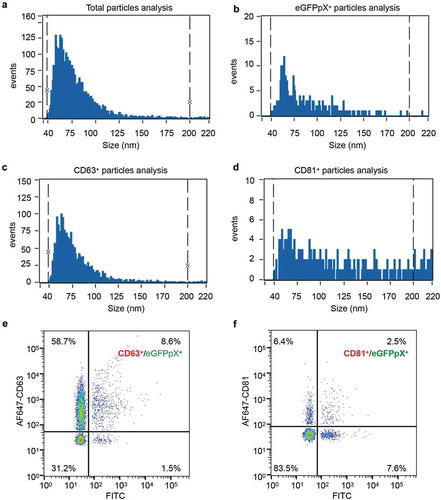
EVs consist of mainly ectosomes and exosomes, which share similar biophysical characteristics, leading to difficulty in their separation [Citation21,Citation22]. However, the biogenesis of ectosomes and exosomes is very different. Ectosomes are assembled at and released from the plasma membrane, while exosomes are released by the exocytosis of MVBs. To elucidate the origin of eGFPpX-loaded EVs, we analysed the intracellular features of eGFP in live cells with a DeltaVision Elite microscope. To our surprise, green puncta were clearly observed in the cytosol of eGFPpX-expressing cells, while the fluorescence for eGFP alone was evenly distributed in both the nucleus and cytosol (), Supplementary Figure 2). The diameter of the eGFPpX puncta ranged from 200 nm to 1000 nm, and the most common eGFPpX puncta diameter was 400 nm ()), similar to that of MVBs. Consistent with this finding, the eGFPpX puncta signal was colocalized with ALIX and CD63, suggesting that eGFPpX is loaded within MVBs (), Supplementary Figure 2). Moreover, immuno-EM showed that eGFPpX was guided into MVBs, while eGFP alone was diffusely distributed ()). In addition, pX did not alter the morphogenesis of MVBs, as the diameters of MVBs and ILVs remained unchanged after the fusion of pX to the C-terminus of eGFP ()). We did not observe accumulation of green fluorescence near the plasma membrane, suggesting the possibility that ectosome-associated eGFPpX secretion is low. These imaging-based analyses indicated that pX can guide eGFP into MVBs, giving rise to eGFPpX-loaded exosome-like EVs.
Figure 3. pX promotes eGFPpX loading into MVBs. (a) pX induces the formation of green eGFP puncta in the cytoplasm. Live cell imaging of eGFP- or eGFPpX-expressing Huh7 cells with a DeltaVision Elite microscope. Images were taken along the Z-axis with a step of 200 nm and rendered by 3D reconstruction or 3D projection (at the right corner). The diameters of the green puncta were calculated and are represented by distribution frequency (bottom). Almost 50 cells were used for the calculation. The scale bar represents 5 μm. (b) eGFPpX colocalized with the ALIX and CD63 in live cells. mCh-ALIX and mCh-CD63 were stably expressed in Huh7-eGFP and Huh7-eGFPpX cells. Images were acquired with a DeltaVision Elite microscope along with the Z-axis with step of 200 nm. The scale bar represents 5 μm. (c) (d) Quantification of colocalization events in (b). (c, d) The percentage of colocalization was calculated by dividing the numbers of colocalized green/red puncta by the total numbers of green or red puncta. (e) Immuno-EM analysis of MVBs with anti-GFP. LR White resin was used for sample preparation. The green arrow indicates cytosolic eGFP, and the yellow arrow indicates the presence of eGFP in MVBs. The gold is 10 nm in diameter. (f) Conventional EM analysis of MVBs. Epoxy resin was used as described in the Methods section. Fiji was used to calculate the diameters of MVBs and ILVs.
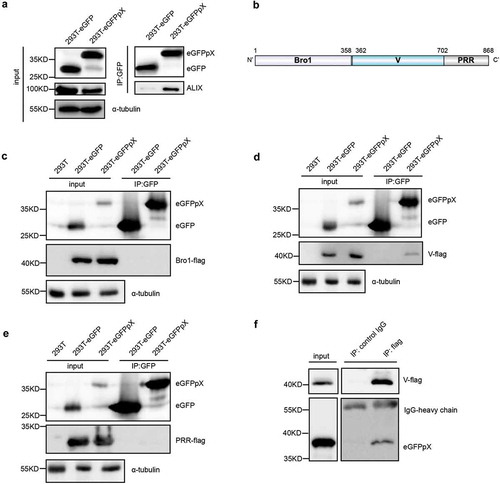
The above data showed that pX alone increased the release of ALIX (), Supplementary Figure 1(a)) and that eGFPpX puncta were colocalized with ALIX aggregates ()). Therefore, we hypothesized that pX directly interacts with ALIX. To test this hypothesis, we performed immunoprecipitation analysis of 293T cells using a GFP-specific antibody. Endogenous ALIX formed a protein complex with eGFPpX ()). To further map the pX-interacting domain in ALIX, we separately expressed the Bro1 domain, V domain and PRR domain of ALIX. pX specifically interacted with the V domain of ALIX ()) and vice versa ()). Collectively, these data indicated that pX facilitates the loading of eGFP into exosomes through the interaction of pX with ALIX.
Figure 4. pX interacts with the V domain of ALIX, an important exosome biogenesis protein. (a) ALIX was coimmunoprecipitated with eGFPpX. Cell lysates of eGFP- or eGFPpX-expressing 293T cells were incubated with anti-GFP nanobody agarose beads as described in the Methods section. Proteins were analyzed by western blotting. (b) Schematic representation of the three domains in ALIX. Numbers indicate the sites of amino acids. (c–e) Identification of the V domain of ALIX that interacts with pX. The C-termini of the Bro1, V and PRR domains of ALIX were tagged with Flag to produce Bro1-Flag (c), V-Flag (d) and PRR-Flag (e), respectively, which were separately expressed in 293T-eGFP and 293T-eGFPpX cells. Co-IP assays were performed with anti-GFP nanobody agarose beads. 293T cells were used as a negative control to confirm rabbit anti-Flag specificity. (f) Co-IP assay with anti-flag to confirm the interaction between the V domain of ALIX and pX. Co-IP was performed using cell lysates of 293T-eGFPpX with anti-flag M2 agarose bead (Thermo Fisher), and mouse-IgG was used as a negative control.
The C-terminal domain of pX determines its interaction with ALIX and exosome-mediated release
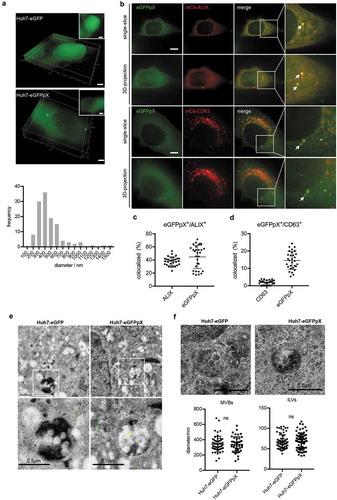
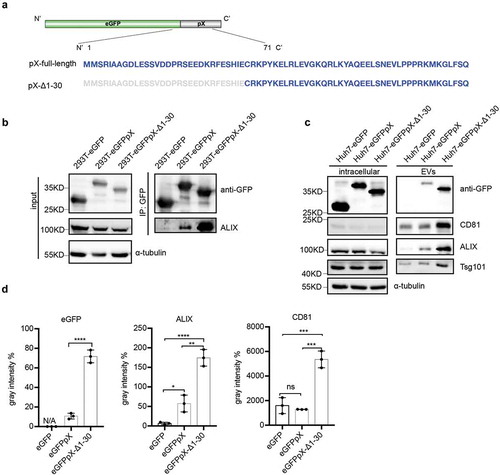
In 2002, Cohen et al. reported that deletion of the N-terminal 40% of pX (aa 1–31) abolished HAV infectivity, whereas deletion of the C-terminal 60% of pX (aa 32–67) resulted in viruses with a small-focus replication phenotype [Citation8]. Ten years later, membrane-enveloped eHAV was discovered, and pX was found to be present in only eHAV but not nonenveloped HAV virions [Citation10]. Here, we assumed that the negligible function of aa 32–67 in pX is responsible for the interaction between pX and ALIX and the acquisition of an exosome-like envelope for eHAV. To confirm this assumption, we overexpressed eGFP fused with pX-Δ1-30 ()). As expected, the C-terminus of pX (aa 31–71) interacted with ALIX and directed eGFP into exosome-like EVs ()). Interestingly, compared to ALIX binding to full-length pX, the deletion of aa 1–30 in pX increased the efficiency of ALIX binding ()) and significantly increased the release of eGFP, ALIX, TSG101 and CD81 via exosome-like vesicles ()). These observations indicated that aa 31–71 in pX are sufficient to guide eGFP into exosome-like vesicles and even more efficient in this function than full-length pX and that aa 1–30 in pX can antagonize its function in foreign protein loading.
Figure 5. The C-terminal 41 aa of pX determines its interaction with ALIX and exosome-mediated release. (a) Schematic representation of the recombinant eGFPpX-Δ1-30 protein. Δ indicates a truncation. Numbers represent the positions of amino acids. (b) Co-IP assays in 293T cells to confirm the interaction between ALIX and pX or the pX mutant. Anti-GFP nanobody agarose beads were used for co-IP as described above. (c) Western blot analysis of EVs released from the indicated cells. EVs were pelleted by ultracentrifugation at 100,000 ×g as described in the Methods section. (d) Relative quantification of the proteins in EVs. The levels of proteins in the western blot shown in (c) were estimated from three independent replicates by their grey intensity using Fiji. Percentages were calculated by dividing by the corresponding intracellular protein level. Data represent the mean of three independent assays.
Truncation of the pX C-terminus in the viral genome abolishes eHAV production
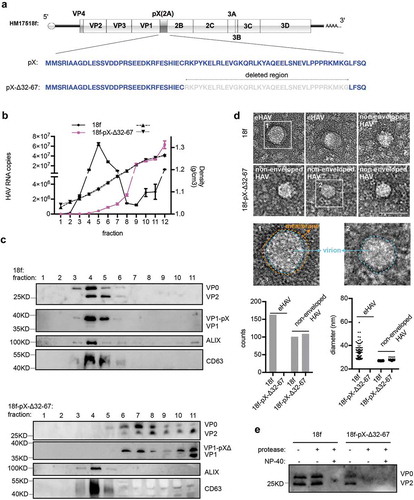
Finally, we tested the biological significance of the C-terminus of pX (aa 31–71) of pX. We constructed an HM175/18f-pX-Δ32-67 infectious clone according to the methods of a previous study [Citation8] in which the C-terminus of pX (aa 32–67) had been deleted ()). Consistent with the findings of Cohen et al., 40 hours after HM175/18f-pX-Δ32-67 viral infection, extracellular infectivity was approximately 4.5-fold lower, while the RNA replication level was approximately 2-fold higher than that following infection with wild-type HM175/18f (Supplementary Figure 3(a)). Notably, at the time of sampling, HM175/18f and HM175/18f-pX-Δ32-67 infection did not result in obvious cell death (data not shown). In addition, aa 32–67 in pX was still absent in the HM175/18f-pX-Δ32-67 genome, which was confirmed by RT-PCR and western blotting (Supplementary Figure 3(b,d)). Next, we performed gradient density centrifugation to separate eHAV from nonenveloped HAV in the cell culture supernatant. Strikingly, RT-qPCR quantification showed that truncation of the C-terminus of pX (aa 32–67) in HM175/18f resulted in an approximately 100-fold decrease in genomic RNA in the fraction containing eHAV (~1.10 g/cm3) and a 10-fold increase genomic RNA in the fraction containing nonenveloped HAV (~1.27 g/cm3) ()). Consistently, the mature extracellular structural protein VP2 and infectious HM175/18f-pX-Δ32-67 virions were located mainly in the fractions corresponding to nonenveloped HAV (~1.27 g/cm3) after centrifugation (), Supplementary Figure 3(c)). VP1-pX was present mainly in eHAV (), upper panel), which was consistent with the results of previous study [Citation10]. In addition, both VP1 and VP1-pX-Δ32-67 were detected in the same fraction of HM175/18f-pX-Δ32-67 (), lower panel). Negative staining analysis showed that eHAV particles (mean diameter of 37 nm) were observed in the viral supernatant from HM175/18f-pX-infected cells but not HM175/18f-pX-Δ32-67-infected cells ()). Finally, a protease digestion protection assay showed that HM175/18f-pX-Δ32-67 could be digested by protease, with or without the presence of NP-40 ()). These data indicated that pX plays a key role in eHAV production. To note, the peak of RNA did not co-exist proportionally with that of VP1-pX, suggesting that a part of genome RNA might be released in different structures.
Figure 6. Truncation of the C-terminal 36 aa of pX in the HAV genome abolishes eHAV release. (a) Schematic representation of the HAV genome. Δ indicates truncation. Numbers represent the positions of amino acids. (b) RT-qPCR quantification of HAV genomic RNA in fractions separated by gradient density centrifugation. HM175/18f and HM175/18-pX-Δ32-67 viruses were used to infect Huh7 cells at a multiplicity of infection (MOI) = 1. Four hours after infection, the cells were washed twice with 1× PBS; 1 day postinfection, the medium was replaced by serum-free DMEM. Forty hours after infection, the cell culture supernatant was harvested for concentration. Due to lower virion production from HM175/18-pX-Δ32-67 virus, 20 ml for HM175/18f and 100 ml for HM175/18-pX-Δ32-67 were applied for ultra-filtration and subsequent gradient density centrifugation. (c) Western blot analysis of fractions separated by gradient density centrifugation. Rabbit anti-VP2 was applied to detect extracellular VP0 and VP2. Rabbit anti-VP1 was used to detect VP1 or pX-bound VP1. VP1-pX-Δ represents VP1-pX-Δ32-67. (d) EM analysis of extracellular HAV virions. HAV virus pelleted at 100,000 ×g was resuspended in 1× PBS and prepared for EM. At least 30 pictures in each group from two independent replicates were used for statistical analysis on the numbers of observed virions. In addition, more than 60% of observed structures were subjected to Fiji software for calculation of the diameters of HAV virions. (e) Protease digestion protection assay to validate the naked form of HM175/18-pX-Δ32-67. Viruses resuspended in 1×PBS were treated with the detergent 0.5%NP-40 and protease (50 μg/ml) at 37°C for 5 minutes.
HAV infection strongly stimulated the release of ALIX, TSG101, CD81 and CD63 in the culture supernatant (Supplementary Figure 4). However, overexpression of eGFPpX alone specifically enhanced the release of ALIX ()), suggesting that ALIX is important for the production of eHAV. To test this hypothesis, we identified the role of ALIX in eHAV production by stably knocking down ALIX in Huh7 cells using shRNAs, among which two shRNAs worked well (S Figure 5(a)). By density gradient centrifugation, we found that ALIX knockdown led to a significant decrease in extracellular HAV RNA and infectivity in the fraction containing eHAV (Supplementary Figure 5(b,c)), which is in agreement with the reduced extracellular VP2 level (Supplementary Figure 5(d)). Furthermore, ALIX knockdown resulted in the reduction of eGFPpX release but appeared not to disturb the release of CD63+ particles and TSG101+ particles (Supplementary Figure 5(e)).
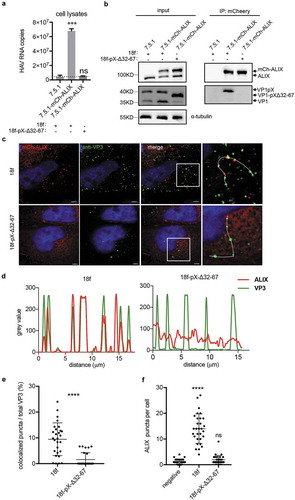
Next, we wanted to determine whether deleting aa 32–67 of pX affects the interaction between HAV virions and ALIX. Co-IP assays using anti-mcherry antibody were performed in cell lysates from mcherry-ALIX expressing Huh-7.5.1 cells ()). Results showed that mCherry-specific antibody could significantly pull down HAV genomic RNA from wild-type HAV-infected cell lysates ()). In contrast, from mutant HAV-infected lysates, we could hardly specifically pull down HAV genomic RNA. Western blot on virus-infected cell lysates also demonstrated the specific interaction between mCherry-ALIX and VP1-pX ()). Additionally, we produced wild-type HAV and mutant HAV from mCherry-ALIX expressing cell line and performed IP on secreted virions using mCherry-antibody (Supplementary Figure 6). In agreement with previous findings [Citation23], NP-40 pre-treatment of secreted wild-type HAV virions led to a significant increase of HAV genomic RNA capture through mCherry-specific antibody. In contrast, either with or without NP40 treatment, mCherry antibody could hardly pull down HAV genomic RNA from mutant HAV preparation. These data suggested that ALIX interacts with VP1-pX on assembled capsids and deletion of the C-terminal of pX abolished this interaction. To corroborate IP results, we performed immunofluorescence (IF) analysis with an antibody specifically recognizing a conformation-based neutralizing epitope in VP3 that reflects the presence of assembled virions. Colocalization between ALIX and VP3 was observed, but deletion of the C-terminal half of pX significantly reduced this colocalization by approximately 5-fold (). In addition, compared to HM175/18f-pX-Δ32-67 infection, HM175/18f infection increased the number of ALIX puncta in the cytoplasm ()), although intracellular ALIX expression was no difference following infection with these two viruses (Supplementary Figure 3(d)). Collectively, these results indicated that the C-terminal half of pX directs HAV virions into exosome-like vesicles and that ALIX plays an important role in this process.
Figure 7. Deletion of the C-terminal half of pX affects the interaction between ALIX and HAV particles. (a)(b) Co-IP assay of the HAV-infected cell lysates using anti-mcherry antibody. Cell lysates were acquired by lysing cells with IP lysis buffer as described in the Methods section. Equivalent amounts of RNA samples were used to perform the co-IP assay. HAV genomic RNA was quantified by qPCR. Dotted line represents the qPCR detection background (a). Data are shown as the mean of three independent assays. Protein was detected by Western blot analysis (b). (c) Immunofluorescence analysis of HAV VP3 and ALIX. mCh-ALIX-expressing Huh7.5.1 cells were infected with HAV virus (MOI=5) as indicated. Forty-eight hours after infection, the cells were fixed with 4% paraformaldehyde as described in the Methods section. Images were acquired along the z-axis (step: 200 nm) with a DeltaVision Elite microscope, and one slice is shown. (d) Colocalization analysis of ALIX and VP3. The intensity of green/red fluorescence along the dotted line in was evaluated by Fiji. (e) Statistical analysis of ALIX/VP3 colocalization. Spots containing colocalized ALIX and VP3 were counted and divided by the total number of VP3 spots to obtain a ratio. (f) Statistic analysis of the numbers of ALIX puncta per cell. ALIX puncta were counted in at least 30 cells in each experimental group.
Discussion
Loading of functional proteins into exosome-like EVs for therapeutic purposes has recently been implemented. Following the discovery of quasi-enveloped eHAV in 2013 [Citation10], there is increasing evidence that many RNA viruses traditionally thought of as nonenveloped undergo nonlytic release from infected cells by enclosing their progeny in membranous vesicles [Citation2,Citation15,Citation16,Citation24]. eHAV biogenesis was reported to be dependent on mainly the ALIX-associated exosome pathway but not other ESCRT-associated pathways, such as those involving TSG101 [Citation10,Citation23]. Since the HAV capsid (a mega protein-RNA complex) can be efficiently loaded into exosome-like EVs, resulting in eHAV, and eHAV robustly infect host cells, we hypothesized that the use of the eHAV loading mode may serve as a novel strategy for functional delivery. In the current study, we aimed to determine the mechanism by which HAV virions are loaded into exosome-like EVs and identify the signal for foreign protein loading. We found that the viral structural protein pX, previously named “2A” according to the nomenclature in the Picornaviridae, interacts with ALIX and facilitates eHAV quasi-envelopment. In addition, tagging eGFP with pX resulted in eGFP sorting into at least 10% of the total EVs, and approximately 85% of the eGFPpX+ EVs were CD63 positive.
The structural proteins of conventional enveloped viruses typically contain “late domains” (e.g. YPXnL, PPXY, P(S/T)AP or GPPX3Y) that mediate interactions with ESCRT complexes [Citation25]. Olga González-López et al. recently demonstrated that two Leu to Ala substitutions (L1,2-A) within the VP2 YPX3L motif disrupt eHAV release during eHAV biogenesis, whereas this substitution did not disrupt capsid assembly and intracellular HAV infectivity [Citation26]. However, Xiangxi Wang et al. demonstrated that the VP2 YPX3L motif is not exposed on the virion surface, indicating the low possibility of an interaction between VP2 and ALIX [Citation11]. Furthermore, we recombinantly overexpressed the eGFP-VP2 protein in Huh7 and 293T cells, similar to the overexpression of eGFPpX. Unfortunately, ALIX was not coimmunoprecipitated with eGFP-VP2 in eGFP-VP2-expressing 293T cells, and the exosome-mediated release of eGFP-VP2 was not detected by western blotting (data not shown). However, pX is located at the five-fold axes of the icosahedron virion as an extension of the C-terminus of VP1 [Citation11]. Therefore, pX is more likely than VP2 to interact with ALIX during and after HAV capsid assembly. Similar to the interaction between the YPXnL “late domain” in HIV-I Gag and the V domain of ALIX [Citation27], the V domain of ALIX could specifically interact with HAV pX. Alanine mapping of the pX C-terminus did not reveal a defined ALIX-binding motif (data not shown), suggesting the presence of a specific tertiary structure contributing to the interaction between pX and ALIX. Removal of the coding region for the pX C-terminus from the HAV genome resulted in a drastic decrease in eHAV production and a corresponding increase in naked HAV release. Partial knockdown of ALIX resulted in a similar decrease in eHAV and naked HAV production, which suggested that ALIX is also important for the release of naked HAV.
A recent investigation suggested that naked nonenveloped HAV results from stripping of the eHAV membrane cloak by the detergent action of bile salts [Citation28]. The ALIX antibody pulled down low levels of the HAV genome without the addition of NP-40, suggesting that the presence of naked HAV particles results from envelope-removed eHAV. Interestingly, from the data in our study, we found that nonenveloped HAV can still be released from infected cells when the C-terminal part of pX is removed. Therefore, we demonstrated an alternative mode for the production of nonenveloped HAV that may be independent of eHAV biogenesis. Although obvious cell death was not observed 40 hours after infection, the possibility that naked HAV is released from a small percentage of dying cells cannot be excluded. Further investigation to decipher the relationship between the biogenesis of eHAV and the release of nonenveloped HAV is needed.
HAV infection produces heterogeneous viral particles such as full particles containing the genomic RNA and empty particles absent of viral RNA [Citation29]. Un-cleaved VP0 is only present in HAV empty particles, while in HAV full particles VP0 is at least partially cleaved [Citation11,Citation30]. Consistent with this, our data ( and supplementary Figure 5) showed that the VP0-enriched fraction does not co-float with genomic RNA-enriched fraction, while mature VP2 was positively correlated with RNA. Removal of C-terminal half part of pX did not ablate the release of HAV empty particles which did not co-float with CD63 and ALIX. Following study will be required to reveal the cellular machinery governing the production of this population. HAV empty particles had been detected in the stool of infected patients about 50 years ago [Citation31], and its biological significance remains unclear. Interestingly, functional significance of virus heterogeneous assembly received growing attention. Independent from infectious HCV lipoviral particles, HCV E2 was released on exosome-like vesicles which assist HCV infection through absorbing neutralizing antibody [Citation32]. Morphological and compositional heterogeneity of Influenza A virus serves as a determinant of viral escape strategy in response to the antivirals [Citation33].
At present, there have been different attempts to use exosome-like vesicles as therapeutic drug carriers. The loading of proteins with high molecular weights has been riddled with several problems: the high cost of protein production and purification, unexpected modifications and tight membrane associations [Citation34,Citation35]. In this study, we found that the C-terminal 41 aa of pX is responsible for the HAV capsid to acquire exosome-like membranes. Expression of pX did not alter the diameters of MVBs, ILVs or EVs, suggesting that pX itself does not significantly modify the exosome-like vesicle production pathway. Compared to an 11 aa peptide containing the YPDL late domain, the C-terminal 41 aa of pX are more efficient in loading foreign proteins (data not shown). Ongoing research in our laboratory will determine whether the loading capacity of pX is equivalent to or higher than that of other reported protein loading methods. After endocytosis, eHAV capsids can be released into the cytosol of infected cells for capsid disassembly and the initiation of translation, which suggests that pX-guided protein cargo in exosome-like EVs might be functionally delivered into target cells. Further study is necessary to clarify this functional delivery.
In conclusion, the HAV structural protein pX plays an essential role in the membrane envelopment of HAV, and the C-terminal domain of pX is sufficient to direct exogenous proteins into exosome-like vesicles. The use of HAV pX might be a new route for large protein complex loading into exosome-like EVs and the functional delivery of these complexes.
Materials and methods
Plasmids and reagents
DNA molecules including eGFP, eGFPpX, eGFPpX-Δ-Deletions, mCh-Alix, mCh-CD63 and Bro1/V/PRR-flag were amplified by PCR and ligated to lentivirus vector pWPI-puro between SbfI and SpeI sites using the EasyGeno Assembly ligation kit (Tiangen). The HM175/18f virus cDNA infectious clone and HM175/p16 virus were a kind gift from Stanley Lemon and Zongdi Feng. HM175/18f-pX-Δ32-67 was created as follows: The region between SacI and EcoRI in the HM175/18f infectious clone was amplified by two rounds of PCR downstream and upstream of the truncated region in pX and then ligated to the SacI/EcoRI-digested HM175/18f plasmid using the EasyGeno Assembly ligation kit. All constructed plasmids were confirmed by sequencing (Sunny Biotechnology). The sh-ALIX target sequences were as follows: target1: AAGGAGGTGTTCCCTGTCTTG; target2: GCTGAGTACCATCAGTCTATC; target3: GGACTTGCAACAAAGCATTGC. Rabbit anti-VP2 antibodies were used as previously described [Citation36]. Rabbit anti-VP1 was prepared by immunizing a rabbit with the synthesized peptide CEQSEFYFPRAPLNS (Genscript). Mouse anti-VP3 antibodies were purchased from LifeSpan BioSciences (catalogue ID: LS-C102930/73034). Mouse anti-tubulin, anti-GFP, and Rabbit anti-ALIX (PICD6IP), anti-TSG101, anti-syntenin-1, anti-CD63, anti-CD81, anti-HSP70, anti-VPS4B, anti-ACTN4 and anti-LC3II antibodies were purchased from Proteintech. Goat anti-rabbit-HRP (ZSGB-Bio), goat anti-mouse-HRP (ZSGB-Bio), goat anti-mouse-10nm gold (Sigma) and rabbit anti-flag (Sigma) antibodies were used as secondary antibodies. The Exo-easy kit was purchased from Qiagen.
Cells and viruses
Huh-7 and Huh-7.5.1-GA cells [Citation36] were cultured in DMEM medium supplemented with 2 mM L-glutamine, nonessential amino acids, 100 U penicillin per ml, 100 μg streptomycin per ml and 10% FBS (complete DMEM). HM175/18f, HM175/18f-pX-N-Δ32-64 and HM175/18f-pX-mut4-9 RNAs were in vitro transcribed by using MEGAscript® Kits (Life Technologies) according to the manufacturer’s protocol. Then, 10 μg RNA was electroporated into 2 × 10⁶ Huh-7.5.1-GA cells suspended in 400 μl cytomix (containing 2 mM ATP and 5 mM glutathione) at 960 mF and 270 V using a GenePulser system (Bio-Rad). Immediately after electroporation, the cells were resuspended in complete DMEM and seeded as required. After Huh-7.5.1-GA cells showed 99% HAV infection, the supernatant was harvested for titration and stored at −80°C. Stable cell lines were constructed as previously described [Citation36]. Briefly, psPAX2 (a packaging plasmid), pMD2.G (a G protein expressing plasmid) and lentiviral vectors were cotransfected into 293T cells; 48 hours later, the supernatants containing lentivirus were harvested and filtered through 0.45-μm filters. Cell lines were selected with puromycin (10 μg/ml).
Quantification of HAV infectivity
Extracellular HAV infectivity was quantified by a limiting-dilution assay as previously described [Citation36,Citation37]. In brief, Huh7.5.1-GA cells were seeded into 96-well plates 1 day prior to infection using serial 6-fold-diluted HAV viruses. At 3 days after infection, the infected cells were observed by fluorescence microscopy to detect GFP translocation into the nucleus.
Quantification of viral RNA by RT-qPCR
Cells were washed with 1× PBS once prior to harvesting. Total cellular RNA was extracted using TRNzol Universal Reagent (Tiangen) according to the manufacturer’s protocol, and the concentration of RNA was determined using a NanoDrop 2000. The extracted RNA was reverse-transcribed into cDNA using ReverTra Ace qPCR RT Kit (Toyobo), and cDNA was subjected to qPCR using an ABI Q6 using SYBR Green (Toyobo). Serial dilution of an equivalent volume of in vitro-transcribed viral genomic RNA was used to create a standard curve for converting Ct values into absolute genome copy numbers. The primers used for qPCR were as follows: GGTAGGCTACGGGTGAAAC; AACAACTCACCAATATCCGC.
Western blot analysis
Western blot was performed as previously described [Citation36]. Briefly, samples were collected in Laemmli buffer, and total proteins were separated by SDS polyacrylamide gel electrophoresis and transferred onto a PVDF membrane. The membrane was blocked using 5% dried milk and incubated with the indicated primary antibody and secondary antibody. Bands were visualized using BeyoECL Moon reagent (Beyotime) under Tanon 4200 or GE AI600, and grey intensity was acquired by using Fiji (NCBI).
Live-cell fluorescence microscopy
For 400× amplification, cells were cultured in a 10-cm dish (Nunc), observed and photographed under an Olympus IX53 fluorescence microscope. For 600× amplification, cells were seed in a glass-bottom dish (Cellvis) and cultured with Fluoro Brite DMEM (Gibco) supplemented with 10% FBS. The dish was placed in a chamber on a Delta-vision Elite (GE) stage, which provides 5% CO₂ and 37°C during image capture. Images were acquired along the Z-axis in 0.2-μm steps and deconvolved by the software provided in Delta-vision Elite. projection and 3D reconstruction were performed by Imaris 9.0.
Immunofluorescence
Virus-infected mCh-ALIX-expressed Huh-7.5.1 cells grown on glass coverslips in a 24-well plate were fixed with 4% paraformaldehyde for 15 minutes, permeabilized with 0.5% Triton-100 and blocked with 5% calf serum. The cells were then incubated with primary antibodies (anti-VP3) for 1 hour at room temperature, washed and incubated with Alexa fluor 488® anti-mouse for 1 hour at room temperature. After washing, the cells were incubated with DAPI (beyotime) for 1 minute. Finally, the glass coverslips were mounted onto glass slides with Fluoromount Aqueous mounting medium (Sigma). Images were acquired along the Z-axis (stepwise: 200 nm) using Delta-vision Elite (GE).
Electron microscopy (EM)
Exosome immune EM was performed according to Cecilia Lasser et al. [Citation38]. In brief, 100 μl concentrated serum-free cell culture supernatant was dropped on a piece of Parafilm, and a carbon-coated nickel grid was placed on the drop for 30–60 minutes. The grid was washed with 1× PBS three times and fixed in 2% paraformaldehyde for 10 minutes. After three washes with 1× PBS, the grid was incubated with the mouse anti-GFP antibody (dilution ratio 1:50) for 40 minutes. After three washes with 1× PBS, the grid was incubated with the goat anti-mouse 10 nm-gold antibody (dilution ratio 1:50) for 40 minutes, postfixed with 2.5% glutaraldehyde for 10 minutes and washed three times with 1× PBS. The sample was treated with 2% uranyl acetate for 30 seconds, washed three times with water and examined by FEI F20 TEM at 200 kV. For EM of supernatant HAV, 50 ml supernatant from infected cells was centrifuged at 2,000 × g to remove cell debris and subsequently centrifuged at 100,000 × g to pellet the virus, which was resuspended in 50 μl 1× PBS. Next, 5 μl PBS-suspended virus was dropped onto a carbon-coated copper grid and allowed to adsorb for 2 minutes; the remaining liquid was removed using a soft tissue, and the grid was stained with 2% uranyl acetate for 30 seconds. The grid was air-dried and disinfected under UV (ultra-violet) before examination by FEI F20 TEM at 200 kV.
For cell immune EM, cells were fixed in a mixture of 0.25% glutaraldehyde and 4% formaldehyde in 0.1 M PB (phosphate buffer pH 7.3) at 4°C overnight. The fixative was removed by washing three times in 0.1 M PB. The cell pellets were dehydrated in a series of increasing concentrations of ethanol, 50%, 70% and 90%, for 15 minutes each. The 90% ethanol was removed, and the sample was infiltrated with a mixture of LR-White and 90% ethanol (1:1) and shaken gently in a rotor for 2 hours. The mixture was removed and replaced by 100% embedding LR-White and incubated overnight at 4°C. The samples were transferred to gelatin capsules filled with LR-White, capped and polymerized in an oven at 55°C for 24 hours. The LR-White blocks were trimmed, and ultrathin sections (70 nm) were obtained with a diamond knife, after which the sections were mounted on a nickel grid. The sample was Immunostained using anti-GFP and goat anti-mouse 10 nm-gold and contrast stained with 2% uranyl acetate, which is similar to the procedure of EM immunostaining. After lead citrate contrast staining for 5 minutes at room temperature, the samples were examined using a Nippon Tekno TEM. For conventional EM, cells were fixed with 4% PFA and 0.1% glutaraldehyde (in PB) overnight at 4°C. After washing with PB, the cells were fixed with 1% osmium tetroxide/PB for 30 minutes, dehydrated through a graded ethanol series, transferred to n-butyl glycidyl ether and embedded in epoxy resin. Ultrathin sections at 70-nm thickness were cut using an ultramicrotome, placed on a nickel grid, etched with 5% sodium metaperiodate for 10 minutes and washed with PBS. The sections were stained with uranyl acetate for 3 minutes and lead citrate for 1 minute and then observed by Nippon Tekno TEM.
Density gradient centrifugation
For eHAV separation, 24 hours after virus infection, cells were washed twice with 1× PBS and cultured in serum-free DMEM. For exosomes separation, cells were cultured in 10% serum DMEM in 15-cm dish and grown to 80% confluence. The culture supernatant was removed, the cells were washed twice with 1× PBS prior to the addition of serum-free DMEM. The next day, the culture was centrifuged at 2,000 × g for 10 minutes at 4°C to remove cells and debris and concentrated by ultrafiltration using Ultracel 100 K (Millipore) at 3,500 × g at 4°C. The resulting concentrated supernatant was loaded onto an 8–40% iodixanol (Opti-Prep) step gradient and centrifuged at 141,000 × g in a P55ST rotor (Himac CP80WX) for 22 h at 4°C. Density was determined with a refractometer.
Differential and ultracentrifugation for extracellular vesicles purification
Cells were cultured in 10% serum DMEM in a 15-cm dish and grown to 80% confluence; the culture supernatant was removed, washed twice with 1× PBS prior to the addition of serum-free DMEM. The culture supernatant was harvested at 24 hours later for EV purification. The supernatant was filtered through a 0.45-μm filter and concentrated using an Ultracel 100 K (Millipore). The concentrated supernatant was differentially centrifuged at 300 × g (10 minutes), 2,000 × g (20 minutes), 10,000 × g (30 minutes) and 100,000 × g (2 hours). For each centrifugation, the pellets were washed once with 1× PBS and centrifuged again; the 100,000 × g ultracentrifugation step was performed using a P55ST rotor (Himac CP80WX) at 4°C.
Immunoprecipitation assay
Cells grown in 10-cm dishes were washed once with 1× PBS and lysed on ice with Western/IP lysis buffer (Beyotime) supplemented with 0.5 mM PMSF. Following 12,000 × g centrifugation at 4°C for 5 minutes, the pellet containing the cell debris was removed and the supernatant was harvested for co-IP. Anti-GFP/mcherry nanobody-coated agarose (KT Health) was used according to the manufacturer’s protocol. Briefly, 600 μl cell lysate supernatant was mixed with 25 μl anti-GFP nanobody suspension and incubated overnight on a rotator at 4°C. The agarose beads were pelleted by centrifugation at 2,400 × g for 1 minute at 4°C and washed three times with the Western/IP lysis buffer on ice. The beads were mixed with 50 μl Laemmli buffer for western blot assay or 500 μl Trizol for qPCR.
Nanoparticle tracking analysis (NTA)
Cells were cultured in 10% serum DMEM in 15-cm dish and grown to 80% confluence; the culture supernatant was removed, and the cells were washed with 1× PBS prior to the addition of serum-free DMEM. The culture supernatant was harvested 24 hours later. Cell debris was removed by centrifugation at 2,000 × g for 20 minutes. The supernatant was filtered through a 0.45-μm filter and concentrated using Ultracel 100 K (Millipore); ZetaView (Particle Metrix, Germany) was used for NTA.
Sizing and molecular profiling of individual EVs via a Flow NanoAnalyzer
For size distribution and particle concentration analysis, a silica nanosphere cocktail (S16M-Exo) was employed as the size standards, and a calibration curve was constructed based on the particle sizes and the side scattering intensities. With this calibration curve, the side scattering intensity of every EV particle could be converted into the corresponding particle size. For surface and intravesicle protein profiling, the EV sample was resuspended in 50 μL of PBS, and 5 μL of Alexa Fluor 647-conjugated anti-CD63 or anti-CD81 Mab was added. The mixture was incubated at 37°C for 30 min and washed twice with PBS via ultracentrifugation at 100,000 g for 17 min at 4°C. The final pellet was resuspended in 100 μL of PBS for analysis with a Flow NanoAnalyzer model type U30 (NanoFCM Inc., Xiamen, China) equipped with a laser (488 nm and 638 nm) and 3 channels (SSC, FITC channel, APC channel).
Statistical analysis
P values were estimated using the t-test/one-way ANOVA in Graphpad Prism 7. Differences at p < 0.05 were considered statistically significant, and the following categories were used: ***, P < 0.001; ****, P < 0.0001.
Author contribution
Conceptualization, W.J.; Methodology, W.J., L.D. and G.L.; Investigation, W.J., P.M., L.D. and Z.L.; Writing-Original Draft, W.J.; Writing-Review & Editing, W.J., L.D., X.L. and G.L.; Supervision, G.L.; Funding Acquisition, G.L.
Conflict of interest
None of the authors has a conflict of interest.
Supplemental Material
Download Zip (3.5 MB)Acknowledgements
We thank Dr. Stanley Lemon (The University of North Carolina, USA) and Dr. Zongdi Feng (Ohio State University, USA) for providing HAV HM175/18f infectious clone and HM175/p16 virus; Yu Kong (Institute of Neuroscience, Chinese Academy of Sciences, Shanghai, China) for the technical support of EM; and Weihua Wu (Institut Pasteur of Shanghai, Chinese Academy of Sciences, Shanghai, China) for the technical support of Delta-Vision Elite microscopy.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- WHO. World Health Organization. Hepatitis A fact sheet; 2017. http://www.who.int/mediacentre/factsheets/fs328/en/.
- Feng Z, Hirai-Yuki A, McKnight KL, et al. Naked viruses that aren’t always naked: quasi-enveloped agents of acute hepatitis. Annu Rev Virol. 2014;1:539–16.
- Scholz E, Heinricy U, Flehmig B. Acid stability of hepatitis A virus. J Gen Virol. 1989;70(9):2481–2485.
- Siegl G, Weitz M, Kronauer G. Stability of hepatitis A virus. Intervirology. 1984;22:218–226.
- Graff J, Richards OC, Swiderek KM, et al. Hepatitis A virus capsid protein VP1 has a heterogeneous C terminus. J Virol. 1999;73:6015–6023.
- Yang X, Cheng A, Wang M, et al. Structures and corresponding functions of five types of Picornaviral 2A proteins. Front Microbiol. 2017;8:1373.
- Harmon SA, Emerson SU, Huang YK, et al. Hepatitis A viruses with deletions in the 2A gene are infectious in cultured cells and marmosets. J Virol. 1995;69:5576–5581.
- Cohen L, Benichou D, Martin A. Analysis of deletion mutants indicates that the 2A polypeptide of hepatitis A virus participates in virion morphogenesis. J Virol. 2002;76:7495–7505.
- Anderson DA, Ross BC. Morphogenesis of hepatitis A virus: isolation and characterization of subviral particles. J Virol. 1990;64:5284–5289.
- Feng Z, Hensley L, McKnight KL, et al. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature. 2013;496:367–371.
- Wang X, Ren J, Gao Q, et al. Hepatitis A virus and the origins of picornaviruses. Nature. 2015;517:85–88.
- Demirov DG, Freed EO. Retrovirus budding. Virus Res. 2004;106:87–102.
- Henne WM, Buchkovich NJ, Emr SD. The ESCRT pathway. Dev Cell. 2011;21:77–91.
- Strack B, Calistri A, Craig S, et al. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114:689–699.
- Bird SW, Maynard ND, Covert MW, et al. Nonlytic viral spread enhanced by autophagy components. Proc Natl Acad Sci USA. 2014;111:13081–13086.
- Chen YH, Du W, Hagemeijer MC, et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell. 2015;160:619–630.
- Takahashi M, Yamada K, Hoshino Y, et al. Monoclonal antibodies raised against the ORF3 protein of hepatitis E virus (HEV) can capture HEV particles in culture supernatant and serum but not those in feces. Arch Virol. 2008;153:1703–1713.
- Emerson SU, Nguyen HT, Torian U, et al. Release of genotype 1 hepatitis E virus from cultured hepatoma and polarized intestinal cells depends on open reading frame 3 protein and requires an intact PXXP motif. J Virol. 2010;84:9059–9069.
- Yin X, Ambardekar C, Lu Y, et al. Distinct entry mechanisms for nonenveloped and quasi-enveloped hepatitis E viruses. J Virol. 2016;90:4232–4242.
- McKnight KL, Xie L, Gonzalez-Lopez O, et al. Protein composition of the hepatitis A virus quasi-envelope. Proc Natl Acad Sci USA. 2017;114:6587–6592.
- Tkach M, Kowal J, Thery C. Why the need and how to approach the functional diversity of extracellular vesicles. Philos Trans R Soc Lond B Biol Sci. 2018;373: 20160479.
- Cocucci E, Meldolesi J. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015;25:364–372.
- McKnight KL, Xie L, González-López O, et al. Protein composition of the hepatitis A virus quasi-envelope. Proc Nat Acad Sci. 2017;7: 201619519–13.
- Robinson SM, Tsueng G, Sin J, et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014;10:e1004045.
- Votteler J, Sundquist WI. Virus budding and the ESCRT pathway. Cell Host Microbe. 2013;14:232–241.
- Gonzalez-Lopez O, Rivera-Serrano EE, Hu F, et al. Redundant late domain functions of tandem VP2 YPX3L motifs in nonlytic cellular egress of quasi-enveloped hepatitis A virus. J Virol. 2018;92:e01308-18.
- Fisher RD, Chung HY, Zhai Q, et al. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell. 2007;128:841–852.
- Hirai-Yuki A, Hensley L, Whitmire JK, et al. Biliary secretion of quasi-enveloped human hepatitis A virus. MBio. 2016;7:e01998-16.
- Ruchti F, Siegl G, Weitz M. Identification and characterization of incomplete hepatitis A virus particles. J Gen Virol. 1991;72(9):2159–2166.
- Bishop NE, Anderson DA. RNA-dependent cleavage of VP0 capsid protein in provirions of hepatitis A virus. Virology. 1993;197:616–623.
- Feinstone SM, Kapikian AZ, Gerin JL, et al. Buoyant density of the hepatitis A virus-like particle in cesium chloride. J Virol. 1974;13:1412–1414.
- Deng L, Jiang W, Wang X, et al. Syntenin regulates hepatitis C virus sensitivity to neutralizing antibody by promoting E2 secretion through exosomes. J Hepatol. 2019;71:52–61.
- Vahey MD, Fletcher DA. Low-fidelity assembly of influenza A virus promotes escape from host cells. Cell. 2018. DOI:10.1016/j.cell.2018.10.056
- Yim N, Choi C. Extracellular vesicles as novel carriers for therapeutic molecules. BMB Rep. 2016;49:585–586.
- Yim N, Ryu SW, Choi K, et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat Commun. 2016;7:12277.
- Jiang W, Muhammad F, Ma P, et al. Sofosbuvir inhibits hepatitis A virus replication in vitro assessed by a cell-based fluorescent reporter system. Antiviral Res. 2018;154:51–57.
- Yang Z, Wang X, Chi X, et al. Neglected but important role of apolipoprotein E exchange in hepatitis C virus infection. J Virol. 2016;90:9632–9643.
- Lasser C, Eldh M, Lotvall J. Isolation and characterization of RNA-containing exosomes. J Vis Exp. 2012;e3037. DOI:10.3791/3037