
ABSTRACT
Background
Withdrawals of drug indications may reveal potential inadequacies in the regulatory approval processes of new drugs. Understanding potential weaknesses of the regulatory approval process is paramount given the increasing use of expedited pathways. In this paper, we focus on three poly-ADP-ribose polymerase inhibitors (olaparib, rucaparib and niraparib) for the treatment of women with heavily pretreated, recurrent ovarian cancer, which were eventually withdrawn.
Methods
We use a comparative case study approach to evaluate the regulatory histories of these drug indications in the US and Europe.
Results
Two drug indications benefited from the FDA’s accelerated approval pathway, which explicitly lowers the bar for evidence of efficacy at the time of approval. Following accelerated approval, manufacturers are mandated to conduct post-marketing studies to confirm clinical benefit. The FDA granted accelerated approval to olaparib and rucaparib based on data on surrogate endpoints and converted the approval to regular approval after the submission of additional data on surrogate endpoints from one of two required confirmatory trials, that is, without data on clinical benefit. Niraparib directly received regular approval based only on data on a surrogate endpoint. By contrast, the EMA granted conditional marketing authorisation to rucaparib and was quicker to restrict usage than the FDA.
Conclusion
The regulatory histories of these drug indications highlight the need to reform the accelerated approval pathway by ensuring that post-marketing requirements are followed, and that regular approval is only based on evidence of clinical benefit.
Introduction
Regulation of pharmaceutical products aims to balance timely patient access to novel therapies with the need to generate robust evidence of clinical efficacy and safety. Over the past three decades, in response to pressures to expedite the development and approval of drugs, regulatory authorities have implemented pathways that lower the regulatory bar for evidence of efficacy at the time of approval in service of earlier market access (Davis & Abraham, Citation2011; FDA, Citation2022; Michaeli et al., Citation2023). As a direct consequence, the likelihood of approval of ineffective or even harmful medications has increased, leading to a higher risk of withdrawals of drugs from the market.
Focusing on a set of recent indication withdrawals, we consider the evolution of available information on the benefits and harms of these drug-indications and how the FDA’s varied approval processes were involved at different steps. Key to understanding these regulatory trajectories are the expedited pathways available. The US Food and Drug Administration’s (FDA) accelerated approval programme and the European Medicines Agency’s (EMA) conditional marketing authorisation are examples of such expedited pathways. Accelerated approval and conditional marketing authorisation allow regulators to approve drugs on the basis of surrogate endpoints that are ‘reasonably likely to predict clinical benefit’ (FDA, Citation2018a; FDA, Citation2023) in the case of the FDA and based ‘on less comprehensive clinical data than normally required’ (EMA, Citation2018) in the case of the EMA. The pathways are intended to bring to market more quickly drugs for which there is significant and unmet medical need. Such approvals are meant to be ‘conditional’; at the time of market entry, pharmaceutical firms are required to document in trials clinical benefit of the approved drugs as the basis for conversion to regular approval.
Expedited pathways such as accelerated approval and conditional marketing authorisation have been criticised for several reasons (Wallach et al., Citation2018). While there is some evidence to suggest that drugs approved using these pathways are more likely to have high therapeutic value (US and Europe),(Hwang et al., Citation2020; Vokinger et al., Citation2022) the evidence is not consistent (Australia) (Lexchin, Citation2022). Cases such as the recent approval of aducanumab by the US FDA have heightened concerns about lack of validity of surrogate measures, especially when drugs have substantial toxicity and high costs (Alexander et al., Citation2021).
In the US, post-marketing requirements of the accelerated approval pathway are often not implemented as the legislation envisioned. Confirmatory trials to verify clinical benefit, when conducted, are often delayed (Office of Inspector General, Citation2022; Shahzad et al., Citation2023). If confirmatory trials also use surrogate endpoints, clinical benefit may never be verified (Gyawali et al., Citation2019). When lack of clinical benefit is confirmed, withdrawal processes are prolonged (Aaron et al., Citation2022; Chang et al., Citation2020) or withdrawal after evidence of lack of benefit may not happen at all (Cliff et al., Citation2023; Gyawali, Rome, et al., Citation2021). Withdrawals themselves may not be indicative of the pathway failing (Woodcock, Citation2018), since higher risk approvals are part of the intention of the pathway. However, delays in the generation of high-quality evidence of clinical benefit and delayed regulatory response to evidence of lack of clinical benefit are concerning.
The EMA counterpart to the accelerated approval pathway, the conditional marketing authorisation, has received similar criticism. For example, researchers have argued that there is a lack of clarity around eligibility of conditions and prolonged periods of time between approval and verification of clinical benefit (Davis et al., Citation2017; Hoekman & Boon, Citation2019). Key questions remain about whether high-quality evidence is ever collected (Banzi et al., Citation2015, Citation2017) and whether benefits of earlier access outweigh the harms associated with approving drugs on the basis of limited evidence (Beaver et al., Citation2018). There is a key difference between the accelerated approval pathway and the EMA’s conditional marketing authorisation programme. In the AA pathway, the FDA may revoke approval on the basis of negative information or in case evidence is not generated in a timely manner. The EMA’s conditional marketing authorisation must be renewed annually until all obligations are met (McPhail et al., Citation2023).
In this analysis, we use a case study approach focused on a set of recent drug indication withdrawals to glean insights into whether there are any modifiable features of the FDA’s approval processes that can be changed to reduce the likelihood of ineffective or harmful drugs being approved. We describe the regulatory histories of 3 poly-ADP-ribose polymerase (PARP) inhibitors (olaparib, rucaparib and niraparib) for treatment of women with heavily pretreated, recurrent ovarian cancer. The drug indications were eventually withdrawn, after trials showed potentially detrimental effects on overall survival (OS) (Matulonis, Citation2022; Worcester, Citation2022). These regulatory trajectories highlight how decisions by the FDA exacerbated risks inherent in the accelerated approval pathway. For these drug indications, EMA decisions differed from those of the FDA.
Materials and methods
Recent media coverage of the withdrawals of three ovarian cancer drugs and associated concerns of clinicians prompted the research project. We aimed to evaluate the approval trajectories of drug indications to understand whether these indications could have been withdrawn earlier or never approved given substantial harmful effects associated with their use.
We used publicly available FDA databases to collect available information at three key FDA decision time points: time of initial approval, time of conversion to regular approval (applicable for drugs approved using the accelerated approval pathway) and time of withdrawal. At each of these time points, we collected information about trial characteristics especially endpoints used, and number and type of patients enrolled. This information was then checked against EMA approvals to both evaluate whether the indication was ever submitted for EMA approval or approved and then withdrawn, and whether information from the same trials considered for FDA approval were used in the EMA’s approval process.
We used the Drugs@FDA database (Drugs@FDA, Citationn.d.) to identify clinical studies that supported FDA drug approval decisions over time. Information about clinical studies is available in review documents published by the FDA when new indications are approved and may contain names of clinical studies, National Clinical Trial (NCT) identifiers used to register a study, and other descriptions of the evidence submitted. Since information available from trials at different points in time may not directly correspond to publications of those trials, information provided in FDA review documents is key for establishing the evidence base available to the regulators at the time when each approval decision was made. We linked each study in FDA review documents to its record in ClinicalTrials.gov (Home—ClinicalTrials.Gov, Citationn.d.) an online database of clinical research studies. We extracted from ClinicalTrials.gov additional information about the design of the pre-approval and post-marketing studies for each of the 3 drug indications. We also use the Drugs@FDA database to extract approval letters published at the time when each drug indication was approved. These letters contain post-marketing requirements and commitments (FDA, Citation2018c) following initial approval which we tracked to assess whether the requirements were fulfilled as detailed.
We separately evaluated the European public assessment reports (EPARs) available through the EMA (Citationn.d.). EPARs provide information on how the EMA assessed a medicine, key evidence supporting EMA decisions such as the clinical trials and the evidence available at the time of approval, and the rationale of the relevant EMA committee for approving or rejecting an application. For each drug, we collected from EPARs information on all indication applications and subsequent approvals or rejections. This allowed us to differentiate indications for which a manufacturer may not have submitted an application for approval from indications with an application that was rejected, to appropriately compare regulatory agency decisions. Of note, we could not study rejected applications by the FDA since those are not publicly reported.
We collated the information from both regulatory agencies and constructed a timeline of regulatory milestones and the evidence supporting each decision of the FDA and EMA.
For all data sources, MS collected information and created a supporting document that provided links and direct text from FDA and EMA documents. To avoid bias, the collated document was reviewed by the team and any questions and differences in interpretation resolved through discussion with the team.
Results
Below we present the approval trajectories () for each drug over time by regulatory agency.
Olaparib
The manufacturer of olaparib initially submitted an application to the FDA as ‘maintenance treatment’ based on a single randomised placebo controlled study (FDA, Citation2014b). The maintenance indication was discussed at a meeting of the Oncology Drugs Advisory Committee (ODAC) in June 2014 where a delay in approval was recommended by an 11-2 vote. After this meeting, the applicant submitted datasets and results for a ‘non-maintenance setting in heavily-pretreated patients with BRCA-positive ovarian cancer’ (FDA, Citation2014b). Olaparib received accelerated approval for this ovarian cancer treatment indication in December 2014 based on a single-arm trial measuring response, a surrogate endpoint (). At the time of the accelerated approval of olaparib, the FDA required ‘progression-free survival and overall survival analyses with datasets from clinical trial D0818C00002, SOLO-2’ and ‘progression-free survival (PFS) and overall survival (OS) analyses with datasets from clinical trial D0816C00010 [SOLO-3]’ (FDA, Citation2014a).
Table 1. Clinical trials supporting regulatory decisions at the FDA and EMA for ovarian cancer indications of olaparib, rucaparib and niraparib.
However, FDA ignored its own requirements when the accelerated approval of olaparib’s treatment indication was converted to regular approval in 2017 after the submission of interim progression-free survival results from only one of the two required confirmatory trials. The FDA argued in the supplementary approval that ‘interim progression-free survival data from SOLO-2 have verified the clinical benefit of olaparib and therefore olaparib capsules for monotherapy in patients with deleterious or suspected deleterious germline BRCA-mutated advanced ovarian cancer who have been treated with three or more prior lines of chemotherapy is granted regular approval’ (FDA, Citation2017b). Of note, in this trial, the drugs were studied as maintenance treatment, that is after complete or partial response to (standard or platinum-based) chemotherapy in patients who were earlier in their disease course, less exposed to chemotherapy and likely healthier.
Additionally, at this time, the maintenance indication was added to olaparib’s label, and the manufacturer was released from the requirement to complete the second trial although the trial did become a post-marketing commitment.
In August 2022, olaparib’s treatment indication was voluntarily withdrawn after results from its originally-required confirmatory trial (SOLO-3) showed a negative impact on patient survival (median 29.4 months with olaparib and 39.4 months with control after 3 or more lines of prior therapy). This trial was one of the requirements at the time of accelerated approval but was later considered by FDA to not be needed for conversion to regular approval in light of interim progression-free survival results of SOLO-2.
The manufacturer of olaparib did not apply for authorisation of the drug for ovarian cancer treatment in Europe. Therefore, by October 2023, EMA had not approved olaparib for the treatment of women with heavily pre-treated ovarian cancer.
Rucaparib
Rucaparib was approved in December 2016 under the accelerated approval pathway for ‘treatment of patients with deleterious BRCA mutation (germline and/or somatic) associated advanced ovarian cancer who have been treated with two or more chemotherapies’ (FDA, Citation2016b). This approval was based on ‘efficacy data from 106 patients … treated on two open-label, single arm trials [Study 10 and ARIEL2]’ (FDA, Citation2016c). The FDA focused on the ‘assessed objective response rate (ORR) of 54% observed in the 106 patients and the supportive 9.2 month duration of response (DOR)’ (FDA, Citation2016c) which it ‘considered reasonably likely to predict clinical benefit’ (FDA, Citation2016c).
Figure 1. Timeline of FDA regulatory events for olaparib, rucaparib and niraparib for the treatment of women with ovarian cancer.
Notes: ORR = overall response rate; PFS = progression-free survival; FDA = Food and Drug Administration; EMA = European Medicines Agency.
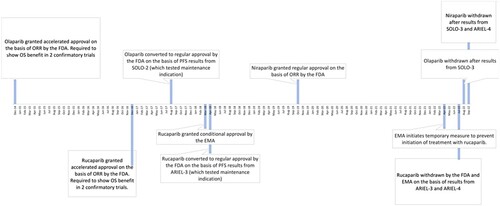
At this time, there were two accelerated approval requirements. The first was to ‘submit the progression-free survival and overall survival analyses with datasets from clinical trial CO-338-014 entitled, ‘Phase 3 Study of Rucaparib as Switch Maintenance After Platinum in Relapsed High Grade Serous and Endometrioid Ovarian Cancer’ (ARIEL3)’ with a final report due by March 2021. The second was to ‘submit the progression-free survival and overall survival analyses with datasets from clinical trial CO-338-043 entitled ‘A Phase 3 Multicenter, Randomised Study of Rucaparib Versus Chemotherapy in Patients With Relapsed, BRCA Mutant, High Grade Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer’ (ARIEL4)’ with a final report due by December 2024 (FDA, Citation2016a).
However, in April 2018 the accelerated approval of rucaparib’s treatment indication was converted to regular approval based on progression-free survival results shown in ARIEL3. ARIEL3 demonstrated a statistically significant improvement in progression-free survival compared to placebo among patients taking rucaparib as maintenance therapy.
At the same time, the applicant was released from the ARIEL4 post marketing requirement and documentation of overall survival benefit was moved to ‘post marketing commitments subject to reporting requirements’ (FDA, Citation2018b).
In July 2022, rucaparib’s treatment indication was voluntarily withdrawn by the manufacturer in the US after results from its originally-required confirmatory trial (ARIEL4) showed potentially harmful impact on overall survival (median 19.4 months with rucaparib and 25.4 months with control after 2 or more lines of prior therapy) (Sternberg, Citation2022).
Rucaparib was approved for ovarian cancer treatment by both FDA and EMA. For rucaparib, the same trials underlying FDA approvals formed the basis for EMA’s conditional marketing authorisation in Citation2018, which was not converted to regular approval. In April 2022, the EMA temporarily restricted new patients from using rucaparib for ovarian cancer treatment after submission of results from ARIEL-4. EMA withdrew rucaparib’s ovarian cancer treatment indication in July 2022.
Niraparib
FDA approved niraparib in March 2017 for ‘the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy’ (FDA, Citation2017a). In October 2019, FDA approved the treatment indication of niraparib. This indication was approved on the basis of a study of ‘98 patients with advanced ovarian cancer with homologous recombination deficiency (HRD) positive tumours in the single-arm QUADRA trial’ (FDA, Citation2019). The main efficacy outcome measures were objective response rate and duration of response. Since the treatment (and the maintenance) indication was approved using the regular approval pathway, no requirements existed to demonstrate clinical benefit post-marketing.
In September 2022, the treatment indication for niraparib was also voluntarily withdrawn.
This decision was made in ‘consultation with the U.S. Food and Drug Administration (FDA) and based on a totality of information from PARP inhibitors in the late line treatment setting in ovarian cancer’ (GSK, Citation2022).
EMA approved niraparib in 2017 for maintenance therapy of ovarian cancer patients. The manufacturer never applied for approval for the treatment indication with the EMA.
Discussion
The regulatory histories of recently withdrawn ovarian cancer treatment indications of 3 PARP inhibitors raise concerns about the FDA’s role in problematic drug approvals. It took more than 7, 5 and 2 years for ovarian cancer treatment indications of olaparib, rucaparib and niraparib to be withdrawn following initial approvals, respectively. During this time, clinicians (Dhruva et al., Citation2024) and patients may have expected survival benefits from potentially detrimental treatments. Prior research has primarily highlighted delays in post-approval evidence that may be driven by manufacturers but has not examined FDA actions that might inadvertently undermine agreements to complete post-marketing requirements.
Post marketing requirements are an essential social contract to mitigate two issues identified with oncology drug approvals. The first issue is the lack of evidence on the predictive validity of surrogate endpoints and the FDA’s overoptimism with regard to their correlation with overall survival (Gyawali et al., Citation2020; Gyawali, D’Andrea, et al., Citation2021; Walia et al., Citation2022). Secondly, short-term trials such as those used for accelerated approval do not generate sufficient safety data (Richardson et al., Citation2022). For the accelerated approval pathway to effectively balance early access to new drugs with uncertain clinical benefits and harms, FDA needs to adhere to the principles of the pathway. The pathway’s regulated post-marketing requirements should be rigorously followed (Office of Inspector General, Citation2022). Additionally, there is no publicly accessible documentation about why the FDA deviated from its own requirements in these cases. With respect to the 3 PARP inhibitors, deviation from required standards proved to be detrimental. Even if the studies had eventually confirmed clinical benefit, such deviations create inconsistencies in the application of FDA approval requirements leading to confusion about evidentiary standards (Janiaud et al., Citation2021).
There are other lessons to be learnt as well. The niraparib approval highlights potential harms of approving drugs based on surrogate endpoints through the regular approval pathway where there is no requirement to generate additional clinical evidence. Prior research has documented that this is not an isolated example and surrogate endpoints are used often for traditional approval of oncology products (Chen et al., Citation2019). The FDA should reserve the regular approval pathway for approvals based on rigorously documented clinical benefits not surrogate endpoints. Notably though, the FDA has begun to place more emphasis on overall survival data for oncology drugs (Brennan, Citation2023).
While the companies eventually voluntarily withdrew ovarian cancer treatment indications in the US, the FDA’s processes to initiate withdrawal of indications approved using the accelerated approval pathway have been cumbersome to implement resulting in a much slower response in cases where manufacturers have disagreed with the FDA’s assessment (Aaron et al., Citation2022; Gyawali, Rome, et al., Citation2021). In the cases studied, the manufacturers agreed to withdraw the indications in question. However, FDA’s actions were still delayed in comparison to the EMA’s. This study underscores the need to also reform indication withdrawal processes to enhance the FDA’s ability to protect patients in the face of post-marketing information documenting lack of clinical benefit or clinical harm.
Earlier research has highlighted differences in the decisions of the FDA and the EMA when limited evidence of efficacy exists although there is no clear indication that one agency has more rigorous standards than the other (Cramer et al., Citation2023; Salcher-Konrad et al., Citation2020; Shah et al., Citation2013). The EMA’s decisions on rucaparib suggest that the agency restricted use faster and never converted the conditional approval to unconditional approval.
This case study also raises an important question for low- and middle-income countries whose regulatory agencies may be facing similar pressures to approve drugs with limited evidence of clinical benefit which are highly priced and take limited resources away from other uses. We caution against other regulatory agencies’ benchmarking their approvals on FDA or EMA approvals of drugs with uncertain clinical efficacy (Ivama-Brummell et al., Citation2023).
This analysis has limitations. Firstly, a case study approach precludes analysis of the prevalence of the issues we identify. Secondly, we rely on publicly available documents from the FDA or EMA websites to ascertain the level of evidence available at each step of the approval process. We may have missed some information that was relevant for regulatory decision making.
Conclusion
This in-depth analysis of the regulatory trajectories of three PARP inhibitors revealed several instances where different decisions by the FDA could have increased consistency in drug approval requirements for manufacturers and in evidence of benefit for providers, and possibly reduced harm to patients. For two of the drugs in the study, rucaparib and olaparib, conversion from accelerated approval to regular approval without verification of overall survival benefit in the indication under consideration signalled a greater level of confidence in the efficacy of the drugs than was warranted by the data. Similarly, directly granting regular approval of niraparib based on a single arm trial with only data on a surrogate endpoint did not include incentives to perform further confirmatory trials. This analysis highlights how discretionary regulatory decisions can exacerbate the risks inherent in expedited approval pathways. A focus on verifying clinical benefit, consistently, should guide regulators as they consider the benefit-risk balance of new therapeutics. Specifically, policies that ensure that regular approval is only granted when clinical benefit is verified, in the population in question, can help mitigate the risks of the accelerated approval pathway.
Ethics approval statement
This study was considered exempt from Harvard Pilgrim Health Care Institutional Review Board since it uses publicly available data.
Acknowledgments
The authors appreciate Dr. Dennis Ross-Degnan’s feedback on this work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Aaron, D. G., Cohen, I. G., & Adashi, E. Y. (2022). The FDA struggle to withdraw Makena: Problems with the accelerated approval process. JAMA, 328(24), 2394–2395. https://doi.org/10.1001/jama.2022.22986
- Alexander, G. C., Knopman, D. S., Emerson, S. S., Ovbiagele, B., Kryscio, R. J., Perlmutter, J. S., & Kesselheim, A. S. (2021). Revisiting FDA approval of aducanumab. New England Journal of Medicine, 385(9), 769–771. https://doi.org/10.1056/NEJMp2110468
- Banzi, R., Gerardi, C., Bertele’, V., & Garattini, S. (2015). Approvals of drugs with uncertain benefit-risk profiles in Europe. European Journal of Internal Medicine, 26(8), 572–584. https://doi.org/10.1016/j.ejim.2015.08.008
- Banzi, R., Gerardi, C., Bertele’, V., & Garattini, S. (2017). Conditional approval of medicines by the EMA. BMJ: British Medical Journal (Online), 357, Article j2062. https://doi.org/10.1136/bmj.j2062
- Beaver, J. A., Howie, L. J., Pelosof, L., Kim, T., Liu, J., Goldberg, K. B., Sridhara, R., Blumenthal, G. M., Farrell, A. T., Keegan, P., Pazdur, R., & Kluetz, P. G. (2018). A 25-year experience of US Food and Drug Administration accelerated approval of malignant hematology and oncology drugs and biologics: A review. JAMA Oncology, 4(6), 849–856. https://doi.org/10.1001/jamaoncol.2017.5618
- Brennan, Z. (2023, October 31). FDA pushes cancer drug developers to include overall survival data more frequently. Endpoints News. https://endpts.com/fda-pushes-cancer-drug-developers-to-include-overall-survival-data-more-frequently/.
- Chang, C. Y., Nguyen, C. P., Wesley, B., Guo, J., Johnson, L. L., & Joffe, H. V. (2020). Withdrawing approval of Makena—A proposal from the FDA center for drug evaluation and research. New England Journal of Medicine, 383(24), Article e131. https://doi.org/10.1056/NEJMp2031055
- Chen, E. Y., Raghunathan, V., & Prasad, V. (2019). An overview of cancer drugs approved by the US Food and Drug Administration based on the surrogate end point of response rate. JAMA Internal Medicine, 179(7), 915–921. https://doi.org/10.1001/jamainternmed.2019.0583
- Cliff, E. R. S., Rome, R. S., Kesselheim, A. S., & Rome, B. N. (2023). National comprehensive cancer network guideline recommendations of cancer drugs with accelerated approval. JAMA Network Open, 6(11), Article e2343285. https://doi.org/10.1001/jamanetworkopen.2023.43285
- Cramer, A., Sorup, F., Christensen, H., Peterson, T., & Karstoft, K. (2023). Withdrawn accelerated approvals for cancer indications in the USA: What is the marketing authorisation status in the EU? The Lancet Oncology, 24(9), e385–e394. https://doi.org/10.1016/S1470-2045(23)00357-1
- Davis, C., & Abraham, J. (2011). Desperately seeking cancer drugs: Explaining the emergence and outcomes of accelerated pharmaceutical regulation. Sociology of Health & Illness, 33(5), 731–747. https://doi.org/10.1111/j.1467-9566.2010.01310.x
- Davis, C., Naci, H., Gurpinar, E., Poplavska, E., Pinto, A., & Aggarwal, A. (2017). Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: Retrospective cohort study of drug approvals 2009-13. BMJ, 359, Article j4530. https://doi.org/10.1136/bmj.j4530
- Dhruva, S. S., Kesselheim, A. S., Woloshin, S., Ji, R. Z., Lu, Z., Darrow, J. J., & Redberg, R. F. (2024). Physicians’ perspectives on FDA regulation of drugs and medical devices: A national survey. Health Affairs, 43(1), 27–35. https://doi.org/10.1377/hlthaff.2023.00466
- Drugs@FDA: FDA-Approved Drugs. (n.d.). Retrieved September 12, 2022, from https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm.
- EMA. (2018, September 17). Conditional marketing authorisation [Text]. European Medicines Agency. https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/conditional-marketing-authorisation
- EMA. (n.d.). Medicines [Text]. European Medicines Agency. Retrieved November 18, 2023, from https://www.ema.europa.eu/en/medicines
- FDA. (2014a). Approval letter for Lynparza. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2014/206162Orig1s000ltr.pdf
- FDA. (2014b, December 16). Summary review for Lynparza. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/206162Orig1s000SumR.pdf
- FDA. (2016a). Approval letter for Rubraca. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2016/209115Orig1s000ltr.pdf
- FDA. (2016b, December). Label for Rubraca 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209115s000lbl.pdf
- FDA. (2016c, December 15). Summary review for Rubraca. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/209115Orig1s000MultiDisciplineR.pdf
- FDA. (2017a). Original Approved Label for Zejula. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208447lbl.pdf
- FDA. (2017b). Summary review for Lynparza 2017. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208558Orig1s000MultidisciplineR.pdf
- FDA. (2018a). Accelerated approval. FDA. https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/accelerated-approval
- FDA. (2018b, April 6). Approval letter for Rubraca maintenance indication. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/209115Orig1s003ltr.pdf
- FDA. (2018c, November 3). Postmarketing requirements and commitments: Introduction. FDA. https://www.fda.gov/drugs/guidance-compliance-regulatory-information/postmarket-requirements-and-commitments
- FDA. (2019, October 23). Label for Zejula. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/208447s014lbl.pdf
- FDA. (2022, June 7). Expedited programs for serious conditions––Drugs and BIOLOGICS. U.S. Food and Drug Administration; FDA. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/expedited-programs-serious-conditions-drugs-and-biologics
- FDA. (2023). Accelerated approval. FDA. https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/accelerated-approval
- GSK. (2022, September 14). Dear Health Care provider letter for Zejula. https://medinfo.gsk.com/5f95dbd7-245e-4e65-9f36-1a99e28e5bba/57e2a3fa-7b9b-432f-a220-5976a509b534/57e2a3fa-7b9b-432f-a220-5976a509b534_viewable_rendition__v.pdf
- Gyawali, B., D’Andrea, E., Franklin, J. M., & Kesselheim, A. S. (2021). A correlation analysis to assess event-free survival as a trial-level surrogate for overall survival in early breast cancer. EClinicalMedicine, 32, Article 100730. https://doi.org/10.1016/j.eclinm.2021.100730
- Gyawali, B., Hey, S. P., & Kesselheim, A. S. (2019). Assessment of the clinical benefit of cancer drugs receiving accelerated approval. JAMA Internal Medicine, 179(7), 906–913. https://doi.org/10.1001/jamainternmed.2019.0462
- Gyawali, B., Hey, S. P., & Kesselheim, A. S. (2020). Evaluating the evidence behind the surrogate measures included in the FDA’s table of surrogate endpoints as supporting approval of cancer drugs. EClinicalMedicine, 21, Article 100332. https://doi.org/10.1016/j.eclinm.2020.100332
- Gyawali, B., Rome, B. N., & Kesselheim, A. S. (2021). Regulatory and clinical consequences of negative confirmatory trials of accelerated approval cancer drugs: Retrospective observational study. BMJ, 374, Article n1959. https://doi.org/10.1136/bmj.n1959
- Hoekman, J., & Boon, W. (2019). Changing standards for drug approval: A longitudinal analysis of conditional marketing authorisation in the European union. Social Science & Medicine, 222, 76–83. https://doi.org/10.1016/j.socscimed.2018.12.025
- Home—ClinicalTrials.gov. (n.d.). Retrieved October 11, 2022, from https://clinicaltrials.gov/ct2/home
- Hwang, T. J., Ross, J. S., Vokinger, K. N., & Kesselheim, A. S. (2020). Association between FDA and EMA expedited approval programs and therapeutic value of new medicines: Retrospective cohort study. BMJ, 371, Article m3434. https://doi.org/10.1136/bmj.m3434
- Ivama-Brummell, A. M., Marciniuk, F. L., Wagner, A. K., Osorio-de-Castro, C. G. S., Vogler, S., Mossialos, E., Tavares-de-Andrade, C. L., & Naci, H. (2023). Marketing authorisation and pricing of FDA-approved cancer drugs in Brazil: A retrospective analysis. The Lancet Regional Health – Americas, 22, Article 100506. https://doi.org/10.1016/j.lana.2023.100506
- Janiaud, P., Irony, T., Russek-Cohen, E., & Goodman, S. N. (2021). U.S. Food and Drug Administration reasoning in approval decisions when efficacy evidence is borderline, 2013–2018. Annals of Internal Medicine, 174(11), 1603–1611. https://doi.org/10.7326/M21-2918
- Lexchin, J. (2022). Use of priority and provisional approval pathways by the Australian therapeutic goods administration in approving new medicines: A cross-sectional study. Australian Health Review: A Publication of the Australian Hospital Association, 46(3), 309–315. https://doi.org/10.1071/AH22008
- Matulonis, U. A. (2022). The rapid evolution of PARP inhibitor therapy for advanced ovarian cancer: Lessons being learned and new questions emerging from phase 3 trial long-term outcome data. Gynecologic Oncology, 167(3), 401–403. https://doi.org/10.1016/j.ygyno.2022.11.018
- McPhail, M., Zhang, H., Bhimani, Z., & Bubela, T. (2023). Lessons from Canada’s notice of compliance with conditions policy for the life-cycle regulation of drugs. Journal of Law and the Biosciences, 10(1), Article lsad008. https://doi.org/10.1093/jlb/lsad008
- Michaeli, D. T., Michaeli, T., Albers, S., Boch, T., & Michaeli, J. C. (2023). Special FDA designations for drug development: Orphan, fast track, accelerated approval, priority review, and breakthrough therapy. The European Journal of Health Economics, https://doi.org/10.1007/s10198-023-01639-x
- Office of Inspector General. (2022, September). Delays in confirmatory trials for drug applications granted FDA’s accelerated approval raise concerns. https://oig.hhs.gov/oei/reports/OEI-01-21-00401.pdf
- Pujade-Lauraine, E., Ledermann, J. A., Selle, F., Gebski, V., Penson, R. T., Oza, A. M., Korach, J., Huzarski, T., Poveda, A., Pignata, S., Friedlander, M., Colombo, N., Harter, P., Fujiwara, K., Ray-Coquard, I., Banerjee, S., Liu, J., Lowe, E. S., Bloomfield, R., … Vergote, I. (2017). Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. The Lancet Oncology, 18(9), 1274–1284. https://doi.org/10.1016/S1470-2045(17)30469-2
- Richardson, N. C., Kasamon, Y., Pazdur, R., & Gormley, N. (2022). The saga of PI3 K inhibitors in haematological malignancies: Survival is the ultimate safety endpoint. The Lancet Oncology, 23(5), 563–566. https://doi.org/10.1016/S1470-2045(22)00200-5
- Rubraca European Public Assessment Report. (2018, March 22). https://www.ema.europa.eu/en/documents/assessment-report/rubraca-epar-public-assessment-report_en.pdf
- Salcher-Konrad, M., Naci, H., & Davis, C. (2020). Approval of cancer drugs with uncertain therapeutic value: A comparison of regulatory decisions in Europe and the United States. The Milbank Quarterly, 98(4), 1219–1256. https://doi.org/10.1111/1468-0009.12476
- Shah, R. R., Roberts, S. A., & Shah, D. R. (2013). A fresh perspective on comparing the FDA and the CHMP/EMA: Approval of antineoplastic tyrosine kinase inhibitors. British Journal of Clinical Pharmacology, 76(3), 396–411. https://doi.org/10.1111/bcp.12085
- Shahzad, M., Naci, H., & Wagner, A. K. (2023). Association between preapproval confirmatory trial initiation and conversion to traditional approval or withdrawal in the FDA accelerated approval pathway. JAMA, 329(9), 760–761. https://doi.org/10.1001/jama.2023.0625
- Sternberg, A. (2022, September 9). ARIEL4 update shows similar OS, better PFS with Rucaparib Vs Chemotherapy in platinum-sensitive BRCA1/2+ relapsed ovarian cancer. Cancer Network. https://www.cancernetwork.com/view/ariel4-update-shows-similar-os-better-pfs-with-rucaparib-vs-chemotherapy-in-platinum-sensitive-brca1-2-relapsed-ovarian-cancer.
- Vokinger, K. N., Kesselheim, A. S., Glaus, C. E. G., & Hwang, T. J. (2022). Therapeutic value of drugs granted accelerated approval or conditional marketing authorization in the US and Europe from 2007 to 2021. JAMA Health Forum, 3(8), Article e222685. https://doi.org/10.1001/jamahealthforum.2022.2685
- Walia, A., Haslam, A., & Prasad, V. (2022). FDA validation of surrogate endpoints in oncology: 2005–2022. Journal of Cancer Policy, 34, Article 100364. https://doi.org/10.1016/j.jcpo.2022.100364
- Wallach, J. D., Ross, J. S., & Naci, H. (2018). The US Food and Drug Administration’s expedited approval programs: Evidentiary standards, regulatory trade-offs, and potential improvements. Clinical Trials (London, England), 15(3), 219–229. https://doi.org/10.1177/1740774518770648
- Woodcock, J. (2018). Expediting drug development for serious illness: Trade-offs between patient access and certainty. Clinical Trials, 15(3), 230–234. https://doi.org/10.1177/1740774518770656
- Worcester, S. (2022, September 23). Drug Companies Withdraw PARPi Indications for Ovarian Cancer. Medscape. https://www.medscape.com/viewarticle/981369.