
Abstract
Background: Poor nutritional status has been shown to be associated with a significant decline in lung function in patients with cystic fibrosis. There are few data published on the lung function decline and the effects of nutritional status in cystic fibrosis (CF) in South Africa.
Aim: To assess anthropometric parameters (weight, height, body mass index Z-score) in relation to lung function parameters in CF patients.
Methodology: A retrospective chart review of clinical records of participants over the age of five years attending the CF clinic at Steve Biko Academic Hospital from 2005 to 2010.
Results: Twenty files were reviewed for lung function, anthropometric measurements, gender and CF-causing mutations. For anthropometric measurements the average changes were –0.8, –0.5 and 2.0 for weight, BMI and height Z-scores, respectively. A decline in FEV1 of –25.3 (95% CI 39.4; –13.3) over the five-year period was noted, with an average decline of 5.3% per year. For FEF25-75, the average change was –22.4 (95% CI-34.6; –10.2) with a decline of 4.5% per year. Using multivariate analysis, the FEV1 was found to be significantly influenced by: age –3.96 (95% CI –7.4; –0.5); p = 0.03, weight 1.8 (95% CI –3.4; –0.9); p = 0.04, BMI Z-score 4.3 (95% CI 5.3; 23.3); p = 0.02 and gender (p = 0.02). The FEF25-75 was significantly influenced by BMI Z-score and gender.
Conclusion: The average lung function decline per year for FEV1 was higher than that seen in developed countries. The decline in FEV1 was related to gender, age, weight and BMI. The decline in FEF25-75 was affected only by BMI Z-score and gender.
Introduction
Cystic fibrosis (CF) is a common life-altering autosomal recessive inherited disorder due to mutations in the CF gene on chromosome 7 that code for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Over 1 900 mutations have been defined in the CFTR gene to date.Citation1 The abnormal protein leads to disruption of chloride secretion in the epithelial cells, with resultant production of abnormal viscid mucus and consequent obstruction of the pulmonary conducting airways and pancreatic ducts. CF occurs in diverse populations with differing rates. In South Africa the incidence is approximately 1 in every 2 800 in Caucasians, 1 in every 10 000 in mixed race and 1 in every 32 000 in black Africans.Citation2 Worldwide p.F508del accounts for around 70% of all CF-causing mutations.Citation3 In South Africa the p.F508del accounts for 74% of chromosomes in Caucasians with the 3120+1G→A being present in 46% of CF causing alleles in the black African population.Citation4–6
Several studies have shown that CFTR genotype has an influence on the phenotype of CF.Citation6,7 Subjects who are homozygous for p.F508del were found to have more severe clinical manifestations as compared with those who are heterozygous for the mutation.Citation6–8 The main difference in the phenotypic presentation is thought to be related to the influence of CFTR genotype on pancreatic function, with p.F508del homozygous subjects having pancreatic insufficiency at an earlier age and having worse nutritional parameters.Citation7–10 The relationship between CFTR genotype and severity of lung disease has been reported previously, where p.F508del homozygous patients were found to have a higher rate of lung function decline when compared with heterozygous subjects.Citation11
The primary cause of morbidity and mortality in patients with CF is progressive obstructive lung disease associated with infection and intense neutrophilic inflammation.Citation8 At an early age, impairment of airway function may have long-term consequences in the disease where the majority of patients die because of pulmonary involvement.Citation8,11 Pulmonary function tests, particularly measurement of forced expiratory volume in 1 second (FEV1) is a practical and objective way of monitoring the severity and progression of CF lung disease. FEV1 has been found to be strongly associated with mortality, while forced expiratory flow during 25–75% of forced vital capacity (FEF25-75) is a sensitive index of small-airway function and is affected earlier in CF lung disease.Citation11 The pattern of lung function decline has been reported to be predictable in developed countries, with an annual rate of decline in the FEV1 of less than 2% predicted for children born after 1980. The annual rate of FEV1 declined in South African children with CF in a relatively short-term longitudinal study, which suggested an average rate of decline in FEV1 of less than 1% per year.Citation12 These values are similar to those of Xu et al., who found that patients born between 1985 and 1990 had an average rate of decline of 0.8%.Citation13 In a subsequent follow-up study by Morrow et al., between 1999 and 2006, lung function tests were found to have increased by 20% over an eight-year period. Predicted FEV1 was 61% in the first quarter of 1999 and 81% in the last quarter of 2006.Citation14 This likely reflects improved care of South African children with CF.
Malnutrition in cystic fibrosis is due to the relationship between nutrient balance and nutrient requirements. CF patients lose weight and fail to grow normally if the intake is less than their total daily energy expenditure. Multiple factors also have the potential to contribute to reduced energy intake including anorexia, gastroesophageal reflux, maldigestion and pancreatic insufficiency leading to reduced intake. Other factors such as lung inflammation may be associated with increase in resting metabolic rate (RMR), which leads to increased energy expenditure. This was proved in previous studies where RMR in CF patients was found to increase as the lung function declines.Citation15 Other parameters such as abnormal CFTR probably affect lung growth, host defences and the ability to repair lung injury. The resulting chronic lung disease may also affect energy expenditure and caloric needs, which in turn impacts on somatic growth.Citation16
Poor nutritional status has been shown to be associated with a significant decline in lung function; children who weigh more and who gain weight at an appropriate rate have better FEV1 Z-scores. Weight-for-age and height-for-age are also positively associated with FEV1% predicted.Citation16–18 In a study conducted by Steinkamp et al. in Germany, the authors revealed that a fall in weight-for-height of 5% predicted or more within one year was associated with a parallel decrease in FEV1, whereas patients with improved nutrition showed constant or even improved FEV1.Citation19 There are few published data on the effects of nutritional status on lung function decline in CF patients in South Africa.
This study was therefore conducted with the primary aim to assess various anthropometric parameters in relation to the lung function over time. The secondary aim is to assess the impact of CF mutations on lung function and finally to determine the rate of lung function decline over a period of five years.
Methods
A retrospective chart review was performed of clinical records of children over five years of age attending the CF clinic at Steve Biko Academic Hospital in Pretoria between 2005 and 2010. For inclusion in the study patients had to have a confirmed diagnosis of CF with two positive sweat tests and/or genetic testing. Participants had to have regular follow-up at the clinic over the five-year period under study. Participants also had to have a minimum of three lung function tests per year during the study period and a record of at least two anthropometric parameters per year during the study period.
Clinical data recorded included age, gender, CF genotype, anthropometric measurements, lung function parameters and use of pancreatic enzyme supplementation. Lung functions (FEV1, FVC, FEV1/FVC and FEF25-75) were measured using the Viasys SpiroPro Jaeger Spirometer (Hoechberg, Germany). Approval of this study was obtained from the University of Pretoria, Faculty of Health Sciences Human Research Ethics Committee.
Statistical analysis
For the statistical analysis, summary statistics reported included mean, 95% confidence intervals and median values for lung function parameters and anthropometric measurements and these were categorised by gender and CF mutation. A two-sample t-test with unequal variances was used to compare lung function parameters and anthropometric measurements, mutations and gender. A multivariate regression model was used to determine which variables had an effect on lung function parameters (FEV1 and FEF25-75).
Results
At the time of the data collection, 37 files were screened and only 20 patients met the eligibility criteria. The majority of the participants were aged between 10 and 20 years of age, (age range: 5–31 years), with more females (12/8) (Table . Information on genotype was available for 18 and 2 had unknown mutations or were unidentified. All participants were pancreatic insufficient and were on pancreatic enzyme supplementation. The mean Z-scores for all the growth parameters were within the normal range for the participants. The mean predicted FEV1 and FEF25-75 were 69% and 48%, respectively.
Table 1: Demographic data for study population at entry (n = 20)
For anthropometric measurements the mean weight by mutation at entry to exit from the study was higher for p.F508del homozygous compared with p.F508del heterozygous participants (Table . There was statistically significant change for weight over time for the whole study population, with a mean change of –19.4 (–35.4; -3.2); (p = 0.02). The mean weight Z-scores declined by –0.8 over the study period. For the height Z-scores the change was –1.15 (–1.69; –0.61) at study entry to 0.86 [–0.07; 1.80] at study exit and this was statistically significant; p = 0.0005. When comparing the mean BMI by mutation, p.F508del heterozygous participants had lower BMI when compared with homozygous participants at both study entry and study exit. The mean BMI Z-score changed from –0.5 (–1.34; –0.33) at study entry to –1.28 (–2.0; –0.56) at study exit; p = 0.25. The change over time for the whole study population from study entry to study end for the observed BMI Z-scores was –0.8 and this was not statistically significant, p > 0.05.
Table 2: Anthropometry measurements in relation to CF mutation
For the lung function parameters, there was no effect of either gender or CF mutation observed for all the changes in relation to FEV1 on FEF25-75 over time (Table . The mean change for FEV1 over five years was –25.3 (–39.4; –13.3); (p = 0.005) and for FEF25-75 the mean change over the same period was –22.4 (–34.6; –10.2); (p = 0.001). The average lung function decline per year for FEV1 and FEF25-75 was 5.3% and 4.5%, respectively. There was a slightly steeper decline in FEV1 for females when compared with males, although this was not statistically significant (Figure ).
Table 3: Comparison of lung function between mutation and gender
Figure 1: Box plot of relative change of FEV1 (light blue), FEF25/75 (medium blue), BMI Z-score (dark blue) and height according to gender.
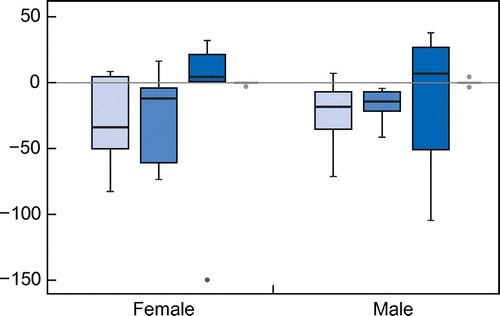
Using multivariate analysis, the FEV1 was found to be significantly influenced by age, weight and BMI Z-score. The correlation coefficients for the parameters were: age –3.96 (–7.4; –0.5); (p = 0.03), weight 1.8 (–3.4; –0.9); (p = 0.04); BMI Z-score 14.3 (5.3; 23.3); (p = 0.02). FEF25-75 was significantly influenced in the multivariate analysis by BMI Z-score (p = 0.03) and gender (p = 0.001). FEF25-75 was not significantly affected by age, weight and height change (all p-values > 0.05). Both FEV1 and FEF25-75 were not found to be significantly affected by mutation, using the same analysis.
Discussion
In this single-centre cystic fibrosis clinic, we found a significant change over time for the anthropometric parameters of these pancreatic-insufficient participants. The weight mean Z-score declined by –0.5 over the five-year study period and this was statistically significant. No statistically significant change was observed in both the mean height and BMI Z-scores over the same period. There was significant decline over time observed for lung function at study entry when compared with study end for both FEV1 and FEF25-75. The average lung function decline per year was 5.3% for FEV1 and 4.5% for FEF25-75. Gender and the type of mutation did not seem to impact on the lung function, although the decline in FEV1 for females was steeper than that in the males but this was not statistically significant. In the multivariate analysis FEF25-75 seemed to be influenced by gender but this is limited by the small numbers in the study population.
In the present study CFTR mutation did not impact on both lung function and anthropometric parameters. This observation supports a result from a previous study conducted at our centre.Citation20 This is contrary to other studies which revealed that a p.F508del homozygous status predicts more severe lung disease when compared with patients who were heterozygous for the p.F508del mutation.Citation11,21 In the studies conducted by Morrow et al. and Corey et al. it was found that the rate of lung function decline was more rapid in p.F508del homozygous patients when compared with those who were heterozygous for the mutation.Citation11,22 The contradictory findings between the previous studies and the current study may be due to factors such as age group and our small population size.
Pancreatic insufficiency carries the risk for maldigestion, malabsorption and low energy intake in relation to increased faecal nutrient losses. In a study conducted by Lucidi et al., weight, height and BMI Z-scores were significantly associated with pancreatic status.Citation23 Lai et al. revealed that prevalence of low weight-for-age and low height-for-age was fairly stable between ages 6 and 12 for boys, whereas for girls there was a sharp increase in the prevalence of growth failure between age 8 and 12 years.Citation24 Zemel et al. also found that growth status was not stable in the mid-childhood in children with CF and progressed with age.Citation18 In this study, the population group had an average age of 16 years (with more females) and all the participants were pancreatic insufficient. We found greater improvement and preservation of stature in comparison with an increasing reduction in mass, with weight Z-scores declining over time. In a multi-centre study in Toronto, patients were prescribed a high-fat and high-calorie diet together with a higher dose of pancreatic enzymes and this was found to be associated with improved stature.Citation25
In the current study, FEV1 decline was found to be higher than the 2% predicted for children born after 1980 in developed countries and higher than the FEV1 decline in South African children with CF, in a relatively short-term longitudinal study where an average rate of decline in FEV1 was less than 1% per year.Citation12 Early reports showed significant association between the degree of malnutrition and the rate of decline in pulmonary function.Citation18,22 Bell et al. and Nir et al. revealed a strong association between FEV1 and BMI.Citation26,27 The current study found that FEV1 is significantly influenced by BMI Z-score, in line with the published literature, but it was found that FEV1 was influenced by other variables such as age and weight. In a study conducted in Germany, by Steinkamp et al., the authors revealed that a fall in weight-for-height of 5% predicted or more within one year was associated with a parallel decrease in FEV1, whereas patients with improved nutrition demonstrated constant or even improved FEV1.Citation19 In the present study, FEV1 and FEF25-75 were significantly affected by BMI, which crudely reflects body fat content and is in agreement with the Steinkamp study.
The strength of the current study is that it has assessed factors which may impact growth and lung function in a CF clinic in a developing country. Our recommendations would be for CF practitioners and other healthcare practitioners to pay special attention to anthropometry and growth in all CF patients, as this impacts on their lung functions and, ultimately, survival. The limitations of this study were the small study population group, which limited the ability to make any conclusions with regard to the impact of the studied variables on specific age categories. All participants were Caucasians and the age at diagnosis was not specified. The study also did not assess the impact of the organisms cultured from the lower respiratory tract.
Conclusion
The average lung function decline per year for FEV1 was higher than that seen in developed countries. The decline in FEV1 was related to gender, age, weight and body mass index. The decline in FEF25-75 was affected only by BMI Z-score and gender.
References
- Cystic Fibrosis Mutation Database. 2011 Apr [cited 2014 Aug 4]. Available from: http://www.genet.sickkids.on.ca/Homehtml.
- Goldman A, Graf C, Ramsay M, et al. Molecular diagnosis of cystic fibrosis in South African populations. S Afr Med J. 2003;93:518–9.
- Mutesa L, Bours V. Diagnostic challenges of cystic fibrosis in patients of African origin. J Trop Pediatr. 2009;55:281–6.10.1093/tropej/fmp064
- Goldman A, Labrum R, Claustres M, et al. The molecular basis of cystic fibrosis in South Africa. Clin Genet. 2001;59:37–41.
- Westwood T. Diagnosing cystic fibrosis in South Africa. S Afr Med J. 2006;96:304–6.
- Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Ann Hum Genet. 2003;67:471–85.10.1046/j.1469-1809.2003.00028.x
- de Gracia J, Mata F, Alvarez A, et al. Genotype-phenotype correlation for pulmonary function in cystic fibrosis. Thorax. 2005;60:558–63.10.1136/thx.2004.031153
- Ranganathan SC, Stocks J, Dezateux C, et al. The evolution of airway function in early childhood following clinical diagnosis of cystic fibrosis. Am J Respir Crit Care Med. 2004;169:929–33.
- Konstan MW, Morgan WJ, Butler SM, et al. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J Pediatr. 2007;151:134–9.10.1016/j.jpeds.2007.03.006
- Navarro J, Rainisio M, Harms HK, et al. Factors associated with poor pulmonary function: cross-sectional analysis of data from the ERCF. Eur Respir J. 2001;18:298–305.10.1183/09031936.01.00068901
- Schaedel C, de Monestrol I, Hjelte L, et al. Predictors of deterioration of lung function in cystic fibrosis. Pediatr Pulmonol. 2002;33:483–91.10.1002/(ISSN)1099-0496
- Morrow BM, Argent AC, Zar HJ, et al. Rate of pulmonary function decline in South African children with cystic fibrosis. S Afr J Child Health. 2009;3:73–6.
- Xu W, Subbarao P, Corey M. Changing patterns of lung function decline in children with cystic fibrosis. J Cystic Fibros. 2004;3:S116–8.
- Morrow BM, Argent AC, Zar HJ, et al. Improvements in lung function of a pediatric cystic fibrosis population in a developing country. J Pediatr. 2008;84:403–9.10.2223/JPED.1829
- Pencharz PB, Durie PR. Pathogenesis of malnutrition in cystic fibrosis, and its treatment. Clin Nutr. 2000;19:387–94.10.1054/clnu.1999.0079
- Konstan MW, Butler SM, Wohl ME, et al. Growth and nutritional indexes in early life predict pulmonary function in cystic fibrosis. J Pediatr. 2003;142:624–30.10.1067/mpd.2003.152
- Callaghan BD, Hoo AF, Dinwiddie R, et al. Growth and lung function in Asian patient with cystic fibrosis. Arch Dis Child. 2005;90:1029–32.10.1136/adc.2004.067264
- Zemel BS, Jawad AF, FitzSimmons S, et al. Longitudinal relationship among growth, nutritional status, and pulmonary function in children with cystic fibrosis: Analysis of the cystic fibrosis foundation national CF patient registry. J Pediatr. 2000;137:374–80.10.1067/mpd.2000.107891
- Steinkamp G, Weidemann B. Relationship between nutritional status and lung function in cystic fibrosis: cross sectional and longitudinal analyses from the German CF quality assurance (CFQA) project. Thorax. 2002;57:596–601.10.1136/thorax.57.7.596
- Pentz A, Becker P, Masekela R, et al. The impact of chronic pseudomonal infection on pulmonary function testing in individuals with cystic fibrosis in Pretoria, South Africa. S Afr Med J. 2014;104:191–194.10.7196/samj.7222
- Johansen HK, Nir M, Koch C., et al. Severity of cystic fibrosis in patients homozygous and heterozygous for ΔF508 mutation. Lancet. 1991;337:631–4.10.1016/0140-6736(91)92449-C
- Corey M, Edwards L, Levison H, et al. Longitudinal analysis of pulmonary function decline in patients with cystic fibrosis. J Pediatr. 1997;131:809–14.10.1016/S0022-3476(97)70025-8
- Lucidi V, Alghisi F, Raia V, et al. Growth assessment of paediatric patients with CF comparing different auxologic indicators: a multicentre Italian study. J Pediatr Gastroenterol Nutr. 2009;49:335–42.10.1097/MPG.0b013e31818f0a39
- Lai HC, Kosorok MR, Sondel SA, et al. Growth status in children with cystic fibrosis based on the national cystic fibrosis patient registry data: evaluation of various criteria used to identify malnutrition. J Pediatr. 1998;132:478–85.10.1016/S0022-3476(98)70024-1
- Corey M, McLaughlin FJ, Williams M, et al. A comparison of survival, growth, and pulmonary function in patients with cystic fibrosis in Boston and Toronto. J Clin Epidemiol. 1988;41:583–91.10.1016/0895-4356(88)90063-7
- Bell SC, Bowerman AR, Davies CA, et al. Nutrition in adults with cystic fibrosis. Clin Nutr. 1998;17:211–5.10.1016/S0261-5614(98)80061-7
- Nir M, Lanng S, Johansen HK, et al. Long-term survival and nutritional data in patients with cystic fibrosis treated in a Danish centre. Thorax. 1996;51:1023–7.