
ABSTRACT
Early dissemination is a unique characteristic and a detrimental process of pancreatic ductal adenocarcinoma (PDAC); however, the underlying mechanism remains largely unknown. Here, we investigate the role of dual-specificity phosphatase-2 (DUSP2)-vascular endothelial growth factor-C (VEGF-C) axis in mediating PDAC lymphangiogenesis and lymphovascular invasion. Expression of DUSP2 is greatly suppressed in PDAC, which results in increased aberrant expression of extracellular vesicle (EV)-associated VEGF-C secretion. EV-VEGF-C exerts paracrine effects on lymphatic endothelial cells and autocrine effects on cancer cells, resulting in the lymphovascular invasion of cancer cells. Tissue-specific knockout of Dusp2 in mouse pancreas recapitulates PDAC phenotype and lymphovascular invasion. Mechanistically, loss-of-DUSP2 enhances proprotein convertase activity and vesicle trafficking to promote the release of the mature form of EV-VEGF-C. Collectively, these findings represent a conceptual advance in understanding pancreatic cancer lymphovascular invasion and suggest that loss-of-DUSP2-mediated VEGF-C processing may play important roles in early dissemination of pancreatic cancer.
Abbreviations: DUSP2: dual-specificity phosphatase-2; VEGF-C: vascular endothelial growth factor-C; EV: extracellular vesicles; PDAC: pancreatic ductal adenocarcinoma; KD: knockdown
Background
More than 90% of pancreatic cancers are originated from the exocrine ducts of the pancreas known as pancreatic ductal adenocarcinoma (PDAC). Early metastatic rate of PDAC is high with an average of 5-year survival. Due to the lack of symptoms, patients are often diagnosed with PDAC when metastasis has already occurred, limiting the choices of treatment. Metastasis is a complex process requiring the gain of multiple abilities of tumour cells. Among which, the ability of tumour cells to enter pre-existing blood/lymphatic vessels, or stimulation of new blood/lymphatic vessels infiltrating into tumours, significantly contribute to the early spreading of cancer cells. Therefore, identifying factors that exacerbate the malignancy of pancreatic cancer, specifically at the early stages of the disease progression may help to design better treatment to improve patient survival.
Vascular endothelial growth factor C (VEGF-C), a member of the VEGF family, was first identified as the master lymphangiogenic factor in embryonic development [Citation1,Citation2]. Overexpression of VEGF-C in cancers has been tightly linked to lymphangiogenesis [Citation3] and is highly associated with lymphatic invasion and metastasis [Citation3,Citation4]. Expression of VEGF-C was detected in 80% of the late stage PDAC, and the expression level is associated with lymph node metastasis and poor 5-year survival rate [Citation5,Citation6]. The underlying mechanism responsible for the aberrant expression of VEGF-C is not well known.
Constitutive activation of MAPK pathway was characterized as one of the features of pancreatic cancer [Citation7]. In a genetic mouse model of pancreatic cancer, MAPK signalling is required to initiate and maintain the pancreatic intraepithelial neoplasia lesions [Citation8]. In normal cells, MAPKs mediated signalling is crucial to various cell functions. Activation of MAPKs is counteracted by dual-specificity phosphatases (DUSPs) [Citation9]. DUSPs are unique protein phosphatases which dephosphorylate both tyrosine and serine/threonine residues on the same substrate [Citation10]. DUSP2, also known as phosphatase of activated cells 1, belongs to the subfamily that predominantly acts in the nucleus and primarily inactivates ERK [Citation11–Citation14]. DUSP2 expresses mainly in the haematopoietic cells and is highly inducible in response to stress and regulates cytokine production or inflammation [Citation15]. In cancer cells, DUSP2 was identified as a transcriptional target of p53, which mediates oxidative damage and nutritional stress-induced apoptosis [Citation16]. Downregulation of DUSP2 and constitutive activation of ERK were detected in acute leukaemia and some solid tumours [Citation17,Citation18], suggesting DUSP2 may have a tumour suppressor function. Previously, we demonstrated that DUSP2 was suppressed by hypoxia and the reduction of DUSP2 contributed to tumour malignancy and drug resistance [Citation18–Citation20]. Considering the early dissemination nature of PDAC and tumour suppressor function of DUSP2, it is important to investigate the pathological effect of DUSP2 in pancreatic cancer progression and to dissect the underlying mechanism.
Extracellular vesicles (EVs)-carried bioactive molecules play an important role in intercellular communication and are involved in pathological processes [Citation21,Citation22]. Emerging studies indicate that tumour cell-derived EVs can promote malignant ability such as epithelial to mesenchymal transition, stemness, chemoresistance and immune suppression [Citation23–Citation26]. However, whether EV biogenesis is regulated by oncogenic signalling remains largely uncharacterized.
In this study, we demonstrate that secreted VEGF-C is associated with EV and that loss-of-DUSP2 not only facilitates VEGF-C processing but also increases the production of EV-associated VEGF-C. Our findings point out the importance and novel regulation of DUSP2/VEGF-C axis in early pancreatic cancer progression.
Methods
Cell culture
Pancreatic ductal adenocarcinoma cell lines, AsPC-1, BxPC3, PANC-1 and MIA PaCa-2 (ATCC), were maintained in RPMI 1640 media containing 10% foetal bovine serum (FBS), glutamineand sodium pyruvate. To collect conditioned media, cells were treated with serum-free RPMI 1640 media, after 24 h, the conditioned media was filtered and stored for subsequent uses. LECs were purchased (PromoCell) and cultured in endothelial cell growth medium MV2 with supplement (PromoCell). HUVECs were cultured in EGM-2/M199/20% FBS (Lonza, EGM-2 BulletKit, CC-3156 and CC-4176). Cell lines used in this study have been authenticated and tested as mycoplasma free. Stable knockdown of DUSP2 was achieved in PANC-1 cells using two different shRNAs (clone TRCN0000355667 and TRCN0000355666, RNAi core lab of Genomics Research Center, Academia Sinica, Taipei, Taiwan) and lentivirus delivery according to manufacturer’s protocol. Stable cells were selected with puromycin (1 μg/ml).
Western blotting
Whole cell lysates were collected by RIPA buffer and subjected to Western blotting as described previously [Citation18]. Antibodies against VEGF-C (GeneTex, GTX113574), DUSP2 (Santa Cruz, sc-32776), Phospho-p44/42 MAPK (Cell Signaling Technology #4370), p44/42 MAPK (Cell Signaling Technology #4696), CD63 (Taiclone, tcba9211), GAPDH (Genetex GTX100118), Furin (Abcam, ab183495), PC5/6 (Abcam, ab40133), PCSK7 (Abcam, ab47925), HSP70 (Santa Cruz, sc-32239), Annexin V (Santa Cruz, sc-74438), SNAP-tag (New England Biolabs, P9310 S) were used for Western blotting.
Cell migration and invasion assays, and transendothelial assay
Cell migration and invasion assays were performed using the 24-well Transwell chamber with a pore size of 8.0 µm (PIEP12R48, Millipore). PANC-1 and MIA PaCa-2 cells were suspended in 200 µl serum-free RPMI media in transwell (upper) chamber. 500 µl 10% FBS RPMI culture media were added to the lower chamber. For LECs migration assay, LECs were suspended in 200 µl basal MV2 media in each chamber. 50 µl conditioned media with 450 µl basal MV2 media were used in the lower chamber. Transwell was pre-coated with Matrigel (BD354230, BD Biosciences) diluted in serum-free media (1:10 dilution) for invasion assay. After 16 h of incubation, migrated cells were fixed and stained with crystal violet. Cell numbers were counted by ImageJ cell counter. For the transendothelial assay, endothelial cells were seeded to a matrigel-coated Transwell. After the endothelial layer was formed, PANC-1 control and DUSP2-KD cells were added to the chamber and transendothelial migration ability was measured after 16 h.
Proprotein convertase activity assay
This assay was adopted from a previous study [Citation27]. Briefly, cell pellets were collected and lysed in 5× lysis/reaction buffer as 107 cells/ml. Protein convertase substrate (R&D pERTKR-AMC # ES013) was pre-warmed (15 min at 37°C) and then incubated with cell lysate in black 96 well plates. Fluorescence intensity was measured immediately (with excitation at 355 nm and emission at 460 nm) every 2 min in a kinetic setting.
Extracellular vesicles isolation and analysis
To extract EVs from PDAC cells, the cell culture medium was replaced with serum-free RPMI medium containing ITS-M (Insulin-Transferrin-Selenium Mixture, Genedirex). MIA PaCa-2 cells and AsPC-1 cells were plated on the first day and medium was changed for SF-for one more day. Conditioned medium (5 ml/6*106 cells) was first centrifugation at 1200 rpm for 10 min and filtered by 0.45 μm to remove cell debris. For ultracentrifugation, microvesicles were pelleted after centrifugation at 16,000 g for 60 min and resuspended in PBS. Supernatants were then centrifuged at 100,000 g for 2.5 h at 4°C (Beckman Coulter, L-90 K). The pelleted exosomes (small EV) were suspended in PBS and supernatant was collected as EV-depleted fraction (Sup). Equal amount of protein was loaded for Western blotting (10 ug).
To perform size exclusion chromatography, conditioned medium (5 ml/6*106 cells) was further concentrated to 500 ul by Amicon-ultra4 (10 kD). Concentrated CM was loaded to Izon’s qEV original columns (IZON science) and fractions were collected according to the manufacturer’s protocol. To measure protein concentration, each fractions were first further concentrated 10 fold by Amicon-ultra 4 (3kD) and employed for DC protein assay (BIO-RAD) according to the manufacturer’s protocol.
EVs were extracted from the cell culture medium (500 μl concentrated CM) or serum of mice using ExoQuick-TC and ExoQuick exosome precipitation solution (SBI system Biosciences) according to the manufacturer’s protocol. Serum-free conditioned medium from control or DUSP-KD PANC-1 cells was under centrifugation to remove debris (500 g, 10 min; and 16,000 g, 30 min, respectively) and were sent to the Centre for Micro/Nano Science and Technology at National Chung Kung University for nanoparticle tracking analysis (NTA).
To determine the topology of VEGF-C, EV was first isolated (by ExoQuick-TC) and resuspend in PBS. 8 ug of protein was treated with PBS (control), proteinase K (1ug/ml), triton X (1%), proteinase K and triton X, trypsin (0.025%) for 60 min at 37°C. Treated samples were added RIPA and sample buffer for Western blotting.
Immunogold labelled protein on purified sample and transmission electron microscope (TEM) inspection
Serum-free conditioned medium from MIA PaCa-2 cells was collected. The EV fraction was purified by ExoQuick-TC. 2 μL of isolated samples was adsorbed onto a glow-discharged nickel grid (EMS CF-200-Ni) for 20 min. The grid containing the samples was incubated in 1% BSA/PBS blocking buffer for 1 h. The excess liquid was removed with a filter paper. Primary antibody against VEGF-C (ProteinTech, #22601) (dilution 1:20) was added to the samples for 2 h. After incubation, the grid with the samples was incubated with the nanogold-conjugated antibody (dilution 1:30) for 1 h. The excess liquid was removed with filter paper and fixed with 1% glutaraldehyde in PBS buffer for 10 min. Finally, the sample was stained with 2% uranyl acetate for 1 min and air-dried. The immunogold labelled samples were inspected by JEM1400 electron transmission microscope.
Immunogold labelled protein in the cell and TEM inspection
Control and DUSP2-KD PANC-1 cells were collected and processed by the Department of Pathology National Chung Kung University Hospital. Cells were pelleted down and fixed by 2.5% glutaraldehyde for 1 h at 4°C. The samples were immersed in H2O2 for 10 min. The grid with samples was blocked for 30 min then incubated with antibodies (VEGF-C ProteinTech, #22601) for 2 hfollowed by incubation with the nanogold-conjugated antibody for 1 h. The sections on the grids were stained with uranyl acetate and lead citrate and investigated under a by JEM1400 electron transmission microscope. The authors gratefully acknowledge the JEM-1400 and Leica EM UC7 belonging to the Instrument Development Center of the National Cheng Kung University.
Cell labelling and tracking
Cells were labelled with PKH67 (PKH67 Fluorescent Cell Linker Kits, Sigma) and plated into the cell holder for the tracking of PKH67 positive particles within the cells. 3D tracking of PKH67 particles in control and DUSP2-KD cells analysed by Imaris software (left). Experiments have been performed two times and represented data are shown in the figure. VEGF-C was cloned into the SNAP-tag vector (NEB) and was stably expressed in control and DUSP2-KD cells under G418 selection for a month. Cells were treated with proprotein convertase inhibitor (20 uM) in serum-free medium for 24 h. After labelling by the fluorescent substrate for SNAP (according to the manufacturer’s protocol), cells were fixed and imaged.
Immunohistochemistry
Tumours from animals were fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 5-μm sections and sections were stained with H&E or immunohistochemistry. Primary antibodies against Lyve-1 (Angiobio 11–034) and CD31 (abcam ab28364) were used for immunohistochemistry staining of lymphangiogenesis and angiogenesis.
Genetically engineered mouse model
LSL-KrasG12D; Trp53flox/flox; Pdx-1-Cre (KPC) mice were provided by Dr. Po-Hsien Huang, National Cheng Kung University. Mice were sacrificed at the age of 5–8 weeks and tumours or pancreases were harvested for IHC analysis. To generate conditional knockout of Dusp2, two LoxP sites were inserted into the intron 1 and intron 2 regions of mouse Dusp2 BAC clone. Then, the CRISPR/Cas9 system was used for replacing endogenous mouse dusp2 by mouse Dusp2 BAC clone carrying two LoxP sites. Conditional knockout Dusp2 mice (Dusp2 F/F) were generated and crossed with Pdx-1-cre recombinase mice to specifically knock out Dusp2 in mouse pancreas. After knockout of Dusp2 for 2–7 months, the histology of the mouse pancreas was examined.
Animal studies
Male SCID mice (6- to 8-weeks of age) were used for pancreas orthotopic or subcutaneous injection using pancreatic cancer cell lines. For orthotopic experiments, 1 × 106 PANC-1 with or without DUSP2 knockdown, suspended in 100 μl of RPMI medium were injected into the pancreas of mice. For subcutaneous inoculation, 1 × 106 cells PANC-1 with or without DUSP2 knockdown suspended in 100 μl of RPMI medium were injected into the left and right flank of mice. To investigate the effects of EVs, PANC-1 and AsPC-1 cells (1 × 105) were subcutaneously injected into left and right flank of male SCID mice. After 1 week, control (PBS) and EVs were given at tumour sites. EVs from AsPC-VEGF-C were isolated by ultracentrifugation. EVs from PANC-1 DUSP2-KD cells were isolated by ExoQuick-TC. 5 ug of EVs was given twice a week for three consecutive weeks (AsPC-1) and five consecutive weeks (PANC-1). Experimental procedures of animal studies were approved by the Institutional Animal Care and Use Committee at the National Chung Kung University.
Clinical samples
Pancreatic tumour tissue arrays were obtained from National Cheng Kung University Hospital with the approval of the institutional review board. Tumour sections were placed in duplicate on the array. Five arrays were used for immunohistochemistry staining of DUSP2 (Genetex).
Statistical analysis
Statistical analyses were performed by GraphPad Prism 5.00. The results were presented as mean ± standard error of the mean. Two-tailed Student’s t-test was employed for comparing two groups while one-way ANOVA with Tukey post-tests was performed for comparing more than two groups. Error bars represent the standard error of the mean from at least three independent experiments. Asterisks denote significant difference from control group *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Results
Secretion of extracellular vesicle associated VEGF-C by pancreatic cancer cells
Lymphangiogenesis is an important process for lymphatic invasion and metastasis of cancer cells. To investigate whether early dissemination of pancreatic cancer cells is mediated by lymphatic vessels, we detected lymphatic vessels in genetically engineered Lox-Stop-Lox (LSL)-KrasG12D; LSL-Trp53R172 H; Pdx1-cre (KPC) mouse model of pancreatic cancer and found that lymphangiogenesis is significantly increased in KPC tumour compared to the pancreas of LSL-Trp53R172 H only littermate (defined as wildtype) (). The expression of master lymphangiogenic factor, Vegf-c, is also increased in the serial section of KPC pancreatic tumour region (). Roles of EVs have been discovered in intercellular communications [Citation28], we thus aimed to characterize whether VEGF-C is associated with EV and promotes metastasis in pancreatic cancer. The expression of VEGF-C was determined in various pancreatic cancer cell lines with MIA PaCa-2 cells expressing the highest level of endogenous VEGF-C (Supplementary Figure 1A). Size exclusion chromatography revealed that secreted VEGF-C from MIA PaCa-2 cells was mainly associated with EV (). We further performed ultracentrifugation methodology to determine the majority of VEGF-C was found in the small EV fraction (Ex) [Citation28] (). Commercial EV precipitating reagent (ExoQuick-TC) was used to isolate EV and shows the enriched expression of VEGF-C in EV fractions (). Transmission electron microscopic examination further confirmed that VEGF-C is associated with EV at sizes between 100 and 150 nm, suggesting that secreted VEGF-C is associated with EV ( and Supplementary Figure 1B). We next determined the topology of EV-VEGF-C and demonstrated that VEGF-C is associated at the surface of EVs ( and Supplementary Figure 1 C).
To determine if exogenous expressed VEGF-C is also associated with EV in pancreatic cancer cells, AsPC1 cells, with very low endogenous VEGF-C levels, were lentivirally transduced with constitutively expressed VEGF-C constructs (). Treatment with EV biogenesis inhibitor GW4869 decreased secreted VEGF-C in conditioned medium (). Secreted VEGF-C was detected in EV isolated by ultracentrifugation fractionation () and exosome precipitation solution (). EVs secreted from pancreatic cancer cells can be uptaken by recipient lymphatic endothelial cells (LECs) (). Functionally, treatment of LECs with EVs isolated from AsPC-1-VEGF-C cells significantly increased LECs proliferation as compared to those treated with EVs from AsPC-1-Ctrl cells (). Furthermore, treatment of AsPC-1 tumours with EVs from AsPC-VEGF-C cells enhanced lymphangiogenesis ().
Taken all together, we provide evidence demonstrating that functional VEGF-C is associated with EV secretion in pancreatic cancer cells.
VEGF-C secretion is mediated by ERK and DUSP2
We have previously demonstrated that DUSP2, a specific phosphatase of MAPK, is downregulated in many solid cancers, which leads to prolonged activation of ERK and aberrant expression of angiogenic genes [Citation18,Citation20]. To investigate the underlying mechanism of VEGF-C overexpression in PDAC, we re-analysed our previous microarray dataset (GSE66656) [Citation20] and found that DUSP2-regulated gene expression is associated with pancreatic neoplasms, angiogenesis and VEGF-C expression (Supplementary Figure 2A-B). Data mining of public microarray datasets (Oncomine) also showed a decrease in DUSP2 mRNA expression in PDAC compared to normal pancreases (Supplementary Figure 2 C). We employed immunohistochemical staining of DUSP2 on 54 pancreatic tumour samples and observed that non-tumour elements (arrowhead) are focally DUSP2-positive while the pancreatic intraepithelial neoplasia (PanIN) and carcinoma (Ca) lack DUSP2 expression (). A similar pattern was observed in KPC tumours, which showed positive Dusp2 expression in benign regions and absence in regions of infiltrating tumour cells or carcinoma (). Consistent with the notion that DUSP2 is an ERK-specific phosphatase, immunohistochemistry staining showed that ERK is activated in the tumour region of KPC mice (). We thus tested whether ERK signalling is involved in EV-VEGF-C expression. Treatment of MIA PaCa-2 cells with ERK inhibitor significantly decreased ERK activation ( left) and EV-VEGF-C expression isolated by reagent precipitation and ultracentrifugation ( middle). Similar results were also observed in AsPC-VEGF-C expressing cells ( right).
To verify whether loss of DUSP2 affects VEGF-C expression, shRNAs targeting DUSP2 were used to knockdown DUSP2 in PANC-1 cells (which express higher levels of endogenous DUSP2). Results showed that knockdown of DUSP2 caused ERK phosphorylation () and VEGF-C but did not cause VEGF-A overexpression (). Treatment with the MEK inhibitor, U0126, to inhibit ERK phosphorylation in DUSP2-knockdown cells suppressed the elevation of VEGF-C (). Forced expression of DUSP2 in MIA PaCa-2 cells (which express low levels of DUSP2) attenuated VEGF-C secretion (). Finally, we tested whether the inverse correlation of DUSP2 and VEGF-C is a common but not specific phenomenon in particular cell type. To this end, we examined the expression of Dusp2 and Vegf-c in several organs from mouse and the result showed that organs with higher Dusp2 levels had lower Vegf-c levels (). Together, these data demonstrated that upregulation of VEGF-C in PDAC is mediated by the loss of DUSP2.
Knockdown of DUSP2 in PDAC promotes lymphangiogenesis and lymphovascular invasion in vivo
We then tested whether knockdown (KD) of DUSP2 in cancer cells promotes lymphangiogenesis. Treatment with conditioned media (CM) from DUSP2-KD PANC-1 cells promoted the proliferation and migration of LECs, which can be reduced by addition of VEGFR2/R3 inhibitor, Lenvatinib (). Then, we conducted a xenograft mouse model to investigate the effect of DUSP2 loss in tumour growth and lymphangiogenesis. Knockdown of DUSP2 led to larger tumour size and increased lymphangiogenesis in the xenograft animal model (Supplementary Figure 3). To further determine the in vivo effects of DUSP2-KD in pancreatic cancer progression, we performed orthotopic injection of control and DUSP2-KD PANC-1 cells into the pancreas of SCID mice. Animals were sacrificed 2 months after the inoculation (to mimic early stage of PDAC) and tumours and pancreases were collected. No obvious difference was observed in the size of primary tumours between control and DUSP2-KD tumours (). Serial section of tumours showed the non-overlapping CD31 and Lyve-1 staining. Intriguingly, there is a marked increase of lymphatic vessels (Lyve-1 positive) in DUSP2-KD tumours () while the number of microvessels (CD31 positive) is low and no significant difference was observed. Evaluation of the primary tumours by histology examination led us to identify a significant increase in lymphovascular invasion (LVI) or nodal metastasis in DUSP2-KD tumours (88.8%) as compared to the control tumours (16.7%) (). These data demonstrate that inhibition of DUSP2 promotes lymphangiogenesis and lymphovascular invasion ability of pancreatic cancer cells via VEGF-C dependent manner.
We generated Dusp2 conditional knockout (KO) in the mouse pancreas ( top). While Dusp2 KO alone is not sufficient to induce tumour formation, increased lymphangiogenesis (in the region of the intralobular duct) can be detected in the Dusp2 KO pancreas when compared to that in the wild type mouse ( bottom). We then generated Kras active and Dusp2 null (KDC) mice. At the 7th month, the KDC mice not only developed cancer in the pancreas but also significantly increased newly formed lymphatic vessels (), a phenomenon similar to what was observed in KPC mice. Furthermore, increased VEGF-C was detected in EV isolated from serum of KDC mice (). Together, our data confirm that loss-of-DUSP2 promoted lymphangiogenesis.
Increased VEGF-C by DUSP2 inhibition mediates autocrine functions of pancreatic cancer cells
While increased lymphangiogenesis is highly associated with LVI, acquisition of migration/invasion ability by cancer cells can also facilitate the invasion into these lymphatic/blood vessels. Since knockdown of DUSP2 makes the cells less adhesive to the neighbouring cells (), we thus investigated whether loss of DUSP2 affects the abilities of cell migration and invasion. Indeed, wound healing migration and Transwell invasion assays demonstrated that DUSP2-KD enhanced cell migration and invasion () and overexpression of DUSP2 decreased migration ability (). Next, we tested whether this effect is mediated via upregulation of VEGF-C. The expression of the main VEGF-C receptor VEGFR3 and co-receptor Neuropilin 2 was detected in human pancreatic cancer cell lines (Supplementary Figure 4A), suggesting an autocrine effect of VEGF-C in cancer cells is possible. We then employed bioinformatic tools to analyse gene expression profiles and biological processes. Results indicated that VEGF-C is involved in cytoskeleton remodelling and cell adhesion pathways (Supplementary Figure 4B). Treatment with VEGF-C significantly increased PANC-1 migration while knockdown of VEGF-C significantly decreased pancreatic cancer cell migration without affecting its proliferation (Supplementary Figure 5). Treatment with Lenvatinib or transient knockdown of VEGF-C abolished DUSP2-KD-promoted cell invasion (), indicating VEGF-C autocrine signalling mediates invasion in DUSP2-KD PANC-1 cells. Finally, we used a transendothelial migration assay to mimic the real situation of extravasation. We found that DUSP2-KD cells also showed enhanced transendothelial migration ability, which can be blocked by Lenvatinib (). These data showed that DUSP2 knockdown resulted in increased malignant characteristics of pancreatic cancer cells, which, in conjunction with increased lymphangiogenesis, promotes early pancreatic cancer cell dissemination.
DUSP2 regulates VEGF-C expression mainly via post-translational regulation
We then aimed to characterize cellular and molecular mechanisms of how DUSP2 regulates VEGF-C expression. As shown in , we observed an approximately two-fold increase of VEGF-C mRNA and a greater than 10-fold increase of protein in DUSP2-KD cell lysates, suggesting there are other mechanisms to regulate VEGF-C biogenesis. Thus, we examined whether mRNA stability would contribute to the marked increase of VEGF-C protein. Treatment with actinomycin D to block gene transcription revealed that half-life of VEGF-C mRNA is comparable in control and DUSP2-KD cells (Supplementary Figure 6), suggesting that mRNA stability is not a factor contributing to the increased of VEGF-C protein. VEGF-C is synthesized as preproprotein (59 kD) and the removal of signal peptide and subsequent proteolytic cleavage generates the functional form of VEGF-C [Citation29]. Therefore, we hypothesized that regulation of VEGF-C processing is a major factor contributing to the marked increase of VEGF-C. Time course study showed that mature form of VEGF-C (30 KD) was markedly increased in the conditioned media of DUSP2-KD cells. However, we also observed gradual accumulation in the control group (), suggesting that VEGF-C is processed faster in DUSP2-KD cells. To study the processing of VEGF-C, AsPC1-VEGF-C cells were treated with cycloheximide to confirm the size of prepro-VEGF-C, pro-VEGF-C and mature form of VEGF-C (). Treatment with brefeldin A (BFA, a compound that blocks protein transportation from endoplasmic reticulum to Golgi apparatus) significantly reduced the mature form of VEGF-C in cell lysate and conditioned media while causing an accumulation of preproVEGF-C and proVEGF-C within the cell (), indicating that protein cleavage may be a critical step for DUSP2-regulated VEGF-C secretion. This finding was further confirmed by transient expression of DUSP2 in AsPC1-VEGF-C expressing cells that the levels of secreted but not preprocessed VEGF-C were significantly decreased ()).
To investigate how DUSP2 affects the processing of VEGF-C, we first checked the expression of proprotein convertases, which have been shown to process VEGF-C. However, the expression level of furin, PC5/6, and PCSK7 was similar in control and DUSP2-KD cells (Supplementary Figure 7). We then detected overall proprotein convertases activity and found it was substantially higher in DUSP2-KD cells (). Treatment of the cells with an irreversible, cell-permeable competitive inhibitor successfully suppressed proprotein convertases activity () and significantly decreased the production of the mature form VEGF-C (30 KD) in DUSP2-KD cells (). In contrast, the unprocessed form of VEGF-C (59 KD) was accumulated (). Pretreatment with proprotein convertase inhibitor in DUSP2-KD cells deceased cell invasion ability, which can be rescued by the addition of recombinant VEGF-C (), confirming the role of the functional form of VEGF-C in meditating cell invasion ability. Taken together, these data show that DUSP2 regulates VEGF-C expression mainly at post-translational level via modulation of proprotein convertase activity.
DUSP2 regulates the secretion of extracellular vesicles which contain VEGF-C
Cell motility has recently been linked to EV secretion [Citation30]. Since we have discovered that secreted VEGF-C is mainly associated with EV () and DUSP2 knockdown promotes cell mobility, we hypothesized that DUSP2 expression regulates the secretion of EV-VEGF-C. Isolation of EV from control and DUSP2-KD cells demonstrated that VEGF-C is increased in EV from DUSP2-KD cells (), which can be diminished by ERK inhibitor ().
To further investigate the cellular process of VEGF-C secretion, a plasmid that encodes VEGF-C-SNAP fusion protein (VE-SNAP) was transiently transfected into cells (Supplementary Figure 8A) to visualize the trafficking of VEGF-C particle. Immunofluorescence imaging shows signals of VE-SNAP were detected in vesicle-like spots within the cell (Supplementary Figure 8B). In addition, VE-SNAP (exogenous VEGF-C), as well as the endogenous mature VEGF-C (30 kD) were detected in EV isolated from various pancreatic cancer cells, respectively (Supplementary Figure 8 C). We observed that VE-SNAP signal can be detected in the secreting-like vesicles in DUSP2-KD cells compared to that in the control cells which shows a more dispersed pattern (). Therefore, we further confirmed that secreted VEGF-C in DUSP2-KD cells is associated with EV by the TEM. As shown, VEGF-C is associated with the double-membraned secreting vesicles adjacent to the plasma membrane (). These data demonstrated that VEGF-C can be secreted via EV and is regulated by DUSP2. Interestingly, we also observed that DUPS2-KD cells have more EVs as compared to control cells (). To quantitatively demonstrate that DUSP2-KD promotes the formation of EVs, we collected conditioned media from control and DUSP2-KD cells for NTA. Our results indicated that DUSP2-KD PANC-1 cells have increased numbers of EVs ranging from 85 to 155 nm ( left and middle), which can be diminished when treated with GW4869 ( right). To further characterize the difference in regard to EV in control and DUSP2-KD cells, we transiently expressed CD63, a transmembrane protein which is associated with intra/extracellular vesicles, in control and DUSP2-KD cells and tracked the signals by time-lapse microscopy. We observed that CD63 positive vesicles moved much faster in DUSP2-KD cells (Supplementary movie). Due to the limitation of 2D tracking, we next measured the vesicles moving using a 3D setting. Control and DUSP2-KD cells were labelled with PKH67 and subjected to 3D tracking by light sheet fluorescence microscopy. Results showed that more vesicles were produced in DUSP2-KD cells and these vesicles move much faster as compared to those in control cells (). CMV-driven VE-SNAP was stably expressed in the cells and proprotein convertase inhibitor was administered to dissect out the effect of DUSP2 in the secretion rate of EV-VEGF-C. Time course detection indicated that DUSP2-KD cells have an increased secretion rate of EV-VEGF-C as compared to control cells (). Last, we demonstrate that EV from DUSP2-KD cells promotes lymphangiogenesis in PANC-1 tumours compared to that treated with control PBS (). Together, we demonstrated that DUSP2 knockdown promotes the secretion of EV-VEGF-C.
Discussion
DUSP2 is a potent tumour suppressor that preferentially inactivates ERK; thus, loss of DUSP2 contributes to the activation of MAPK signalling, which results in accelerating cancer progression and malignancy [Citation18,Citation19]. Herein, we unravel a novel mechanism demonstrating that DUSP2 dysfunction may contribute to early dissemination of pancreatic cancer. We report here that loss-of-DUSP2 promotes early metastasis via increasing lymphovascular invasion in both orthotopic and genetic mouse models of PDAC. Suppression of DUSP2 enhances lymphangiogenesis and cancer cell invasiveness via paracrine and autocrine effects of VEGF-C. As early dissemination is a unique feature that significantly increases the mortality rate of PDAC, this finding is of particular importance.
Increased expression of VEGF-C is highly associated with lymphangiogenesis and lymph node metastasis in many types of cancer. In the rat model of endocrine pancreatic β cells tumours, overexpression of VEGF-C promoted lymphangiogenesis and lymph node metastasis of non-metastatic β cells [Citation31]. As we showed in this study, knockdown of DUSP2 in pancreatic cancer cells increased the mature form of VEGF-C, which can enhance the lymphangiogenic properties of LECs. Pathway analysis of four public pancreatic cancer datasets led us to speculate that VEGF-C and correlated genes may involve in cytoskeleton remodelling, a phenomenon associated with cell migratory ability. Since we also detected VEGF-C-related receptors in pancreatic cancer cells, we reasoned there is an autocrine function of VEGF-C signalling on cancer cells. Indeed, the migratory and transendothelial abilities of DUSP2-KD pancreatic cancer cells are significantly increased, which can be abolished by treating with VEGFR2/3 inhibitor. These data strongly suggest that VEGF-C signalling in the tumour environment not only increases lymphangiogenesis but also promotes the ability of tumour cells to invade into the lymphatic/vascular vessels concomitantly.
Expression of VEGF-C in cancer cells can be controlled at different levels [Citation32–Citation38], but few studies focus on its post-translational regulation [Citation39–Citation41]. As a secreted growth factor, the function and receptor specificity of VEGF-C are determined by proteolytic cleavage via different modes [Citation29,Citation41]. The 30 kD VEGF-C promotes mainly lymphangiogenesis while the fully processed 21 kD VEGF-C stimulates both lymphangiogenesis and angiogenesis [Citation4]. Therefore, it is critical to determine the level of secreted VEGF-C in conditioned medium of cancer cells. In our study, we found that DUSP2-KD not only affects the mRNA level of VEGF-C but most importantly, significantly enhances the level of secreted and functional forms of VEGF-C. The 30 kD form of VEGF-C was markedly elevated in the conditioned media and EVs of DUSP2-KD cells, which is consistent with the known function of VEGF-C and the observation that lymphangiogenesis is elevated in murine and human pancreatic cancers. Although the expression level of proprotein convertases such as furin, PC5/6, and PCSK7, is similar, overall proprotein convertase activity is increased in DUSP2-KD cell lysates. On the topic of regulation of proprotein convertase activity, little is known besides that autocleavage and correct subcellular location are required [Citation42,Citation43]. Thus, a detailed mechanism of how DUSP2 regulates proprotein convertase activity remains to be investigated.
One of the most intriguing findings of the current study is the identification that secreted VEGF-C is mainly associated with EVs and loss-of-DUSP2 increases EV-VEGF-C secretion. Secreted proteins, such as growth factors or cytokines, were recovered with EVs [Citation28] while the mechanism of how those secreted factors are loaded to EVs is still largely unknown. Heparan sulphate proteoglycans (HSPGs) exist on cell-surface and in the extracellular matrix plays an important role in regulating growth factors action and distribution; thus, the presence of HSPGs on EVs may tether growth factors and mediate their function. The association of VEGF-C to EVs is not heparinase II sensitive in our study (supplementary 9), suggesting the association is not mediated via HSPGs. Interestingly, it was recently found that the latent form of transforming growth factor beta 1 (TGFβ1), not the active TGFβ1, is associated with EVs via heparin and heparan sulphate[Citation44]. Therefore, active and inactive form of growth factors may be carried on EVs via different mechanisms. Chaperone Hsp90 was found associated with EV-VEGF-A [Citation45], thus VEGF-C may attach to EVs via similar mechanism albeit remains to be characterized. By transmission electron microscopy and NTA analysis, we demonstrated that DUSP2-KD PANC-1 cells have increased numbers of EVs. Furthermore, 3D tracking for the fast-moving vesicles by inverted light sheet fluorescence microscopy provides evidence that vesicles’ moving speed is accelerated in DUSP2-KD cells. Our data are consistent with the previous finding that hypoxic stress promotes EV secretion in breast cancer cells [Citation46]. Since we have previously demonstrated that hypoxia suppresses DUSP2 expression, it is likely that hypoxia induces EV secretion is mediated by downregulation of DUSP2. Although the cellular mechanism of EV biogenesis remains largely unknown, it was recently found that actin network, the driving force for cell migration, and small GTPase, Rab7 and 27a, are involved in the secretion of EVs [Citation24,Citation47]. Therefore, it is likely that DUSP2 knockdown affects the cytoskeleton dynamics which results in enhancing migration ability and secretion of EVs. Selective inhibitors of EV biogenesis and secretion also inhibited the activation of ERK [Citation48], implying the involvement of ERK signalling. Therefore, sustained ERK signalling due to DUSP2 suppression may alter phosphorylation status of proteins participate in EV biogenesis. Tetraspanin proteins are not only EV markers but also functionally involve in EV secretion, biogenesis, and cargo sorting [Citation49]. Glycosylation and palmitoylation have been shown as post-translational modification of tetraspanin proteins [Citation50], while the correlation of phosphorylation and EV-related functions is not yet investigated. Therefore, it will be interesting to further study the impact of tetraspanin protein phosphorylation on cargo loading and the involvement of DUSP2.
In conclusion, a model of early dissemination of pancreatic cancer cells was proposed based on our current findings (). Downregulation of DUSP2 in pancreatic cancer cells increases VEGF-C mRNA transcription and enhances proprotein convertase activity, which promotes the production of the functional form of VEGF-C. DUSP2 downregulation also increases the amount and moving ability of EV-carried VEGF-C, which enhances the secretion of functional VEGF-C into the tumour microenvironment. Secreted VEGF-C can then bind to receptors on LECs to promote lymphangiogenesis and to receptors expressed in pancreatic cancer cells to increase the invasive ability, allowing tumour cells to enter the blood and lymphatic vessels. As a result, downregulation of DUSP2 in pancreatic cancer cells will facilitate the lymphovascular invasion of tumour cells. Our data further reveal that blocking EV-carried VEGF-C production may be a new approach to inhibit PDAC early dissemination and cancer malignancy.
Figure 1. VEGF-C is associated with extracellular vesicles. (a) Representative immunohistochemical staining images (serial section) show expression of Lyve-1 and VEGF-C in the pancreas of Lox-Stop-Lox (LSL)-Trp53R172 H (WT) and in the tumour of LSL-KrasG12D; LSL-Trp53R172 H; Pdx1-cre (KPC) transgenic mouse. (b) Serum-free conditioned medium from MIA PaCa-2 cells was collected and fractions were isolated based on size exclusion chromatography according to the manufacturer’s protocol. Expression of VEGF-C, CD63 and HSP70 was detected in vesicle-associated fractions by Western blotting (upper). Three fractions (as indicated) were sent for NTA analysis (bottom left). Protein concentration in each fraction was measured (bottom right). (c) VEGF-C is highly expressed in EV fraction. Conditioned medium from MIA PaCa-2 cells was collected and ultracentrifugation was performed to isolate microvesicles (MV), exosomes (Ex) and supernatant (Sup). Western blotting was performed to detect the expression of VEGF-C, CD63 and ALBUMIN (ALB) in whole cell lysate (WCL), MV, Ex and Sup with equal amount of protein. (d) EV was isolated by ExoQuick-TC from CM of MIA PaCa-2 cells. Equal protein amount was loaded to compare VEGF-C expression in WCL, EV and CM. CD63, TSG101 and HSP70 were used as EV markers. ALB was detected to demonstrate the purity of EV (left). EV isolated by ExoQuick-TC was sent for NTA analysis (right). (e) Representative transmission electron microscopic images show that VEGF-C is associated with EV. VEGF-C (dark particles) was stained with gold particle-labelled anti-VEGF-C antibody. (f) VEGF-C is associated with surface of EV. Isolated EV (by ExoQuick-TC) was treated with proteinase K, triton X, proteinase K plus triton X and trypsin. Western blotting was performed to detect VEGF-C and GAPDH. (g) Expression of VEGF-C in AsPC1 cells. VEGF-C cDNA was cloned into pCDH lentivirus vector under the control of CMV promoter (upper panel). Expression of secreted VEGF-C is decreased by treatment with GW4869 (40 μM). (h) VEGF-C is enriched in the small EV fraction. Conditioned medium from AsPC-VEGF-C cells was collected and the EV fraction (MV and Ex) was purified by ultracentrifugation. CD63 was detected as a marker for EV. (i) NTA analysis (left) and Western blotting (right) show particle numbers and VEGF-C expression in AsPC-Ctrl and AsPC-VEGF-C cells. EV was isolated by ExoQuick-TC. (j) LECs uptake EV from MIA PaCa-2 cells. Mia PaCa-2 cells were labelled with PKH67 and serum-free conditioned medium from PKH67 labelled Mia PaCa-2 cells were collected and isolated by ExoQuick-TC. LECs were treated EVs for 6 h and fixed for image taken. (k) Quantification of LECs proliferation treated with EV from AsPC-control and AsPC-VEGF-C cells. Ki67 staining was performed as the indicator of the proliferation of LECs. **P < 0.01. (L) Schematic of experimental design to evaluate the effect of EV from AsPC-VEGF-C cells (left). Representative immunohistochemical images show the increase of lymphatic vessels (Lyve-1) in AsPC-1 tumours treated with EV isolated from AsPC-VEGF-C cells (by ultracentrifugation isolation).
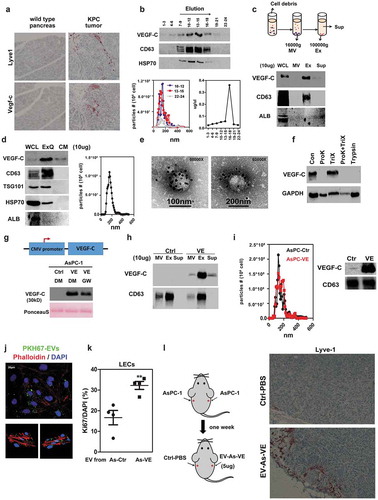
Figure 2. Inhibition of DUSP2 increases VEGF-C expression. (a) Representative pictures of DUSP2 immunostaining in pancreatic tumours. PanIN: pancreatic intraepithelial neoplasia, Ca: invasive carcinoma, arrowhead: non-tumour elements. (b) Representative pictures of DUSP2 immunostaining in KPC tumours. Arrows: infiltrating cancer cells; circles: carcinoma (Ca). (c) Representative immunohistochemical staining images show the expression of ERK1/2 phosphorylation in LSL -Trp53R172 H (WT) pancreas and KPC tumours. (d) Representative Western blots show the expression of phosphorylated ERK (left), EV-VEGF-C (middle) in MIA PaCa-2 cells treated without (DMSO) or with ERK inhibitor, SCH 772984 (1 μM, SCH) for 24 h. EVs were isolated by ultracentrifugation (Ultra) or ExoQuick-TC (ExoQ) exosome precipitation reagent. The right panel shows EV-VEGF-C in AsPC-1-Ctrl and AsPC-1-VEGF-C cells treated with DMSO or SCH 772984 (1 μM, SCH). EV in AsPC-1 was isolated by ExoQuick-TC. Annexin V and CD63 were used as markers of EVs in MIA PaCa-2 and AsPC-1 cells, respectively. (e) Representative image shows levels of DUSP2 in human pancreatic cancer cell lines PANC-1 and MIA PaCa-2 (left) and levels of DUSP2, total and phospho-ERK in control and DUSP2-KD PANC-1 cells (right). DU#1 and DU#2 indicate two stable clones of PANC-1 cells with DUSP2 knockdown. β-actin is a loading control. (f) VEGF-A and VEGF-C were measured in control and DUSP2-KD PANC-1 cells by RT-qPCR. Data represent mean and SEM of three experiments using different batches of cells (left). Representative image and the quantitative result (right) show the increase of secreted VEGF-C in DUSP2-KD PANC-1 cells. Ponceau S staining was employed to examine the equal loading of proteins. (g) RT-PCR and Western blotting for the detection of VEGF-C mRNA and secreted protein in control and DUSP2-KD PANC-1 cells treated with U0126 (10 μM). (h) Representative Western image shows levels of DUSP2 and VEGF-C in cells with or without transient expression of DUSP2 in MIA PaCa-2 cells. Secreted VEGF-C was quantified in control and DUSP2 transfected MIA PaCa-2 cells (right). (i) Inverse expression of DUSP2 and VEGF-C in mouse tissues. Various tissues from male BALB/c mice were collected and homogenized. Western blotting was performed to measure the expression of DUSP2 and VEGF-C. *P < 0.05; **P < 0.01; ***P < 0.001.
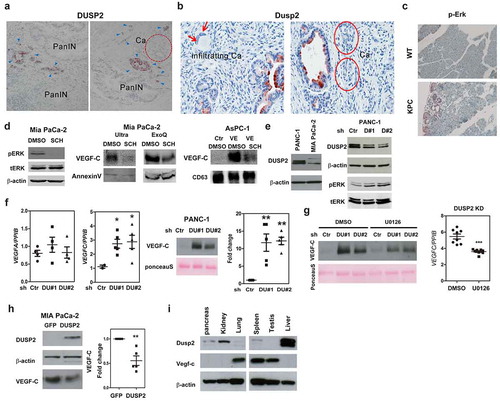
Figure 3. DUSP2 knockdown promotes lymphangiogenesis and lymphovascular invasion in the orthotopic pancreatic cancer mouse model. (a) Conditioned media from DUSP2-KD PANC-1 cells enhanced the proliferation of LECs. Conditioned media from control and DUSP2-KD PANC-1 cells were used to treat LECs with or without the addition of VEGFR2/R3 inhibitor, Lenvatinib (10 nM). Data represent mean and SEM of three independent experiments using conditioned media collected from different batches of cells. (b) Representative pictures (left) and quantitative result (right, n = 3) show conditioned media from DUSP2-KD PANC-1 cells promoted migration of LECs. *P < 0.05; ***P < 0.001. (c) Representative picture of tumours developed in the pancreas of mice when injected with PANC-1 control and DUSP2-KD cells. (d) Representative images show the increase of lymphatic vessels in DUSP2-KD orthotopic injected tumours compared with that in control tumours. Lymphatic marker Lyve-1 was used to identify lymphatic vessels. CD31 was used to identify blood vessels. Original magnification, ×200. (e) Example of lymphovascular invasion and lymph node metastasis by H&E stain. No tumour cells were present in the lymphatic/blood vessels in mice injected with control PANC-1 cells while tumour emboli were seen in mice injected with DUSP2-KD PANC-1 cells. Asterisk represents tumour cells. Fisher’s exact test (two sided) indicates a significant increase in the number of mice with lymphovascular invasion (LVI) and nodal metastasis (LNM) if they are bearing DUSP2-KD tumours (P = 0.01). Tumours were sliced as serial sections and H&E-stained histologic sections were scored in a blinded manner by a pathologist (bottom right). (f) Schematic drawings illustrate the strategy of generating conditional knockout of Dusp2 in the pancreas of mice (top). Representative image of genotyping result by PCR (middle). Representative immunohistochemical staining images show expression of Lyve-1 in the pancreas of 4 months old wild type (WT) and Dusp2 knockout (Dusp2−/-) mice (bottom). Original magnification, ×200. (g) Representative immunohistochemical staining images show expression of phosphorylated ERK, Lyve-1 and CD31 in serial sections in the pancreas of 7 months old wild type (WT) and LSL-KrasG12D; LSL-Dusp2; Pdx1-cre (KDC) mice. Original magnification, ×200. (h) The expression of VEGF-C is increased in EV isolated from serum in KDC mice. Western blotting was performed for the detection of VEGF-C and CD63. Ponceau S staining was used as control for equal amount of protein loading.
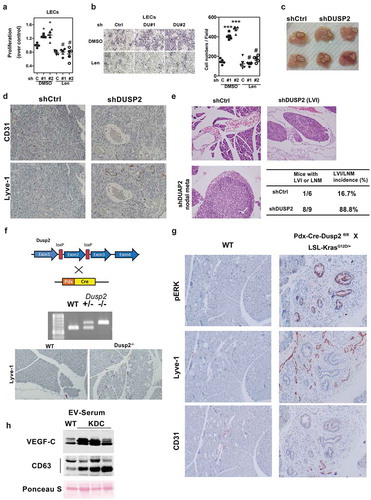
Figure 4. Knockdown of DUSP2 mediates autocrine function in pancreatic cancer cells via VEGF-C signalling. (a) Representative image shows the morphology of control and DUSP2 knockdown in PANC-1 cells. (b) Representative image (left) and the quantitative result (right, n = 3) show the migration ability in DUSP2-KD PANC-1 cells. Control (ctrl) and DUSP2-KD (DU#1 and DU#2) PANC-1cells were seeded in the 24-well plate to confluent and a straight scratch was made by pipette tips. Images were recorded and analysed after 24 h. (c) Representative image (left) and the quantitative result (right, n = 3) show cell invasion ability in control and DUSP2-KD PANC-1 cells. (d) Expression of DUSP2-GFP in MIA PaCa-2 cells inhibited cell migration through transwell. MIA PaCa-2 cells were transfected with GFP or DUSP2-GFP, equal numbers of cells were plated on top of transwell. Data represent mean and SEM from two independent experiments (performed in triplicate) using different batches of cells. (e) Representative images (left) and the quantitative result (right, n = 3) show increased invasion ability in DUSP2-KD PANC-1 cells was suppressed when treated with VEGFR2/R3 inhibitor, Lenvatinib (10 nM). *P < 0.05 compared to control, # P < 0.05 compared to DUSP2-KD. (f) Representative images (left panels) and quantitative result (right, n = 3) show knockdown of VEGF-C abolished loss-of-DUSP2-mediated increased invasion ability. VEGF-C was transient knocked down in control and DUSP2-KD PANC-1 cells. Cell invasion ability was measured in matrigel-coated transwell. *P < 0.05 compared to control, # P < 0.05 compared to DUSP2-KD. (g) Schematic diagram (top) and quantitative result (bottom, n = 3) show increased transendothelial migration assay in control and DUSP2-KD PANC-1 cells were suppressed when treated with VEGFR2/R3 inhibitor. Endothelial cells were seeded to a matrigel-coated transwell. After the endothelial layer was formed, PANC-1 control and DUSP2-KD cells were added to the chamber and transendothelial migration ability was measured after 16 h. *P < 0.05 compared to control, #P < 0.05 compared to DUSP2-KD. *P < 0.05; **P < 0.01.
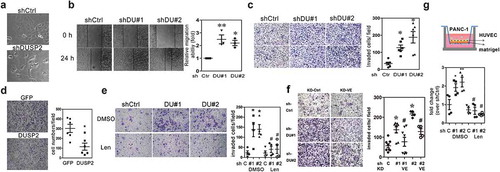
Figure 5. DUSP2 regulates VEGF-C expression mainly via post-translational modification. (a) Representative Western blots show expression of VEGF-C in conditioned media of control and DUSP2-KD PANC-1 cells at different time points. Angiogenin (ANG) was used as a loading control in conditioned medium. (b) AsPC1-VEGF-C cells were treated with cycloheximide (CHX) and BFA to block protein synthesis and protein secretion. The size of prepro-VEGF-C, pro-VEGF-C and processed VEGF-C was detected as indicated. (c) AsPC1-VEGF-C cells were transiently transfected with GFP or DUSP2-GFP plasmids and levels of DUSP2, VEGF-C in whole cell lysate (WCL) and conditioned medium (CM) were detected. (d) Proprotein convertase activity in control and DUSP2-KD PANC-1 cells. Cell lysates of control and DUSP2-KD cells were incubated with two different concentrations of fluorogenic proprotein convertase substrate. Fluorescent was measured in a kinetic manner with Ex: 360–380 nm, Em: 440–460 nm. Representative quantification (of three independent experiments) is shown. (e) Proprotein convertase activity was measured in control and DUSP2-KD PANC-1 cells treated with proprotein convertase inhibitor for 24 h. (f) Representative images (left) and the quantitative result (n = 3, right) show loss-of-DUSP2-enhanced VEGF-C secretion was inhibited by treatment with proprotein convertase inhibitor. Control and DUSP2-KD PANC-1 cells were treated with proprotein convertase inhibitor (20 μM) in serum-free RPMI medium. After 24 h, serum-free conditioned media were collected and VEGF-C expression was measured. ** P < 0.01 compared to control, # P < 0.05 compared to DUSP2-KD. (g) Control and DUSP2-KD cells were pre-treated with proprotein convertase inhibitor and plated for invasion ability. Recombinant VEGF-C was treated in the upper chamber of transwell. *P < 0.05.
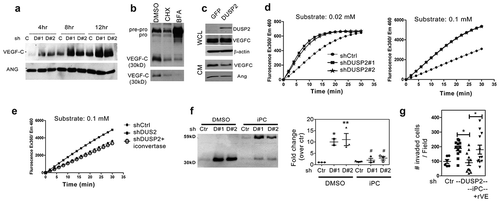
Figure 6. DUSP2 regulates the secretion of EV-VEGF-C in pancreatic cancer cells. (a) Representative (upper) and quantification (bottom) of VEGF-C and HSP70 expression by Western blotting in EV (isolated by ExoQuick-TC) from control and DUSP2-KD PANC-1 cells. (b) Representative Western blots show VEGF-C expression in WCL, EV and CM in DUSP2-KD PANC-1 cells (left). Representative Western blots show VEGF-C expression in EV isolated from DUSP2-KD PANC-1 cells treated with DMSO (control) and SCH 772984 (1uM) for 24 h (right). CD63 and HSP70 were detected as EV markers. ALB was detected to demonstrate the purity of EV. Equal amount of protein (20 ug) was loaded. EV was isolated by ExoQuick-TC. (c) Images of control and DUSP2-KD PANC-1 cells transfected with VEGF-C-SNAP tag for 24 h. Cells were treated with proprotein convertase inhibitor in serum-free medium for another 24 h after transfection. After labelling by the fluorescent substrate for SNAP, cells were fixed and imaged. (d) Images of control and DUSP2-KD PANC-1 cells taken by the transmission electron microscope. Red arrows indicate positive staining by 10 nm gold labelled anti-VEGF-C antibody. (e) Nanoparticle Tracking Analysis (NTA) was performed to detect secreted particles from control and DUSP2-KD cells. Serum-free conditioned medium was collected and centrifuged to remove debris and were sent for NTA. The X-axis represents the size of particles and the Y-axis represents the number of particles secreted per cell. DUSP2-KD cells have increased numbers of EV ranging from 85 to 155 nm (middle panel). EV secreted from DUSP2-KD cells was diminished if treated with GW4869 (20 μM) (right panel). (f) Inverted light sheet microscope (Luxengo) was used to track fast-moving particles in control and DUSP2-KD cells. Cells were labelled with PKH67 and plated into the cell holder for the tracking of PKH67 positive particles within the cells. 3D tracking of PKH67 particles in control and DUSP2-KD cells analysed by Imaris software (left). Particles in DUSP2-KD cells have increased speed (upper right) and track length (bottom right). Experiments have been performed two times and represented data are shown. (g) DUSP2-KD cells have an increased rate of EV-VEGF-C secretion. VE-Snaptag was stably expressed in control and DUSP2-KD cells. Cells were treated with proprotein convertase inhibitor (20 uM) in serum-free RPMI. EV was isolated from the medium at a different time point. Western blotting was performed to detect unprocessed VEGF-C (VE-Snap) in EV (upper). Expression of EV-VEGF-C in control and DUSP2-KD cells by fold change analysis over 2 h (lower). (h) Schematic of experimental design to investigate the function of EV from DUSP2-KD PANC-1 cells (left). Representative immunohistochemistry images show the increase of lymphatic vessels (Lyve-1) in PANC-1 tumours treated with EV isolated from DUSP2-KD cells.
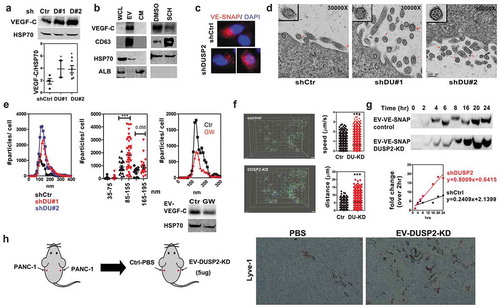
Figure 7. A proposed model shows the role of DUSP2-VEGF-C axis in promoting pancreatic cancer progression. Downregulation of DUSP2 in pancreatic cancer cells leads to ERK phosphorylation which enhances proprotein convertase activity. Therefore, the production of a functional form of VEGF-C is increased. On the other hand, the amount and moving ability of EV are increased in DUSP2 knockdown cells, which enhance the secretion of functional VEGF-C to the tumour microenvironment. Increased EV-VEGF-C can promote lymphangiogenesis and enhances pancreatic cancer cell invasive ability, leading to lymphovascular invasion.
Authors’ Contributions
CA Wang designed and performed experiments, analyzed data and wrote the initial draft of the manuscript. IH Chang, PC Hou, YJ Tai, WN Li and PL Hsu performed in vitro experiments (Western blotting and IHC staining). S Wu contributed to the TEM experiment and CF Li scored the pathology of mouse and human sections. WT Chiu performed the 2D vesicle tracking experiment. YS Shan provided human pancreatic cancer tissues and intellectual framework. SJ Tsai conceived the project and provided the critical review of the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Declarations
All authors declare no conflict of interest.
Ethical Approval
The clinical specimens were conducted with permission from the institutional review board approval from the National Cheng Kung University Hospital, Tainan. Experimental procedures of animal studies were approved by the Institutional Animal Care and Use Committee at the National Chung Kung University.
Supplemental Material
Download Zip (10.1 MB)Acknowledgments
We thank Dr. Jonathan Jou (University of Illinois) for critical reading and editing of the manuscript. Human lymphatic endothelial cell line (LECs) was kindly provided by Dr. Wen-Chun Hung, National Health Research Institutes and human umbilical vein endothelial cells (HUVECs) was a kind gift from Dr. Chia-Ching Wu’s laboratory in Department of Cell Biology and Anatomy, College of Medicine, National Cheng Kung University.
We like to thank Yi-Shan Ya, Shu-Chen Hung, Yi-Chen Tang, Yi-Jou Chung and Yen-Yu Lai for the technical support in immunohistochemistry staining, animal experiments, and bioinformatics analysis. We thank Taiwan Bioinformatics Institute Core Facility for the assistance on using Oncomine (National Core Facility Program for Biotechnology (MOST 105-2319-B-400-002). We thank the technical services provided by the “Bioimaging Core Facility of the National Core Facility for Biopharmaceuticals, Ministry of Science and Technology, Taiwan”. We thank the Center for Genomic Medicine and Center for Micro/Nano Science and Technology for Bioinformatic analysis and NTA analysis, respectively.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Haiko P, Makinen T, Keskitalo S, et al. Deletion of vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol Cell Biol. 2008;28:4843–17.
- Kukk E, Lymboussaki A, Taira S, et al. VEGF-C receptor binding and pattern of expression with VEGFR-3 suggests a role in lymphatic vascular development. Development. 1996;122:3829–3837.
- Zhao YC, Ni XJ, Wang MH, et al. Tumor-derived VEGF-C, but not VEGF-D, promotes sentinel lymph node lymphangiogenesis prior to metastasis in breast cancer patients. Med Oncol. 2012;29:2594–2600.
- Skobe M, Hawighorst T, Jackson DG, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7:192–198.
- Tang RF, Itakura J, Aikawa T, et al. Overexpression of lymphangiogenic growth factor VEGF-C in human pancreatic cancer. Pancreas. 2001;22:285–292.
- Kurahara H, Takao S, Maemura K, et al. Impact of vascular endothelial growth factor-C and -D expression in human pancreatic cancer: its relationship to lymph node metastasis. Clin Cancer Res. 2004;10:8413–8420.
- Furukawa T. Impacts of activation of the mitogen-activated protein kinase pathway in pancreatic cancer. Front Oncol. 2015;5:23.
- Collins MA, Yan W, Sebolt-Leopold JS, et al. MAPK signaling is required for dedifferentiation of acinar cells and development of pancreatic intraepithelial neoplasia in mice. Gastroenterology. 2014;146:822–834 e827.
- Lang R, Hammer M, Mages J. DUSP meet immunology: dual specificity MAPK phosphatases in control of the inflammatory response. J Immunol. 2006;177:7497–7504.
- Jeffrey KL, Camps M, Rommel C, et al. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov. 2007;6:391–403.
- Rohan PJ, Davis P, Moskaluk C, et al. PAC-1: a mitogen-induced nuclear protein tyrosine phosphatase. Science. 1993;259:1763–1766.
- Rios P, Nunes-Xavier CE, Tabernero L, et al. Dual-specificity phosphatases as molecular targets for inhibition in human disease. Antioxid Redox Signal. 2014;20:2251–2273.
- Ward Y, Gupta S, Jensen P, et al. Control of MAP kinase activation by the mitogen-induced threonine/tyrosine phosphatase PAC1. Nature. 1994;367:651–654.
- Zhang Q, Muller M, Chen CH, et al. New insights into the catalytic activation of the MAPK phosphatase PAC-1 induced by its substrate MAPK ERK2 binding. J Mol Biol. 2005;354:777–788.
- Lu D, Liu L, Ji X, et al. The phosphatase DUSP2 controls the activity of the transcription activator STAT3 and regulates TH17 differentiation. Nat Immunol. 2015;16:1263–1273.
- Yin Y, Liu YX, Jin YJ, et al. PAC1 phosphatase is a transcription target of p53 in signalling apoptosis and growth suppression. Nature. 2003;422:527–531.
- Kim SC, Hahn JS, Min YH, et al. Constitutive activation of extracellular signal-regulated kinase in human acute leukemias: combined role of activation of MEK, hyperexpression of extracellular signal-regulated kinase, and downregulation of a phosphatase, PAC1. Blood. 1999;93:3893–3899.
- Lin SC, Chien C-W, Lee J-C, et al. Suppression of dual-specificity phosphatase-2 by hypoxia increases chemoresistance and malignancy in human cancer cells. J Clin Invest. 2011;121:1905–1916.
- Hou PC, Li Y-H, Lin S-C, et al. Hypoxia-induced downregulation of DUSP-2 phosphatase drives colon cancer stemness. Cancer Res. 2017;77:4305–4316.
- Lin SC, Hsiao KY, Chang N, et al. Loss of dual-specificity phosphatase-2 promotes angiogenesis and metastasis via up-regulation of interleukin-8 in colon cancer. J Pathol. 2017;241:638–648.
- Lobb RJ, Lima LG, Moller A. Exosomes: key mediators of metastasis and pre-metastatic niche formation. Semin Cell Dev Biol. 2017;67:3–10.
- Kosaka N, Yoshioka Y, Tominaga N, et al. Dark side of the exosome: the role of the exosome in cancer metastasis and targeting the exosome as a strategy for cancer therapy. Future Oncol. 2014;10:671–681.
- Li Z, Jiang P, Li J, et al. Tumor-derived exosomal lnc-Sox2ot promotes EMT and stemness by acting as a ceRNA in pancreatic ductal adenocarcinoma. Oncogene. 2018;37:3822–3838.
- Dorayappan KDP, Wanner R, Wallbillich JJ, et al. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: a novel mechanism linking STAT3/Rab proteins. Oncogene. 2018;37:3806–3821.
- Lobb RJ, van Amerongen R, Wiegmans A, et al. Exosomes derived from mesenchymal non-small cell lung cancer cells promote chemoresistance. Int J Cancer. 2017;141:614–620.
- Wen SW, Sceneay J, Lima LG, et al. The biodistribution and immune suppressive effects of breast cancer-derived exosomes. Cancer Res. 2016;76:6816–6827.
- Bourne GL, Grainger DJ. Development and characterisation of an assay for furin activity. J Immunol Methods. 2011;364:101–108.
- Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750.
- Joukov V, Sorsa T, Kumar V, et al. Proteolytic processing regulates receptor specificity and activity of VEGF-C. EMBO J. 1997;16:3898–3911.
- Sung BH, Ketova T, Hoshino D, et al. Directional cell movement through tissues is controlled by exosome secretion. Nat Commun. 2015;6:7164.
- Mandriota SJ, Jussila L, Jeltsch M, et al. Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. EMBO J. 2001;20:672–682.
- Wang CA, Tsai SJ. The non-canonical role of vascular endothelial growth factor-C axis in cancer progression. Exp Biol Med (Maywood). 2015;240:718–724.
- Wang CA, Jedlicka P, Patrick AN, et al. SIX1 induces lymphangiogenesis and metastasis via upregulation of VEGF-C in mouse models of breast cancer. J Clin Invest. 2012;122:1895–1906.
- Zhang H, Muders MH, Li J, et al. Loss of NKX3.1 favors vascular endothelial growth factor-C expression in prostate cancer. Cancer Res. 2008;68:8770–8778.
- Ristimaki A, Narko K, Enholm B, et al. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J Biol Chem. 1998;273:8413–8418.
- Li G, Yang F, Gu S, et al. MicroRNA-101 induces apoptosis in cisplatin-resistant gastric cancer cells by targeting VEGF-C. Mol Med Rep. 2016;13:572–578.
- Zhou XU, QI L, TONG S, et al. miR-128 downregulation promotes growth and metastasis of bladder cancer cells and involves VEGF-C upregulation. Oncol Lett. 2015;10:3183–3190.
- Morfoisse F, Kuchnio A, Frainay C, et al. Hypoxia induces VEGF-C expression in metastatic tumor cells via a HIF-1alpha-independent translation-mediated mechanism. Cell Rep. 2014;6:155–167.
- Siegfried G, Basak A, Cromlish JA, et al. The secretory proprotein convertases furin, PC5, and PC7 activate VEGF-C to induce tumorigenesis. J Clin Invest. 2003;111:1723–1732.
- Bui HM, Enis D, Robciuc MR, et al. Proteolytic activation defines distinct lymphangiogenic mechanisms for VEGFC and VEGFD. J Clin Invest. 2016;126:2167–2180.
- Jha SK, Rauniyar K, Karpanen T, et al. Efficient activation of the lymphangiogenic growth factor VEGF-C requires the C-terminal domain of VEGF-C and the N-terminal domain of CCBE1. Sci Rep. 2017;7:4916.
- Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. 2002;3:753–766.
- Jones BG, Thomas L, Molloy SS, et al. Intracellular trafficking of furin is modulated by the phosphorylation state of a casein kinase II site in its cytoplasmic tail. EMBO J. 1995;14:5869–5883.
- Shelke GV, Yin Y, Jang SC, et al. Endosomal signalling via exosome surface TGFβ-1. J Extracell Vesicles. 2019;8:1650458.
- Feng Q, Zhang C, Lum D, et al. A class of extracellular vesicles from breast cancer cells activates VEGF receptors and tumour angiogenesis. Nat Commun. 2017;8:14450.
- King HW, Michael MZ, Gleadle JM. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer. 2012;12:421.
- Gangoda L, Mathivanan S. Cortactin enhances exosome secretion without altering cargo. J Cell Biol. 2016;214:129–131.
- Datta A, Kim H, McGee L, et al. High-throughput screening identified selective inhibitors of exosome biogenesis and secretion: A drug repurposing strategy for advanced cancer. Sci Rep. 2018;8:8161.
- Andreu Z, Yanez-Mo M. Tetraspanins in extracellular vesicle formation and function. Front Immunol. 2014;5:442.
- Termini CM, Gillette JM. Tetraspanins function as regulators of cellular signaling. Front Cell Dev Biol. 2017;5:34.