
ABSTRACT
Polymyositis (PM) and dermatomyositis (DM) are subtypes of autoimmune inflammatory myopathies. Interstitial lung disease (ILD) involvement is common in PM/DM. There is no evidence base for immunosuppression in DM/PM-ILD and current evidence is based on case stories and expert opinions. We present a 63-year-old male with severe respiratory failure due to PM-ILD who was treated successfully with intravenous immunoglobulin, recovered the acute phase and survived more than 4 years.
Background
Polymyositis (PM) and dermatomyositis (DM) are subtypes of autoimmune inflammatory myopathies [Citation1]. The clinical feature is symmetric proximal muscle weakness. The prevalence of PM is 10 per 100.000 [Citation2]. PM/DM can involve the lungs with interstitial lung disease (ILD) in 5–64% of cases [Citation3,Citation4]. The available evidence on treatment efficacy is based on expert experience, case reports [Citation4,Citation5,Citation6–10] and extrapolated from treatment of PM/DM without ILD [Citation1,Citation2,Citation11]. Glucocorticosteroid (GCS) therapy is often used as a first-line treatment, as in the case of patients with myositis without ILD. Unfortunately, the high doses of GCS required over longer periods are often associated with severe side effects. Other immunosuppressants and immunomodulatory therapies as azathioprine, methotrexate, cyclophosphamide and cyclosporine A are often tried and have been reported to be beneficial in patients with ILD [Citation3,Citation5,Citation7,Citation12,Citation13,Citation14].
Here, we present a patient with GCS-resistant polymyositis-ILD treated with intravenous immunoglobulin (IVIG).
Case report
A 63-year-old male complained of arthralgia in the hands and wrists, generalized muscle pain, morning muscle stiffness and progressive shortness of breath during the past three weeks and had been bed-bound the last 4 days.
He had a history of hypertension, hypercholesterolemia and myocardial infarction. There was no family history of respiratory- or rheumatic diseases. He was ex-smoker (35 pack-years).
On examination, he had a temperature of 38.3°C, saturation of 92%, BP 141/59 mmHg and pulse 88 beat/min. Physical examination showed bilateral basal crackles upon auscultation, grade 4 proximal muscle weakness and no objective signs of arthritis.
Blood tests () showed elevated inflammatory parameters, signs of myositis and compromised renal function.
Table 1. A: C-reactive protein, b: alanine aminotransferase, c: Lactate dehydrogenase, d: creatine kinase, e: partial pressure of oxygen, f: partial pressure of carbon dioxide, g: bicarbonate, h: saturation on ABG
Arterial blood gas (ABG) analysis () showed hypoxemia.
Sjögren-syndrome A-Ab (IgG), Jo1 and 52KDA RO-protein antibody were all positive; muscle biopsy revealed polymyositis. An echocardiography was normal without signs of pulmonary hypertension.
High resolution computed tomography (HRCT) scan () was consistent with a combination of organizing pneumonia (OP) and non-specific interstitial pneumonia (NSIP).
Figure 1. HRCT with severe interstitial findings with peribronchial infiltrates and ground-glass opacities (GGO) in all lung fields, especially in the upper lobes. There was mediastinal lymphadenopathy
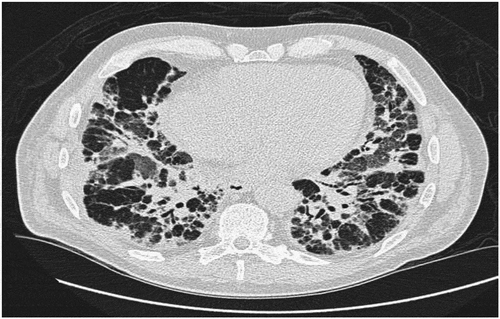
Based on symptoms, clinical findings, autoimmune serology and HRCT, a diagnosis of Anti-Jo1-antibody-positive polymyositis with OP/NSIP was made. Broad spectrum antibiotics and IV methylprednisolone pulse therapy of 500 mg daily for 3 days was initiated with continuation of 75 mg prednisolone daily resulting in a significant improvement of the muscle and joint pain with decreasing muscle enzymes. However, the respiratory symptoms worsened and the patient developed respiratory failure and was moved to the intensive care unit (ICU) where he was intubated and needed high flow oxygen therapy.
Figure 2. Second HRCT six weeks after first HRCT with progression of the interstitial changes
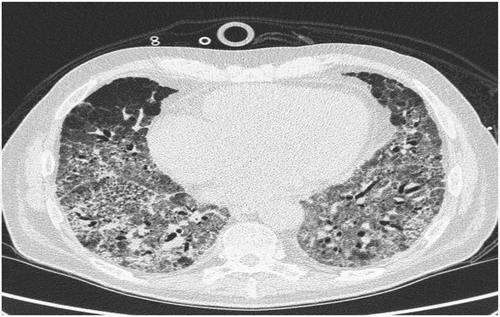
No relevant infectious agents were found in culture or PCR analysis of bronchoalveolar lavage or blood samples. During the next 6 weeks, he was treated with methylprednisolone pulse therapies every second week without respiratory improvement. A new HRCT showed progression of the interstitial changes ().
As treatment with cyclophosphamide was considered risky, treatment with intravenous immunoglobulin (IVIG) 0.4 mg/kg (30 mg) daily for 5 days was initiated. 10 days after, another methylprednisolone pulse course was given, and the patient improved with lower requirement of oxygen, was extubated and transferred to the Rheumatology department 30 days after the initiation of IVIG.
A new HRCT () showed regression of the interstitial findings. After a second IVIG infusion of 30 g, he was discharged from the hospital 4 months after the admission with continuous oxygen therapy and 25 mg daily oral prednisolone.
Figure 3. Regression of the interstitial findings after initiation of IVIG
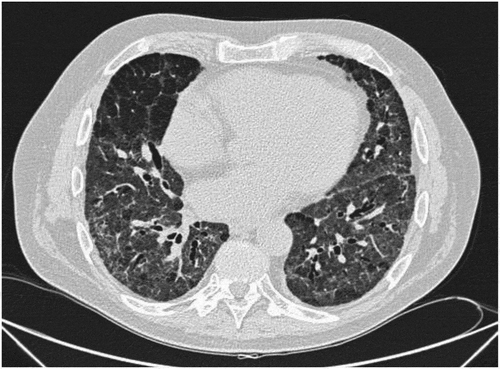
The patient was regularly seen in the outpatient clinic and the reported respiratory symptoms improved.
Initial 6MWT distance (6MWTD) on 12 L of continous oxygen therapy was 315 m with desaturation from 100 to 82%. Pulmonary function test (PFT) showed restriction and a severely reduced diffusion capacity (). The following months, PFT and 6MWTD improved. Due to muscle and joint pain and increasing CK, azathioprin 150 mg daily was initiated. During the next 4 years he was kept on maintenance therapy with IVIG of 30 g as a single dose every third month in combination with azathioprin and low dose prednisolone..
Table 2. A: x indicates administration of 30 g IVIG, b: daily dosis of prednisolone in mg after the patient were discharged, c: Forced Expiratory Volume in 1 s in liters. d: % of predicted, e: Forced Vital Capacity, f: Diffusing capacity for carbon monoxide, g: 6 minute walk test distance, h: 315 m with 12-l O2/min of continous oxygen therapy, i: saturation during 6MWT with highest saturation – lowest saturation, j: creatine kinase
The respiratory symptoms and lung function were stable until he developed pulmonary hypertension with right ventricular failure, peripheral edema and worsening of the respiratory symptoms 4 years after the diagnosis. Few months later he died from a combination of cardiac and respiratory failure.
Discussion
IVIG is produced from pooled plasma from a large number of healthy blood donors. The primary component is IgG [Citation15,Citation17]. IVIG is an effective treatment of several autoimmune and inflammatory disorders [Citation16–19] and has previously been used for treatment of PM and DM successfully [Citation20–22]
There are no prospective randomized controlled trials on the effect of IVIG in inflammatory myopathy associated ILD and the level of evidence is based on case series. lists case reports on inflammatory myopathy associated ILD treated with IVIG.
Table 3. Case reports on inflammatory myopathy-ILD patients treated with IVIG
In a retrospective study of 17 patients with ILD associated to antisynthetase syndrome lung function was stabilized after initiation of IVIG and in some cases mean FVC and DLCO increased [Citation23]. Suzuki et al reported five patients with PM and amyopathic dermatomyositis (ADM) associated ILD refractory to high doses GCS and cyclosporin A and/or cyclophosphomide [Citation24]. Two of the patients of whom one had PM responded to IVIG and survived the acute phase and stayed alive 3 years after the treatment initiation.
Miyazaki et al [Citation25] reported a patient with ADM-ILD who recovered from respiratory failure after treatment with cyclosporine followed by two cycles of pulse cyclophosphamide and IVIG (400 mg/kg for 5 days). Similarly, as shown in the table, other cases show improvement after IVIG treatment.
In most case IVIG was initiated as salvage treatment in combination with or after other immunosuppressive treatments had failed [Citation23–26]. IVIG has only been used as first-line therapy in ILD associated to DM/PM in a single case [28].
IVIG was administered with different doses ranging from 0.2 to 0.5 mg/kg/day and with different time intervals. Moreover, the ILD pattern on HRCT in the cases was different, as well as the patients had different subtypes of inflammatory myopathies with different serology. Thus, the existing evidence makes it difficult to choose the right patient for IVIG and the right treatment combination.
In our case, IVIG significantly improved the ILD based on the symptoms, lower oxygen demand, increasing lung function and regression of HRCT findings. Other immunosupressants could potentially also have been used.
ILD associated with autoimmune inflammatory myopathies are rare and robust data guiding the treatment is lacking. However, IVIG seems to be a valid treatment option.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Notes on contributors
Soran Peshbahar
Soran Peshbahar, MD, is a Specialist Registrar at Diagnostic Centre, Silkeborg Regional Hospital and The Department of Respiratory Diseases and Allergy, Aarhus University Hospital, Denmark. He is specializing in pulmonary Diseases and focuses on interstitial lung diseases and allergy.
Elisabeth Bendstrup
Elisabeth Bendstrup, MD, PhD, is a consultant at The Department of Respiratory Diseases and Allergy, Aarhus University Hospital, Denmark. She is a specialist in pulmonary diseases, and her main research focus is interstitial lung diseases.
References
- Schmidt J. Current classification and management of inflammatory myopathies. J Neuromuscul Dis. 2018;5(2):109–6.
- Furst DE, Amato AA, Iorga ŞR, et al. Epidemiology of adult idiopathic inflammatory myopathies in a U.S. managed care plan. Muscle Nerve. 2012;45(5):676–683.
- Schnabel A, Reuter M, Biederer J, et al. Interstitial lung disease in polymyositis and dermatomyositis: clinical course and response to treatment. Semin Arthritis Rheum. 2003;32(5):273–284.
- Fathi M, Lundberg IE. Interstitial lung disease in polymyositis and dermatomyositis. Curr Opin Rheumatol. 2005;17(6):701–706.
- Kashiwabara K, Ota K. Rapidly progressive interstitial lung disease in a dermatomyositis patient with high levels of creatine phosphokinase, severe muscle symptoms and positive anti-Jo-1 antibody. Intern Med. 2002;41(7):584–588.
- Oddis CV. Update on the pharmacological treatment of adult myositis. J Intern Med. 2016;280(1):63–74.
- Mimori T, Nakashima R, Hosono Y. Interstitial lung disease in myositis: clinical subsets, biomarkers, and treatment. Curr Rheumatol Rep. 2012;14(3):264–274.
- Morisset J, Johnson C, Rich E, et al. Management of myositis-related interstitial lung disease. Chest. 2016;150(5):1118–1128.
- Wallace B, Vummidi D, Khanna D. Management of connective tissue diseases associated interstitial lung disease: a review of the published literature. Curr Opin Rheumatol. 2016;28(3):236–245.
- Gordon PA, Winer JB, Hoogendijk JE, et al. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev. 2012;8:CD003643.
- Marie I, Dominique S, Rémy-jardin M, et al. Interstitial lung diseases in polymyositis and dermatomyositis. Rev Med Interne. 2001;22(11):1083–1096.
- Tubéry M, Lauque D, Murris M, et al. Course of interstitial lung diseases associated with inflammatory myopathies. Apropos of 5 cases. Ann Med Internet (Paris). 1997;148(1):2–10.
- Tomsic M, Sifrer F. Acute respiratory distress syndrome in a polymyositis patient with the anti-Jo-1 antibody. Wien Klin Wochenschr. 2000;112(15–16):728–731.
- Quick A, Tandan R. Mechanisms of action of intravenous immunoglobulin in inflammatory muscle disease. Curr Rheumatol Rep. 2011;13(3):192–198.
- Dalakas MC. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA. 2004;291(19):2367–2375.
- Kamali S, Cefle A, Sayarlioglu M, et al. Experience with monthly, high-dose, intravenous immunoglobulin therapy in patients with different connective tissue diseases. Rheumatol Int. 2005;25(3):211–214.
- Dalakas MC. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA. 2004;291(19):2367–2375.
- Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329(27):1993–2000.
- Miyasaka N, Hara M, Koike T, et al. Effects of intravenous immunoglobulin therapy in Japanese patients with polymyositis and dermatomyositis resistant to corticosteroids: a randomized double-blind placebo-controlled trial. Mod Rheumatol. 2012;22(3):382–393.
- Saito E, Koike T, Hashimoto H, et al. Efficacy of high-dose intravenous immunoglobulin therapy in Japanese patients with steroid-resistant polymyositis and dermatomyositis. Mod Rheumatol. 2008;18(1):34–44.
- Huapaya JA, Hallowell R, Silhan L, et al. Long-term treatment with human immunoglobulin for antisynthetase syndrome-associated interstitial lung disease. Respir Med. 2019;154:6–11.
- Suzuki Y, Hayakawa H, Miwa S, et al. Intravenous immunoglobulin therapy for refractory interstitial lung disease associated with polymyositis/dermatomyositis. Lung. 2009;187(3):201–206.
- Miyazaki E, Ando M, Muramatsu T, et al. Early assessment of rapidly progressive interstitial pneumonia associated with amyopathic dermatomyositis. Clin Rheumatol. 2007;26(3):436–439.
- Murota H, Muroi E, Yamaoka T, et al. Successful treatment with regimen of intravenous gamma globulin and cyclophosphamide for dermatomyositis accompanied by interstitial pneumonia, opportunistic infection and steroid psychosis. Allergol Int. 2006;55(2):199–202.
- Diot E, Carmier D, Marquette D, et al. IV immunoglobulin might be considered as a first-line treatment of severe interstitial lung disease associated with polymyositis. Chest. 2011;140(2):562–563.
- Bakewell CJ, Raghu G. Polymyositis associated with severe interstitial lung disease: remission after three doses of IV immunoglobulin. Chest. 2011;139(2):441–443.