Abstract
The extension of neurites by bovine chromaffin cells in culture represents an opportunity to analyse the factors contributing to shape secretion from a round endocrine to a polarized neuronal model. Neurite-emitting chromaffin cells indeed present a different cytoskeletal organization characterized by smaller cytoskeletal cages when compared to round cells and also show a higher density of P/Q-type calcium channels at the neurite tips. Furthermore, these cells present faster and more transient secretory kinetics at the neurite processes when compared with the secretory activity found in round cells. These results were taken into account when proposing a stochastic mathematical model to analyse the impact of different factors in intracellular calcium elevations and the resulting secretory kinetics. Thus, our modelling studies predict that the accumulation of calcium channels at the neurite tips is a major cause of accelerated secretory kinetics, whereas reducing the space for calcium diffusion consequence of smaller cytoskeletal cages enhances the sensitivity of secretion to lower calcium concentrations when assuming a porosity factor for calcium crossing F-actin structures.
Introduction
The exocytotic fusion of the vesicles containing neurotransmitters and hormones with the plasma membrane is the key event to understand the function of the neuronal and endocrine cellular systems. In neurons, this process take place in specialized regions called “active zones” (Heuser et al. Citation1974), concentrating the molecular constituents of the secretory machinery such as the ubiquitous SNARE proteins (Bennett & Scheller Citation1993). Most of our knowledge of the exocytotic process comes from the work with neuroendocrine models, such chromaffin cells, where many of the molecular elements have been found and implicated in secretion (Roth & Burgoyne Citation1994; Gutierrez et al. Citation1995; Gil et al. Citation2002). Although these cells are located outside the nervous system, they are derived from the neural crest and present active-site like structures suggested by the precise localization of discrete releasing sites with calcium entry zones or “hot spots” (Robinson et al. Citation1995).
Interestingly, chromaffin cells in culture spontaneously emit neurite-like processes that are very actively concentrating the secretory response at their tips (Gutierrez et al. Citation1998), as measured by the probability of finding individual fusions by using narrow carbon fibre amperometric electrodes. Among other possible factors, the accumulation of P/Q type voltage-dependent calcium channels has been described as a major influence determining the preferential localization of the secretory response in these neurites (Gil et al. Citation2001), which is in agreement with the preferential association of these channels with neurotransmitter release in many types of neurons (Catterall Citation1998). Thus, the study of the secretory organization in neurite-alike structures in chromaffin cells appeared interesting to understand the fine-tuning of exocytosis when the endocrine organization evolves to a pseudoneuronal system. In addition to studying such organization, we use stochastic models of secretion (Gil et al. Citation2000; Torregrosa-Hetland et al. Citation2010, Citation2011; Villanueva et al. Citation2010) to study the influence of calcium channel accumulation and cytoskeletal organization in determining the kinetic differences in secretion found between neurite terminals and the cell body. Our study enriches the variety of roles assumed by the cortical cytoskeleton when governing the secretory process (Gutierrez Citation2012).
Materials and methods
Chromaffin cell preparation, culture and treatment
Chromaffin cells were isolated from bovine adrenal glands by collagenase digestion and were further separated from the debris and erythrocytes by centrifugation on Percoll gradients as described elsewhere (Gutierrez et al. Citation1995; Gil et al. Citation2002). The cells were maintained as monolayer cultures in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 10 μM cytosine arabinoside, 10 μM 5-fluoro-2′-deoxyuridine, 50 IU/ml penicillin and 50 μg/ml streptomycin. The cells were harvested at a density of 150,000 cells/cm2 in 22 mm diameter coverslips coated with polylysine and were used between the third and sixth day after plating.
For experiments, cell culture medium was replaced with Krebs/HEPES (K/H) basal solution containing 134 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgCl2, 2.5 mM CaCl2, 11 mM glucose, 0.56 mM ascorbic acid and 15 mM HEPES, at pH 7.4. Cells were stimulated for 1 min using a depolarizing solution with 59 mM high potassium (obtained by replacing isosmotically NaCl with KCl) in K/H basal solution.
Confocal dynamic images of the F-actin cytoskeleton
Transmitted light images were investigated using an Olympus Fluoview FV300 confocal laser system mounted on a IX-71 inverted microscope incorporating a 100× PLAN-Apo oil-immersion objective with 1.45 n.a. Excitation was achieved using Ar and HeNe visible light lasers. This system permits z-axis reconstruction (0.5–0.55 μm theoretical z slice) and dynamic time-lapse studies, with a time resolution ranging from 0.1 s and with 200×150 pixel image acquisition (adequate for regional studies) to about 0.6 s for images of 400×300 pixels (for visualization of the entire cell). Simultaneous acquisition of transmitted light images provided information of the distribution of the F-actin cytoskeleton and were obtained using the channel implemented in the confocal microscope as well as using bright field optics (the theoretical depth of field is 0.55–0.60 μm, see Giner et al. Citation(2005)). The intensity of this channel was adjusted to avoid saturation of the subcortical region and to visualize the lighter cytoplasmic structures. These images were employed to calculate cage size and cell area increase during stimulation, using macros of the public domain program ImageJ. Cytoskeletal size dimensions were obtained from transmitted light images analyzed using ImageJ after applying channel threshold to generate particles covering the spaces of the dark cage interiors followed by particle area measurements. Similarly, area increase after depolarization was obtained by image threshold detecting the cell limits and area measurements in individual frames from time-lapse experiments.
Graphics were obtained with IgorPro, Graphpad Prism (GraphPad software, San Diego, CA, USA) and Adobe Photoshop 7.0. The Student t-test for paired samples or the two-way ANOVA test were used to establish statistical significance among the experimental data (samples were considered significantly different when p<0.05). Non-Gaussian data distributions were analyzed using the non-parametric Mann–Whitney U test. Data were expressed as mean±SEM from experiments performed in a number (n) of individual cells, vesicles, or fusion events from at least three different cultures.
Mathematical models of calcium maps and secretion
The simulation algorithm for computing calcium profiles and secretion events is an extension of a Monte Carlo code for the simulation of calcium-triggered exocytotic processes developed by Gil et al. Citation(2000). This algorithm includes mathematical models for calcium ions entering through L and P/Q calcium channels (see , 3D diffusion of mobile particles, endogenous buffering of calcium ions and kinetics for vesicle fusion. All these mechanisms are considered as stochastic processes taking place in a cylindrical domain representing a portion of the cell body or the neurite terminal. A 3D orthogonal grid approximately maps the domain in cubes of volume (Δ x)3, where Δ x is the spatial resolution. In the first slice of the grid (representing the cell submembrane region), calcium channels and docked vesicles are placed.
Results and discussion
Cytoskeletal organization in neurite-emitting chromaffin cells
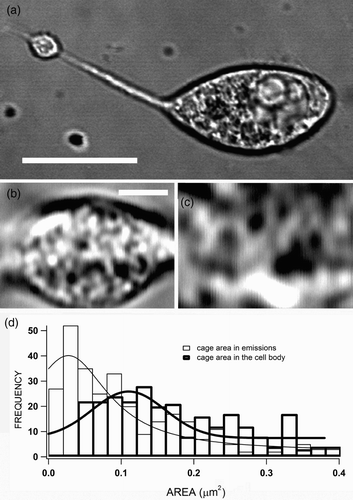
The use of scanning light microscopy in confocal microscopes allowed us to study the cytoskeletal organization and its changes during the secretion of “typical” round chromaffin cells (Giner et al. Citation2005, Citation2007) and therefore we were prompted to study these changes during the secretion of neurite-emitting cells. An example of such cells could be observed in ; the long neurite emission ended in a engrossed tip that has been shown to accumulate secretory vesicles (Gutierrez et al. Citation1998). Interestingly, an inspection at higher magnification revealed the structural organization in cytoskeletal cages in both the neurite terminal ( and the cell body cortex (, as it could be observed the smaller size of these cytoskeletal cages visualized in the neurite terminal. A quantification of the size of the dark interior of these cages in eight neurite-emitting cells shows different cage size distributions for the terminal and body areas (, with an average area of 0.075±0.005 and 0.17±0.01 μm2 for the neurite emission and the cell body cortex, respectively. Thus, assuming circular cages vesicles move and are imprisoned in structures of 310-nm radius in the neurite terminals, and with 465-nm radius in the cell body cortex, results in an almost four-fold difference in volume. These differences in the organization of the cytoskeleton appear to be important for the function of different cytoplasmic regions in our neuroendocrine chromaffin cells, as it has been described that the cortical cytoskeleton is composed of smaller cages in comparison with the structures found in the interior of the cells (Giner et al. Citation2007).
Figure 1. Cytoskeletal organization in neurite-emitting bovine chromaffin cells. Bovine chromaffin cells were isolated and cultured (as described in “Materials and methods” section); after 3–5 days of plating, many cells emitted neurites as the example cell observed by the transmitted light channel implemented in a confocal microscope (a). Parts (b) and (c) present high-magnified images of the neurite tip and a part of the cell body, respectively, evidencing the presence of cytoskeletal cages (dark spaces) with different sizes in both areas of the cell. These dark spaces were measured for eight cells and the size distribution plotted for neurites and the cell body (d). The estimated average area of the cages are 0.075±0.005 and 0.17±0.01 μm2 for the neurite emission and the cell body, respectively. Bars represent 10 μm in (a) and 1 μm in (b).
Different secretory kinetics characterizes secretion at the neurites and cell body
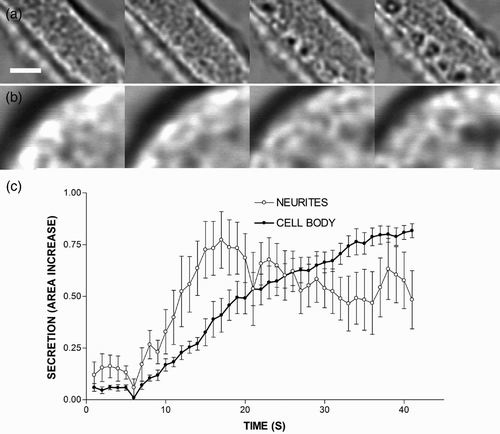
Interestingly, the organization of cytoskeletal cages suffers major changes during secretion in the neurite terminals. As can be observed in the example depicted in , cell stimulation by depolarization causes the formation of open spaces, which are devoid of cytoskeletal structures (submembranal dark spaces), 10 s after the initiation of the stimulus and increases the area of the neurite as a result of the incorporation of the granular membrane during exocytosis. This increase in the neurite area could be measured in several cells to estimate the secretory kinetics at the neurite terminals (. As can be observed in this figure, secretion is faster and stops earlier in the neurite endings as compared to the cell body ( and ). Thus, it appears that secretory kinetics is markedly different in diverse areas of neurite-emitting cells, and it is possible that the cytoskeletal organization or the accumulation of other molecular factors could enhance the secretory kinetics at the neurite terminals. In agreement with this finding, it has been reported that the density of secretory amperometric spikes appear to be enhanced in these neurite terminals, thus giving a higher probability of response when compared with the secretory behaviour observed in the cell body (Gutierrez et al. Citation1998).
Figure 2. Cytoskeletal alterations and secretory behaviour in the terminals and the cell body of neurite-emitting chromaffin cells. The figure shows transmitted light images of a neurite process (a) and the cell body of a round cell (c) stimulated by perfusion with a depolarizing solution (KCl 59 mM). Time-lapse images were separated by 5-s intervals, showing that the increase in the area occurred after stimulation. The increase in the area was estimated in nine cells to measure secretory kinetics in both the terminals of neurite-emitting cells and the cell body of round cells (c). Bar represents 1 μm.
Modelling calcium channel influence in the secretion of neurite-emitting chromaffin cells
To quantify the effect of the factors influencing secretory kinetics in neurite-emitting chromaffin cells, we used a mathematical model for computing calcium concentrations and secretory responses in the cell body cortex and the neurite terminal. The model takes into account (i) the entry of calcium ions through L- and P/Q-type calcium channels, (ii) the 3D diffusion of mobile particles in the submembrane cellular domain, (iii) the kinetic reactions between the endogenous buffer and calcium, (iv) the kinetic reactions between calcium ions and secretory vesicles and (v) a (possible) porosity of the cytoskeletal cages. Our simulations are performed in a cylinder representing a portion of either a neurite terminal or the cell body cortex.
We started our analysis focusing on the role of the calcium channels in the secretory response. We compared calcium concentrations and secretory responses obtained in response to a short depolarizing pulse (lasting 20 ms), taking into account first the different density of P/Q type voltage-dependent calcium channels estimated in neurite terminals and the cell body cortex – a factor approximately twice of P/Q channels in neurites with respect to the cell body when these channels are labelled with specific antibodies coupled to fluorescent probes (Gil et al. Citation2001). We took the same density of L-type calcium channels because fluorescence estimations show no significant variation from neurites to the cell body. For simulating these configurations, we considered several random distributions of clusters of calcium channels formed by 1 L and 1 P/Q channels in the case of cell body and clusters of 1 L and 2 P/Q calcium channels in the case of neurite terminals. We also assumed, during our analysis, that secretory vesicles are co-localized with calcium channels in both cases. Calcium currents through L- and P/Q-type channels are simulated according to the I–V curves shown in . These I–V curves agree with the opening potentials and peak voltages reported for chromaffin cells by Perez-Alvarez et al. Citation(2008) and Hernandez et al. Citation(2011).
Figure 3. Modelling calcium elevations and secretion in the terminals and the cell body of neurite-emitting chromaffin cells. (a) Current to voltage relationships simulated for the P/Q- and L-type Ca2+ channels. (b) Upper panel: computed calcium concentrations from 0 to 50 nm to the cellular membrane in response to a depolarizing pulse from−80 to 0 mV lasting 20 ms. The results are shown for the simulated portion of neurites (green line) and the cell body cortex (red line). The same density of clusters of calcium channels is considered in both cases but with a different number of calcium channels per cluster: 1 L and 1 P/Q in the cell body cortex, and 1 L and 2 P/Q in neurites. Lower panel: normalized accumulated secretory responses obtained in neurites and the cell body. A non-cooperative kinetic scheme for the binding of calcium to secretory vesicles is considered in the computations.
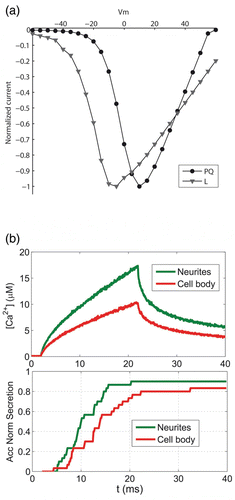
The upper panel of shows the comparison of average calcium concentrations obtained for a 50-nm-thick layer underneath the plasma membrane for neurites and the cell body. As can be seen, the higher P/Q calcium channel density in neurites is translated into a calcium peak 40% larger than the corresponding calcium concentration in the simulated portion of the cell body. This higher calcium concentration has a significant impact on secretion, as shown in the lower panel of where the normalized accumulated secretory responses for neurites and cell body are compared. As can be seen, the secretory response is both faster and steeper in neurites, in agreement with the experimental observations. In any case, this mathematical approach gives theoretical support to the concept that preferential localization of specific calcium channel subtypes in neuronal cells facilitates a coordinated strong exocytotic response in the neurite terminals (Catterall Citation1998) or a more efficient coupling of channel subtypes with the secretory response in neuroendocrine cells (Lopez et al. Citation1994; Garcia et al. Citation1998). In addition, even if there is secretory response that is proportional to the overall calcium elevation rather than the presence of specific channels subtypes (Lukyanetz & Neher Citation1999), the asymmetric distribution of calcium channel subtypes may influence the voltage dependence of secretory inactivation (Villarroya et al. Citation1999), the posterior process of endocytosis (Rosa et al. Citation2011), the clustering of SNAREs forming the secretory machinery (Lopez et al. Citation2007) or directly influence the exocytotic machinery (Seagar et al. Citation1999; Atlas et al. Citation2001).
Introducing the cytoskeletal organization as a factor influencing secretion in mathematical models of neurite-emitting chromaffin cells
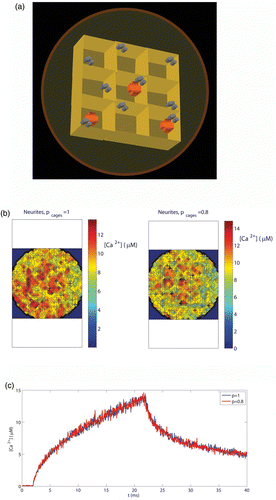
As we have described, the cytoskeletal organization could be another factor influencing the secretory behaviour in the neurites and the cell body (Torregrosa-Hetland et al. Citation2010; Villanueva et al. Citation2010; Gutierrez & Gil Citation2011; Torregrosa-Hetland et al. Citation2011). The active role of the cytoskeletal cages was incorporated in our model because these cages act as diffusion barriers for mobile particles in the intracellular medium. For this purpose, a “porosity parameter” (p) was assigned to the cytoskeletal cages. We simulated the effect of an arrangement of nine cytoskeletal cages, as represented in . To simplify, the volume of the cages was taken as 300×300×300 nm3 in neurites and 450×450×450 nm3 for the cytoskeletal cages in the cell body, although real data distributions will result in larger variability among the different domains of the cells. The cages are part of a cylindrical simulation domain of radius R CB=1.2 μm (cell body) and R N=0.8 μm (neurites) and height h=1.2 μM. The porosity parameter of the cages will simulate how densely packed the cytoskeletal cages are. The porosity (p) will be determined by the probability of a given ion of crossing through when encountering the barrier. In our discrete simulation, a particle encounters a barrier when it tries to make a move connecting two compartments that are separated by this barrier. The value of the porosity is taken to be the same through the five walls of each barrier and the barriers are considered to display the same porosity for incoming or outgoing particles. We assume that clusters of voltage-dependent calcium channels of the L and P/Q subtypes are located in the cages with the arrangement shown in . As before, we will take for the calcium clusters 1 L and 1 P/Q channels for the cell body and 1 L and 2 P/Q channels for the neurite terminal. The calcium channels will be placed near the border of the cytoskeletal cage, as experimental data suggest (Villanueva et al. Citation2010; Torregrosa-Hetland et al. Citation2011). We simulate the calcium current corresponding to a short depolarizing pulse to 0 mV lasting 20 ms. As an example of the impact of such diffusion barriers on the calcium concentrations, shows maps of local calcium concentrations in the submembrane domain (0–50 nm, corresponding to our spatial resolution) at t=25 ms for two values of the porosity parameter (p=1, 0.8) and for the barrier configuration corresponding to neurites. A porosity value of 1 represents a fully porous wall and so no geometrical obstacles for calcium diffusion are considered in the medium (the barriers do not enter into play). A porosity value of 0.8 represents a reduction of a 20% in the probability of crossing one of the walls of the cages. Therefore, the lower the value of the porosity parameter, the higher will be the reduction in the probability for the mobile particles of crossing the walls. As can be seen in , for the porosity value of 0.8 there is an increase in the local calcium in the medium, which, as we next show, will have a measurable impact on the secretory response. It is important to note that this is not an average effect of the system but a local one – the same value for the computed average calcium concentration in the whole domain will be obtained with different values of the porosity (see .
Figure 4. A model to study cytoskeletal influence in calcium elevations and secretory responses. (a) Representation of the arrangement of nine cytoskeletal cages considered in the simulations. The volume of the cages is taken as 300×300×300 nm3 in neurites and 450×450×450 nm3 for the cytoskeletal cages in the cell body cortex. In the figure, cylinders represent calcium channels and crosses represent secretory vesicles. (b) Calcium maps obtained in neurites when simulating the same depolarizing pulse as in and for two values of the porosity parameter: p=1, 0.8. (c) Average calcium concentrations obtained in neurites from 0 to 50 nm to the cellular membrane when simulating the same depolarizing pulse than in and for two values of the porosity parameter: p=1, 0.8.
To compare the secretory responses obtained in neurites and the cell body for configurations having these diffusion barriers, we have considered random distributions of vesicles inside the cages. In this way, different distances of the vesicle sensor to the clusters of calcium channels can be taken into account. shows the accumulated secretory responses obtained both in neurites (upper panel) and the cell body (middle panel) when the values of the porosity are taken as p=1 and p=0.5 (representing this last value is a reduction of 50% in the diffusion of particles when arriving at a wall). The lower panel shows the relative increase in secretion obtained for neurites and the cell body. As can be seen, when reducing the porosity of the walls there is an increase in secretion in the cell body but not in neurites. This result seems somehow counterintuitive, given that the cytoskeletal cages are larger in the case of the cell body. Then, one would expect that confining calcium in a smaller box (as is the case of the cytoskeletal cages in neurites) will be translated into a larger increase in secretion. This is because for the depolarizing protocol (from −80 mV to 0 mV) that we are simulating, the relative effect of the local calcium concentrations versus the wall effect is much more important in neurites than in the cell body. Therefore, the nonlinear relation between calcium levels and the size/porosity of the cytoskeletal cages seems to be a key point for analyzing the response of the system.
Figure 5. Cytoskeletal influence in calcium elevations and secretion in both the cell body and the neurite terminals. (a) Upper panel: temporal dependence of the normalized accumulated secretory response obtained in neurites when the calcium influx corresponding to a depolarizing pulse from−80 to 0 mV lasting 20 ms is simulated. The results obtained for two values of the porosity parameter (p) are plotted: p=1 (solid line) and p=0.5 (dotted line). Middle panel: same as upper panel, but for the cell body cortex. Lower panel: relative increase in secretion obtained when the porosity parameter value decreases from p=1 to p=0.5. The results are shown for neurites (green line) and the cell body cortex (red line). (b) Same as (a) but for a depolarizing pulse from−80 to 40 mV.
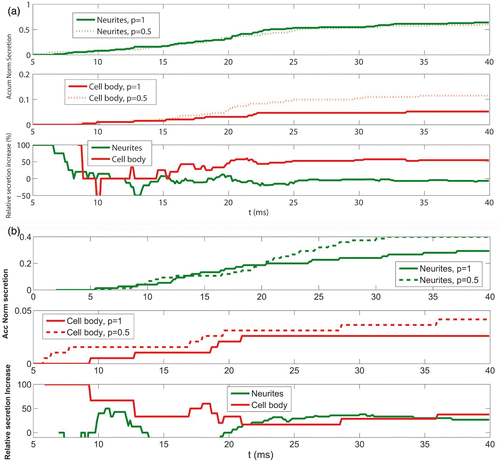
This hypothesis can be tested by considering an alternative depolarizing protocol (this time from −80 to 40 mV). According to the I–V curve for L and P/Q calcium channels in chromaffin cells plotted in , the expected calcium currents will be lower both in the neurite terminal and the cell body. In this situation, one would expect that the effect of diffusion barriers would now play a more important role in neurites than it played in the previous depolarizing protocol. This is shown in , where the temporal dependence for the relative increase in the accumulated secretion obtained for this new simulated protocol is plotted. As can be seen, both curves converge to a very similar value.
The study of the impact on secretion of the cytoskeletal cage size by itself has to be then performed considering the same calcium channel distribution in neurite terminals and the cell body. For this purpose, we performed the following numerical experiment: we computed the number of secretory events by assuming a single cluster formed by two channels (of types L and P/Q) located in a cytoskeletal cage of variable size and porosity. We found that when the value of the porosity is reduced from p=1 to p=0.5, there is an increase in secretion of about a 4% more for a typical cytoskeletal cage in neurite terminals in comparison to the results obtained for a soma cage. This effect is much more evident when the porosity parameter is reduced to p=0.1. In this case, the secretion in the soma cage increases a 130% (with respect to secretion values obtained for p=1), whereas for a typical cage in neurite terminals, the secretion increases to a 420%. This means that for small values of the porosity parameter, the reduction in the size of a cytoskeletal cage is a factor that strongly enhances the probability of secretion.
A more general view of the voltage dependence of the exocytotic response can be obtained from numerical simulations where the voltage of the depolarizing is also varied (see the I–V curve shown in . In these numerical experiments, the value of the porosity parameter is also varied (p=1, 0.5, 0.1). The results indicate that the largest relative increase in secretion obtained in neurites, when the cytoskeletal cages act as diffusion barriers with porosity coefficients 0.5 and 0.1, is obtained for the depolarizing pulse to −20 mV. In contrast, for this voltage there is no increase in secretion in the cell body because the local calcium concentrations are below the threshold for triggering secretion even when barriers enter into play. For the rest of tested voltages, the effect of barriers in the secretory response is shown to be more important in the cell body than in neurites. The results obtained suggest that the changes in the cytoskeletal organization when evolving to a neuron-like structure result in a larger sensitivity to low calcium concentrations. As a whole, the cytoskeletal organization appears to be a factor to take into account when explaining the voltage dependence of the excitability in different parts of cells that extend neurite processes.
Acknowledgements
This study was supported by grants from the Spanish Ministerio de Ciencia e Innovación (MICINN) (BFU 2008-0073) and the Spanish Ministerio de Economia y Competitividad (BFU 2011-25095) to Luis M. Gutiérrez. Cristina J. Torregrosa-Hetland is a recipient of a FPU predoctoral studentship from the MICINN of Spain. Virginia González-Vélez thanks CONACyT for financial support given through postdoctoral fellowship.
References
- Atlas , D , Wiser , O and Trus , M . 2001 . The voltage-gated Ca2+ channel is the Ca2+ sensor of fast neurotransmitter release . Cell Mol Neurobiol. , 21 : 717 – 31 . (doi:10.1023/A:1015104105262)
- Bennett , M K and Scheller , R H . 1993 . The molecular machinery for secretion is conserved from yeast to neurons . Proc Natl Acad Sci USA. , 90 : 2559 – 63 . (doi:10.1073/pnas.90.7.2559)
- Catterall , W A . 1998 . Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release . Cell Calcium. , 24 : 307 – 23 . (doi:10.1016/S0143-4160(98)90055-0)
- Garcia , A G , Albillos , A , Cano-Abad , M F , Garcia-Palomero , E , Hernandez-Guijo , M , Herrero , C J , Lomax , R B and Gandia , L . 1998 . Calcium channels for exocytosis in chromaffin cells . Adv Pharmacol. , 42 : 91 – 4 . (doi:10.1016/S1054-3589(08)60703-6)
- Gil , A , Gutierrez , L M , Carrasco-Serrano , C , Alonso , M T , Viniegra , S and Criado , M . 2002 . Modifications in the C terminus of the synaptosome-associated protein of 25 kDa (SNAP-25) and in the complementary region of synaptobrevin affect the final steps of exocytosis . J Biol Chem. , 277 : 9904 – 10 . (doi:10.1074/jbc.M110182200)
- Gil , A , Segura , J , Pertusa , J A and Soria , B . 2000 . Monte Carlo simulation of 3-D buffered Ca(2+) diffusion in neuroendocrine cells . Biophys J. , 78 : 13 – 33 . (doi:10.1016/S0006-3495(00)76569-6)
- Gil , A , Viniegra , S , Neco , P and Gutierrez , L M . 2001 . Co-localization of vesicles and P/Q Ca2+-channels explains the preferential distribution of exocytotic active zones in neurites emitted by bovine chromaffin cells . Eur J Cell Biol. , 80 : 358 – 65 . (doi:10.1078/0171-9335-00168)
- Giner , D , Lopez , I , Villanueva , J , Torres , V , Viniegra , S and Gutierrez , L M . 2007 . Vesicle movements are governed by the size and dynamics of F-actin cytoskeletal structures in bovine chromaffin cells . Neuroscience. , 146 : 659 – 69 . (doi:10.1016/j.neuroscience.2007.02.039)
- Giner , D , Neco , P , Frances , M M , Lopez , I , Viniegra , S and Gutierrez , L M . 2005 . Real-time dynamics of the F-actin cytoskeleton during secretion from chromaffin cells . J Cell Sci. , 118 : 2871 – 80 . (doi:10.1242/jcs.02419)
- Gutierrez , L M . 2012 . New insights into the role of the cortical cytoskeleton in exocytosis from neuroendocrine cells . Int Rev Cell Mol Biol. , 295 : 109 – 37 . (doi:10.1016/B978-0-12-394306-4.00009-5)
- Gutierrez , L M and Gil , A . 2011 . Modeling F-actin cortex influence on the secretory properties of neuroendocrine cells . Commun Integr Biol. , 4 : 413 – 5 .
- Gutierrez , L M , Gil , A and Viniegra , S . 1998 . Preferential localization of exocytotic active zones in the terminals of neurite-emitting chromaffin cells . Eur J Cell Biol. , 76 : 274 – 8 . (doi:10.1016/S0171-9335(98)80005-8)
- Gutierrez , L M , Quintanar , J L , Viniegra , S , Salinas , E , Moya , F and Reig , J A . 1995 . Anti-syntaxin antibodies inhibit calcium-dependent catecholamine secretion from permeabilized chromaffin cells . Biochem Biophys Res Commun. , 206 : 1 – 7 . (doi:10.1006/bbrc.1995.1001)
- Hernandez , A , Segura-Chama , P , Jimenez , N , Garcia , A G , Hernandez-Guijo , J M and Hernández Cruz , A . 2011 . Modulation by endogenously released ATP and opioids of chromaffin cell calcium channels in mouse adrenal slices . Am J Physiol-Cell Ph. , 300 : C610 – 23 . (doi:10.1152/ajpcell.00380.2010)
- Heuser , J E , Reese , T S and Landis , D M . 1974 . Functional changes in frog neuromuscular junctions studied with freeze-fracture . J Neurocytol. , 3 : 109 – 31 . (doi:10.1007/BF01111936)
- Lopez , I , Giner , D , Ruiz-Nuno , A , Fuentealba , J , Viniegra , S , Garcia , A G , Davletov , B and Gutierrez , L M . 2007 . Tight coupling of the t-SNARE and calcium channel microdomains in adrenomedullary slices and not in cultured chromaffin cells . Cell Calcium. , 41 : 547 – 58 . (doi:10.1016/j.ceca.2006.10.004)
- Lopez , M G , Villarroya , M , Lara , B , Martinez , S R , Albillos , A , Garcia , A G and Gandia , L . 1994 . Q- and L-type Ca2+ channels dominate the control of secretion in bovine chromaffin cells . FEBS Lett. , 349 : 331 – 7 . (doi:10.1016/0014-5793(94)00696-2)
- Lukyanetz , E A and Neher , E . 1999 . Different types of calcium channels and secretion from bovine chromaffin cells . Eur J Neurosci. , 11 : 2865 – 73 . (doi:10.1046/j.1460-9568.1999.00707.x)
- Perez-Alvarez , A , Hernandez-Vivanco , A , Cano-Abad , M and Albillos , A . 2008 . Pharmacological and biophysical properties of Ca2+ channels and subtype distributions in human adrenal chromaffin cells . Pflugers Arch. , 456 ( 6 ) : 1149 – 62 . (doi:10.1007/s00424-008-0492-7)
- Robinson , I M , Finnegan , J M , Monck , J R , Wightman , R M and Fernandez , J M . 1995 . Colocalization of calcium entry and exocytotic release sites in adrenal chromaffin cells . Proc Natl Acad Sci USA. , 92 : 2474 – 78 . (doi:10.1073/pnas.92.7.2474)
- Rosa , J M , Torregrosa-Hetland , C J , Colmena , I , Gutierrez , L M , Garcia , A G and Gandia , L . 2011 . Calcium entry through slow-inactivating L-type calcium channels preferentially triggers endocytosis rather than exocytosis in bovine chromaffin cells . Am J Physiol Cell Physiol. , 301 : C86 – C98 . (doi:10.1152/ajpcell.00440.2010)
- Roth , D and Burgoyne , R D . 1994 . SNAP-25 is present in a SNARE complex in adrenal chromaffin cells . FEBS Lett. , 351 : 207 – 10 . (doi:10.1016/0014-5793(94)00833-7)
- Seagar , M , Leveque , C , Charvin , N , Marqueze , B , Martin-Moutot , N , Boudier , J A , Boudier , J L , Shoji-Kasai , Y , Sato , K and Takahashi , M . 1999 . Interactions between proteins implicated in exocytosis and voltage-gated calcium channels . Philos Trans R Soc Lond B Biol Sci. , 354 : 289 – 97 . (doi:10.1098/rstb.1999.0380)
- Torregrosa-Hetland , C J , Villanueva , J , Giner , D , Lopez-Font , I , Nadal , A , Quesada , I , Viniegra , S , Exposito-Romero , G , Gil , A Gonzalez-Velez , V . 2011 . The F-actin cortical network is a major factor influencing the organization of the secretory machinery in chromaffin cells . J Cell Sci. , 124 : 727 – 34 . (doi:10.1242/jcs.078600)
- Torregrosa-Hetland , C J , Villanueva , J , Lopez-Font , I , Garcia-Martinez , V , Gil , A , Gonzalez-Velez , V , Segura , J , Viniegra , S and Gutierrez , L M . 2010 . Association of SNAREs and calcium channels with the borders of cytoskeletal cages organizes the secretory machinery in chromaffin cells . Cell Mol Neurobiol. , 30 : 1315 – 9 . (doi:10.1007/s10571-010-9565-1)
- Villanueva , J , Torregrosa-Hetland , C J , Gil , A , Gonzalez-Velez , V , Segura , J , Viniegra , S and Gutierrez , L M . 2010 . The organization of the secretory machinery in chromaffin cells as a major factor in modeling exocytosis . HFSP J. , 4 : 85 – 92 . (doi:10.2976/1.3338707)
- Villarroya , M , Olivares , R , Ruiz , A , Cano-Abad , M F , Lomax , R B , Lopez , M G , Mayorgas , I , Gandia , L and Garcia , A G . 1999 . Voltage inactivation of Ca2+ entry and secretion associated with N- and P/Q-type but not L-type Ca2+ channels of bovine chromaffin cells . J Physiol. , 516 ( Pt 2 ) : 421 – 32 . (doi:10.1111/j.1469-7793.1999.0421v.x)