
ABSTRACT
Adipose tissue dysfunction correlates with the development of diabetes. Mice with an adipocyte-specific deletion of the SUMO-specific protease SENP1 develop symptoms of type-1 diabetes mellitus (T1DM). Peri-pancreatic adipocytes (PATs) exert both systemic and paracrine effects on pancreases function. Our recent studies report that PATs of SENP1-deficient mice have increased proinflammatory cytokine production compared with other adipose depots. Proinflammatory cytokines produced from PATs not only have direct cytotoxic effects on pancreatic islets, but also increase CCL5 expression in adjacent pancreatic islets, which induces persistent inflammation in pancreases by acquisition of Th1 and Th17 effector T cell subsets. Small ubiquitin-like modifier (SUMO) can post-translationally conjugate to cellular proteins (SUMOylation) and modulate their biological functions. Several components in SUMOylation associate with T1DM susceptibility. We find that SUMOylation of NF-κB essential molecule NEMO augments NF-κB activity, NF-κB-dependent cytokine production and pancreatic inflammation. NF-κB inhibitor should provide therapeutic approach to block PAT inflammation and ameliorate the T1DM phenotype. We further propose that adipocytes in PATs may play a primary role in establishing pancreatic immune regulation at onset of diabetes, providing new insights into the molecular pathogenesis of type 1 diabetes.
Diabetes and its complications are a growing concern, given the increases in its worldwide prevalence. The distinct etiology of the two types of diabetes suggests that different causal mechanisms are involved. Type 1 diabetes (T1DM) arises from the autoimmune destruction of the pancreatic islet β cells, leading to an absolute lack of insulin.Citation1 In contrast, Diabetes mellitus type 2 (T2DM) is a metabolic disorder that is characterized by hyperglycemia in the context of insulin resistance and relative lack of insulin.Citation2 Inflammatory processes are implicated in the onset of diabetes and the progression of complications.Citation3 Adiposity, and in particular the release of inflammatory cytokines and other mediators from adipose cells, is associated with diabetes.Citation4,5 Most research focus on adipocyte function and type 2 diabetes. However, whether adipocyte inflammation plays a role in T1DM is still unclear.
Role of adipocytes in type 1 diabetes
Adipose tissue in glucose metabolism, lipodystrophy and insulin resistance are well known.Citation4,6 Recent studies indicate that adipose tissue is not simply the organ that stores fat and regulates lipid metabolism, but also is the largest endocrine organ with immune functions.Citation7 Adipocytes produce several mediators, such as adiponectin, resistin, IL-6, TNF-α, leptin, monocyte chemotactic protein-1 (MCP-1 or CCL2), and IL-1β.Citation8 Cytokines appear to be major regulators of adipose tissue metabolism. Therapeutic modulation of cytokine systems offers the possibility of major changes in adipose tissue behavior. Gene expression profiling studies show that adipocytes can synthesize both tumor necrosis factor α (TNF-α) and several interleukins (ILs), notably IL-1beta and IL-6. Other adipocyte products with ‘immunological’ actions include complement system products and macrophage colony-stimulating factor.
In this regard, emerging evidence suggests that separated adipose tissue depots are originated from distinct precursor cells and are functionally diverse.Citation9,10 Pancreatic adipose tissue (PAT) has been traditionally classified as WAT. However, it is now accepted that PAT is more harmful and associated with diabetic because of its greater inflammatory characteristics and high expression brown marker such as UCP-1.Citation11-13 PAT adipocytes exert both systemic and local (paracrine) effects on pancreases function and insulin sensitivity. Our results suggest that SENP1 deletion in the PAT adipocytes induces a reduced state of adipocytes differentiation, and a remarkably higher level of proinflammatory cytokine expression prior to the onset of immune cell activation in the pancreases and diabetic phenotype, as compared with adipocytes from other regional depots. This observation is consistent with the report that a low degree of adipocyte differentiation is associated with a high level of cytokine production.Citation14 Moreover, we observe that mouse PAT is strikingly responsive to high-fat feeding. Thus, by virtue of their unique functional and biochemical properties, we propose that adipocytes in PATs may play a primary role in establishing pancreases immune regulation in diabetes; proinflammatory cytokines overproduction from PATs in early phase of disease could serve as a main driver for immunologic tolerance break and immune cell expansion. Indeed, adipocyte-specific SENP1-deficient mice spontaneously develop TIDM. Pancreases in these mice exhibit increased NRP-V7 autoantigen specific CD8+ T cells, targeting a peptide from islet-specific glucose-6-phosphatase catalytic subunit-related protein.Citation15 Autoantibodies, mainly insulin autoantibody (IAA), along with other clinical information, aid in the diagnosis of T1DM.Citation16,17 CRP and β-hydroxybutyrate are important clinical parameters for the diagnosis of diabetes.Citation18 Our work indicates that SENP1-deficient mice have increased levels of IAA, CRP and β-hydroxybutyrate in the blood. Our observations suggest that SENP1-deficient adipocytes may gain immune cell-like functionCitation19 and this needs to be careful examined in the future.
Proinflammatory cytokines alter immune balance in pancreases
How proinflammatory cytokine original from PATs break immune tolerance in pancreases? First, proinflammatory cytokines have direct effects on islet immunogenicity and islet β-cell survival and autoantigens are presented in the context of a pro-inflammatory environment. Culture supernatant from adipocytes of SENP1-dificient mice strongly induces β-cell death and pancreatic disruption. We also observe massive apoptosis of β cells in our mouse model, which accompanying with infiltrations of CD8+ and CD4+ T cells. Indeed, we detect IAA in the SENP1-aP2KO mice at old ages, indicative of autoimmune disease T1DM in these mice. Moreover, islet-specific autoimmune NRP-V7-positive CD8+ T cells could be detected in the pancreatic lymph nodes and the spleen of SENP1-deficient mice. In other diabetic models, dead β-cells could be detected in pancreatic lymph nodes from where isolated dendritic cells could stimulate the TCR transgenic diabetogenic T-cell, suggesting that a role of β-cell death in the priming of autoreactive T-cells.Citation20 β-Cell injury and death induced by proinflammatory cytokines may also facilitate the spreading of specificities of pathogenic autoimmune cells during progression of disease by exposing new epitopes.Citation21 Second, chemokines are induced in islet cells in response to proinflammatory stimuli. It is reported that CCL5 binds to the chemokine receptors CCR1/3/5 may contribute to type 1 diabetic development. CCL5 receptor (CCR5) expression has also been detected in NOD mouse spleen and islet lymphocytes.Citation22 Our results indicate that proinflammatory cytokine from adipocytes significant up-regulation CCL5 expression in islets. We show that the high concentrations of PAT-derived cytokines directly induce expression of CCL5 and other chemokines (such as CCL2, CCL21, CXCL9 and CXCL10) in adjacent pancreatic islets, chemokines responsible for recruitment of immune cells. The number of activated DCs, T and B cell are increased in the lymph node of pancreases in SENP1-KO mice at the onset of T1DM. It needs to further investigate if and how CCL5 and other chemokines directly contribute to T1DM progression. Third, proinflammatory cytokines have direct effects on various immune cell types, particularly T cells. These immune cells further release more innate inflammatory cytokines, which damage β cells and break tolerance through activation of adaptive responses. It has been reported that many cytokines act directly on regulatory T cell, influencing their differentiation and stability and function. In the mouse, persistent inflammation leads to Foxp3 instability, loss of suppression and acquisition the functions of IFNγ producing Th1 cells. IL-6 and IL-1β together also lead islet autoantigen-specific regulatory T cell lose suppressor function and become inflammatory Th17 cells.Citation23 We observe that Th1 and Th17 effector T-cell subsets expanded and regulatory T cell number decreased by proinflammatory cytokine environment in the pancreases in SENP1-deficient mice (). Taken together, our study provides a strong mechanistic link between adipose secreted proinflammatory cytokines and T1DM. It will be important to test whether blockade of proinflammatory cytokines or cytotoxic T cells could ameliorate T1DM pathogenesis development in the SENP1-deficient mouse model.
Role of SUMOylation in adipocyte regulating type 1 diabetes
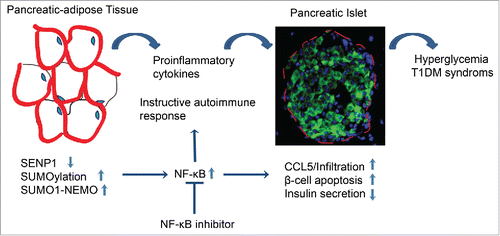
SUMOylation is a dynamic process that is mediated by activating, conjugating, and ligating enzymes and is readily reversed by a family of deSUMOylating proteases SENPs.Citation24,25 As a post-translational modification, SUMOylation is involved in various cellular processes, SUMOylation of proteins by the small ubiquitin-like modifier (SUMO) has also emerged as an important mechanism contributing to the dynamic regulation of protein function. SUMOylation has been linked to the pathogenesis of a variety of disorders including Alzheimer disease (AD), Huntington'sdisease (HD), and type 1 diabetes.Citation26 Advances in our understanding of the role of SUMOylation suggested a novel regulatory mechanism for the regulation of immune responsive gene expression. Interestingly, several components in SUMOylation have been identified as candidate genes implicated in T1DM susceptibility.Citation27,28 SUMO4, in the IDDM5 interval on chromosome 6q25, and present strong genetic and functional evidence as a T1DM susceptibility gene.Citation29 The expression of cytokine and other target genes that participate in host immune responses is controlled by transcriptional regulators of the NF-κB family.Citation30 It is reported several component of NF-κB are regulated by SUMOylation.Citation31 SUMO1 and SUMO4 are able to modify IκBα to become resistant to proteasome-mediated ubiquitylation and thus to inhibit NF-κB activity.Citation26 It has been shown that only SENP1 and SENP2, but not other SENPs, can efficiently deSUMOylate the NF-κB essential molecule, NEMO, and enhance NF-κB activation induced by DNA damage in a HEK 293T overexpression system.Citation32,33 Our data indicate that SENP1 expression is six-fold higher than SENP2 in mouse adipose tissues. Mechanistic studies show that SENP1 deletion in adipocytes (SENP1-apKO) enhances SUMOylation of NEMO at lysine 277/309, leading to increased NF-κB activity, cytokine production and pancreatic inflammation.Citation19 Although a genetic mutation of SENP1 has not been identified thus far, our study suggests that reduced SENP1 expression and enhanced NF-κB activity in adipose tissues may represent a common mechanism for the role of SUMOylayion in the pathogenesis of T1DM (). Of clinical significance of our current work, we observe an age-dependent reduction of SENP1 expression in PATs of the diabetic NOD mice, correlating with the diabetic progression in these mice.
NF-KB inhibitor to treat diabetes
The release of inflammatory cytokines and other mediators from adipose cells is associated with the development of diabetes, through activation of the NF-κB pathway. Importantly, the inhibitor of NF-κB p65 nuclear translocation JSH-23 could reduce cytokine levels and ameliorate the type 1 diabetes phenotypes. It worth mentioning that type-1 and type-2 diabetes are systemically linked. Our data demonstrate that SENP1-deficient mice have strong T1DM phenotypes concomitant with mild T2DM, which can be exacerbated by high-fat diet (HFD). Specifically, HFD augments PAT morphological change, cytokine production and pancreatic damage in SENP1 deficient mice, consistent with the effects of HFD on observed NF-κB and cytokine signaling in adipocytes. It is interesting to know whether the HFD alters beige precursor phenotype and if this alteration plays a key role in the diabetic phenotype of obesity mice. If so, inhibiting the inflammatory aspects of beige precursors with NF-κB inhibitor can protect obesity people from diabetes phenotype.Citation34-36
In summary, our studies have defined the role of the SENP1-mediated protein SUMOylation in pancreatic immune responses, β-cell damages and consequently diabetes progression. Our studies provide therapeutic approaches by delivery of NEMO inhibitory peptides or NF-κB inhibitors for treatment of type 1 diabetes. Further studies need to examine pancreatic adipocyte immune function and unique differentiation pattern of PAT adipocytes in the SNEP1-KO mice. The broad implications of understanding SENP1-mediated SUMOylation in pancreatic adipocytes may provide new insights into the molecular pathogenesis of type 1 diabetes.
Figure 1. SENP1 deletion augments PAT inflammation and islet immunogenicity. PATs from SENP1-aP2KO mice expressed high levels of IL-6, TNF-a and IL-1β. Proinflammatory cytokines have direct effects on islet immunogenicity and islet β-cell survival. We show that the high concentrations of PAT-derived cytokines directly induce expression of CCL5 and other chemokines (such as CCL2, CCL21, CXCL9 and CXCL10) in adjacent pancreatic islets, chemokines responsible for recruitment of immune cells. Second, proinflammatory cytokines have direct effects on various immune cell types, specifically T cells. These immune cells further release more innate inflammatory cytokines, which damage β cells and break tolerance through activation of adaptive responses. This is a Th1 and Th17 effector T-cell subsets expanded and concomitant with reduction of regulatory T-cell subset in the pancreatic lymph nodes of SENP1deficient mice. Third, the inflammatory cytokines have direct cytotoxic effects on islet β cells. proinflammatory cytokines and cytotoxic T cells triggers β-cell turnover, resulting in the release of autoantigens and endogenous ‘danger signals’ capable of promoting pathologic self-antigen presentation, suggesting a strong mechanistic link between adipose secreted proinflammatory cytokines and T1DM.
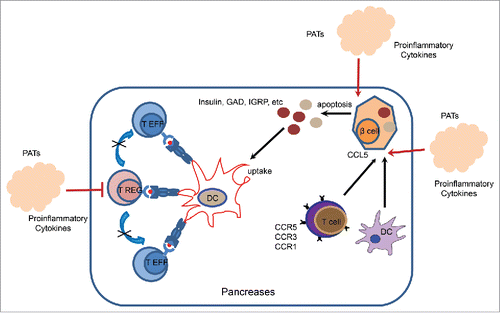
Figure 2. Model for the role of SENP1 in T1DM. SENP1 keeps NF-κB and inflammation in quiescent in adipose tissues. Here we show that adipocyte specific SENP1 deletion induces NEMO SUMOylation, NF-κB activation and NF-κB-dependent proinflammatory cytokine production in adipose tissues, profoundly in the peri-pancreatic adipose tissue (PAT). These cytokines induce high levels of chemokine CCL5 expression in adjacent islets to recruit CCR5+ immune cells. Subsequently, the cytokines and activated immune cells, especially CD8+ cells, attack the pancreases, leading to the chronic destruction of the islet structures, damaged β cells, autoantibody onset and type-1 diabetes progression in the SENP1-deficient mice. Therefore, NF-κB inhibitors block inflammation and ameliorate diabetes progression in the SENP1-deficient mice. Our current study demonstrates that reduced SENP1 expression and enhanced NF-κB activity in PATs may represent a common mechanism for the role of protein SUMOylayion in the pathogenesis of T1DM.
Reference
- Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol 2010; 10:501-13; PMID:20577267; http://dx.doi.org/10.1038/nri2787
- American Diabetes, A. Diagnosis and classification of diabetes mellitus. Diabetes Care 2011; 34 Suppl 1:S62-69; PMID:21193628
- Navarro-Gonzalez JF, Mora-Fernandez C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol 2008; 19:433-42; PMID:18256353; http://dx.doi.org/10.1681/ASN.2007091048
- Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 2006; 6:772-83; PMID:16998510; http://dx.doi.org/10.1038/nri1937
- Greenberg AS, Obin MS. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr 2006; 83:461S-465S; PMID:16470013
- Odegaard JI, Chawla A. Connecting type 1 and type 2 diabetes through innate immunity. Cold Spring Harbor Perspectives Med 2012; 2:a007724; http://dx.doi.org/10.1101/cshperspect.a007724
- Rocha VZ, Folco EJ. Inflammatory concepts of obesity. Int J Inflam 2011; 2011:529061; PMID:21837268; http://dx.doi.org/10.4061/2011/529061
- Chandran M, Phillips SA, Ciaraldi T, Henry RR. Adiponectin: more than just another fat cell hormone? Diabetes Care 2003; 26:2442-50; PMID:12882876; http://dx.doi.org/10.2337/diacare.26.8.2442
- Gesta S, Blüher M, Yamamoto Y, Norris AW, Berndt J, Kralisch S, Boucher J, Lewis C, Kahn CR. Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc Natl Acad Sci U S A 2006; 103:6676-81; PMID:16617105; http://dx.doi.org/10.1073/pnas.0601752103
- Tchkonia T, Lenburg M, Thomou T, Giorgadze N, Frampton G, Pirtskhalava T, Cartwright A, Cartwright M, Flanagan J, Karagiannides I, et al. Identification of depot-specific human fat cell progenitors through distinct expression profiles and developmental gene patterns. Am J Physiol Endocrinol Metab 2007; 292:E298-307; PMID:16985259; http://dx.doi.org/10.1152/ajpendo.00202.2006
- Franco-Pons N, Gea-Sorli S, Closa D. Release of inflammatory mediators by adipose tissue during acute pancreatitis. J Pathol 2010; 221:175-82; PMID:20217859; http://dx.doi.org/10.1002/path.2691
- Heni M, Machann J, Staiger H, Schwenzer NF, Peter A, Schick F, Claussen CD, Stefan N, Häring HU, Fritsche A. Pancreatic fat is negatively associated with insulin secretion in individuals with impaired fasting glucose and/or impaired glucose tolerance: a nuclear magnetic resonance study. Diabetes/Metab Res Rev 2010; 26:200-5; http://dx.doi.org/10.1002/dmrr.1073
- Rippe C, Berger K, Mei J, Lowe ME, Erlanson-Albertsson C. Effect of long-term high-fat feeding on the expression of pancreatic lipases and adipose tissue uncoupling proteins in mice. Pancreas 2003; 26:e36-42; PMID:12604926; http://dx.doi.org/10.1097/00006676-200303000-00024
- Chatterjee TK, Stoll LL, Denning GM, Harrelson A, Blomkalns AL, Idelman G, Rothenberg FG, Neltner B, Romig-Martin SA, Dickson EW, et al. Proinflammatory phenotype of perivascular adipocytes: influence of high-fat feeding. Circ Res 2009; 104:541-9; PMID:19122178; http://dx.doi.org/10.1161/CIRCRESAHA.108.182998
- Wang J, Tsai S, Shameli A, Yamanouchi J, Alkemade G, Santamaria P. In situ recognition of autoantigen as an essential gatekeeper in autoimmune CD8+ T cell inflammation. Proc Natl Acad Sci U S A 2010; 107:9317-22; PMID:20439719; http://dx.doi.org/10.1073/pnas.0913835107
- Pihoker C, Gilliam LK, Hampe CS, Lernmark A. Autoantibodies in diabetes. Diabetes 2005; 54 Suppl 2:S52-61; PMID:16306341; http://dx.doi.org/10.2337/diabetes.54.suppl_2.S52
- van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev 2011; 91:79-118; PMID:21248163; http://dx.doi.org/10.1152/physrev.00003.2010
- Hu FB, Meigs JB, Li TY, Rifai N, Manson JE. Inflammatory markers and risk of developing type 2 diabetes in women. Diabetes 2004; 53:693-700; PMID:14988254; http://dx.doi.org/10.2337/diabetes.53.3.693
- Shao L, Zhou HJ, Zhang H, Qin L, Hwa J, Yun Z, Ji W, Min W. SENP1-mediated NEMO deSUMOylation in adipocytes limits inflammatory responses and type-1 diabetes progression. Nat Commun 2015; 6:8917; PMID:26596471; http://dx.doi.org/10.1038/ncomms9917
- Hoglund P, Mintern J, Waltzinger C, Heath W, Benoist C, Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J Exp Med 1999; 189:331-9; PMID:9892615; http://dx.doi.org/10.1084/jem.189.2.331
- Akirav E, Kushner JA, Herold KC. Beta-cell mass and type 1 diabetes: going, going, gone? Diabetes 2008; 57:2883-8; PMID:18971435; http://dx.doi.org/10.2337/db07-1817
- Carvalho-Pinto C, García MI, Gómez L, Ballesteros A, Zaballos A, Flores JM, Mellado M, Rodríguez-Frade JM, Balomenos D, Martínez AC. Leukocyte attraction through the CCR5 receptor controls progress from insulitis to diabetes in non-obese diabetic mice. Eur J Immunol 2004; 34:548-57; PMID:14768060; http://dx.doi.org/10.1002/eji.200324285
- Zhang Y, Bandala-Sanchez E, Harrison LC. Revisiting regulatory T cells in type 1 diabetes. Curr Opin Endocrinol Diabetes Obesity 2012; 19:271-8; http://dx.doi.org/10.1097/MED.0b013e328355a2d5
- Li SJ, Hochstrasser M. A new protease required for cell-cycle progression in yeast. Nature 1999; 398:246-51; PMID:10094048; http://dx.doi.org/10.1038/18457
- Gill G. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev 2004; 18:2046-59; PMID:15342487; http://dx.doi.org/10.1101/gad.1214604
- Li M, Guo D, Isales CM, Eizirik DL, Atkinson M, She JX, Wang CY. SUMO wrestling with type 1 diabetes. J Mol Med (Berl) 2005; 83:504-13; PMID:15806321; http://dx.doi.org/10.1007/s00109-005-0645-5
- Guo D, Li M, Zhang Y, Yang P, Eckenrode S, Hopkins D, Zheng W, Purohit S, Podolsky RH, Muir A, et al. A functional variant of SUMO4, a new I kappa B α modifier, is associated with type 1 diabetes. Nat Genet 2004; 36:837-41; PMID:15247916; http://dx.doi.org/10.1038/ng1391
- Aribi M. Candidate genes implicated in type 1 diabetes susceptibility. Curr Diabetes Rev 2008; 4:110-21; PMID:18473758; http://dx.doi.org/10.2174/157339908784220723
- Wang CY, Podolsky R, She JX. Genetic and functional evidence supporting SUMO4 as a type 1 diabetes susceptibility gene. Ann N Y Acad Sci 2006; 1079:257-67; PMID:17130563; http://dx.doi.org/10.1196/annals.1375.039
- Hayashi T, Faustman D. NOD mice are defective in proteasome production and activation of NF-kappaB. Mol Cell Biol 1999; 19:8646-59; PMID:10567588; http://dx.doi.org/10.1128/MCB.19.12.8646
- Mabb AM, Miyamoto S. SUMO and NF-kappaB ties. Cell Mol Life Sci 2007; 64:1979-96; PMID:17530464; http://dx.doi.org/10.1007/s00018-007-7005-2
- Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell 2003; 115:565-76; PMID:14651848; http://dx.doi.org/10.1016/S0092-8674(03)00895-X
- Lee MH, Mabb AM, Gill GB, Yeh ET, Miyamoto S. NF-kappaB induction of the SUMO protease SENP2: A negative feedback loop to attenuate cell survival response to genotoxic stress. Mol Cell 2011; 43:180-91; PMID:21777808; http://dx.doi.org/10.1016/j.molcel.2011.06.017
- Qiao Z, Wang W, Wang L, Wen D, Zhao Y, Wang Q, Meng Q, Chen G, Wu Y, Zhou H. Design, synthesis, and biological evaluation of benzodiazepine-based SUMO-specific protease 1 inhibitors. Bioorg Med Chem Lett 2011; 21:6389-92; PMID:21930380; http://dx.doi.org/10.1016/j.bmcl.2011.08.101
- Chen X, Lu J, Bao J, Guo J, Shi J, Wang Y. Adiponectin: A biomarker for rheumatoid arthritis? Cytokine Growth Factor Rev 2012; 24(1):83-9; PMID:22910140; http://dx.doi.org/10.1016/j.cytogfr.2012.07.004
- Madu IG, Chen Y. Assays for investigating deSUMOylation enzymes. Curr Protoc Mol Biol 2012; Chapter 10, Unit10. 30: 1-13; PMID:22870856