
ABSTRACT
Obesity-associated low-grade inflammation underlies insulin resistance and associated metabolic comorbidities, such as type 2 diabetes (T2D) and nonalcoholic fatty liver disease. Excessive ectopic fat deposition in obesity causes disorders of energy homeostasis and low-grade chronic inflammation in metabolic tissues. In particular, obesity-induced recruitment and activation of adipose tissue macrophages play a key role in the pathogenesis of insulin resistance and T2D. Therefore, treatment options for energy metabolism and macrophage polarization in obese subjects are needed. Sodium-glucose cotransporter (SGLT) 2 inhibitors increase urinary glucose excretion by inhibiting renal glucose reabsorption, thereby having subsequent anti-hyperglycemic effects and reducing body weight. We recently reported that the SGLT2 inhibitor empagliflozin increases fat utilization and browning in white adipose tissue and attenuates obesity-induced inflammation and insulin resistance by activating M2 macrophages. Thus, this review focuses on the beneficial effects of empagliflozin in energy homeostasis and obesity-related inflammation and insulin resistance.
Safety and tolerability of the SGLT2 inhibitor empagliflozin
The sodium-glucose cotransporters (SGLTs) SGLT1 and SGLT2 are responsible for glucose reabsorption in the kidneys. SGLT1 is a high-affinity, low-capacity transporter that is expressed in the distal proximal convoluted tubule, where it acts as a transporter for dietary glucose and galactose, and accounts for approximately 10% of glucose reabsorption. By contrast, SGLT2 is a low-affinity, high-capacity transporter that is expressed exclusively in renal proximal tubules and reabsorbs 90% of the glucose from urine [Citation1]. Mutations in the gene encoding SGLT2 have been associated with familial renal glucosuria, and SGLT2-deficient mice show higher urinary glucose excretion (UGE) than wild-type mice [Citation2-Citation4]. These observations suggest that inhibiting SGLT2 function may be effective for treating hyperglycemia, obesity, and type 2 diabetes (T2D).
Data from clinical studies have demonstrated that oral administration of SGLT2 inhibitors induces UGE, improves hyperglycemia, and reduces the body weight of patients with T2D [Citation5-Citation7]. These SGLT2 inhibitors were developed based on the structure of phlorizin [Citation8]. Currently, several members of SGLT2 inhibitors are approved (empagliflozin, dapagliflozin, canagliflozin, etc.) and some others are in development (ipragliflozin, tofogliflozin, ertugliflozin etc.) [Citation8,Citation9]. Among of these SGLT2 inhibitors, empagliflozin is characterized by highly selective and potent inhibitor of SGLT2 (), and which had already approved in the EU and US in 2014 [Citation9,Citation10]. Linear pharmacokinetics indicate that the half-maximum inhibitory concentration (IC50) of empagliflozin is 3.1 nM (pIC50 ± S.E. 8.5 ± 0.02 nM), and its selectivity for SGLT2 is more than 2,500-fold and 5,800-fold higher than that for SGLT1 in humans and mice, respectively () [Citation11,Citation12]. The high selectivity of empagliflozin for SGLT2 suggests that the renal pharmacological response to empagliflozin treatment is mediated solely by SGLT2. Several studies have demonstrated that empagliflozin is safe for rodents and well tolerated [Citation13,Citation14]; doses of empagliflozin up to 800 mg/day do not cause clinically significant safety concerns in healthy male subjects [Citation15]. Taken together, these findings indicate that empagliflozin is a potent and competitive SGLT2 inhibitor with an excellent selectivity profile that has potential as a treatment for insulin resistance and T2D.
Table 1. Comparison of each SGLT2 inhibitor in the potency on SGLT2 and selectivity over SGLT1 in the kidney.
Macrophage polarization and insulin resistance
Obesity is characterized by excessive fat accumulation and is highly correlated with the incidence and prevalence of insulin resistance, T2D, and nonalcoholic fatty liver disease (NAFLD). Obesity is closely associated with low-level chronic inflammation, which is characterized by abnormal cytokine and chemokine production and activation of inflammatory pathways that interfere with insulin signaling (), including mitogen-activated protein kinases, IκB-kinase β (IκKβ)/nuclear factor κB (NF-κB), and mammalian target of rapamycin/S6 kinase [Citation16]. Although the mechanism of this inflammatory response remains unclear, increasing evidence reveals that obesity-induced inflammation is mediated primarily by immune cells, such as macrophages and T lymphocytes, in metabolic tissues [Citation17,Citation18]. Tissue macrophages are phenotypically heterogeneous and are characterized according to their activation/polarization state as M1 (classically activated, proinflammatory) or M2 (alternatively activated, anti-inflammatory) macrophages [Citation19]. M1/M2 polarization of macrophages is a highly dynamic process, and the phenotype of polarized macrophages can be reversed under certain physiological and pathological conditions (). These subsets can be triggered by in vitro incubation with interferon gamma, tumor necrosis factor (TNF)-α, and lipopolysaccharide (LPS) or interleukin-4 (IL-4), respectively [Citation20,Citation21]. The polarization of M1-type macrophages in obesity is enhanced, which leads to increased production of various proinflammatory cytokines, such as TNF-α and IL-6, which induce insulin resistance via IκKβ- and JNK-mediated inhibitory serine phosphorylation of insulin receptor substrate proteins. By contrast, M2-polarized macrophages generate anti-inflammatory cytokines, such as IL-10 and IL-1 receptor antagonist, which are suppressed in obese subjects [Citation17,Citation22].
Figure 1. Obesity-related macrophage polarization and insulin resistance. In a lean state, M2 macrophages are the primary resident macrophages and maintain insulin sensitivity. In contrast, excess calories or a sedentary lifestyle cause adipocyte hypertrophy, which initiates secretion of CCL2 and CCL5, leading to the recruitment of circulating monocytes in adipose tissues. Subsequently, CCR2+ macrophages accumulate and presumably maintain inflammation as M1 macrophages in obese adipose tissue. Once these ATMs are present and active, they maintain a vicious cycle involving ATM recruitment and the production of inflammatory cytokines, such as TNF-α, IL-6, and IL-1β, in conjunction with adipocytes and other infiltrated immune cells. These secreted proinflammatory cytokines subsequently cause inflammation and insulin resistance in adipose tissue, liver, and skeletal muscle.
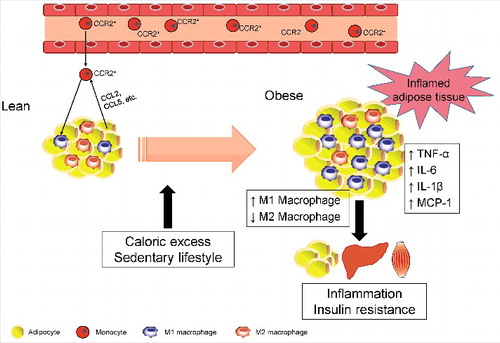
Emerging lines of evidence show that adipose tissue macrophages (ATMs) release proinflammatory cytokines similar to classically activated M1-type macrophages that directly contribute to insulin resistance or T2D [Citation23]. A study by Hotamisligil et al. identified adipocytes as a source of TNF-α in white adipose tissue (WAT) that ultimately impairs insulin signaling in obesity [Citation24]. Moreover, findings by Xu et al. demonstrated that mainly the stromal vascular fraction of obese WAT expresses inflammatory cytokines [Citation25]. Adipose tissue in lean mice is populated with M2 ATMs and governs adipocyte lipid metabolism by secreting factors such as IL-10 and catecholamines. The M2 ATMs cooperate with regulatory T cells and innate type 2 lymphoid cells to maintain the anti-inflammatory WAT environment [Citation26,Citation27]. During these processes, anti-inflammatory cytokines, such as IL-4, IL-13, and IL-33, in lean adipose tissue assist the ATMs in an anti-inflammatory state and restrain the progression of insulin resistance. ATM polarization in the obese state is shifted toward the proinflammatory M1 macrophage phenotype that expresses the surface marker CD11c () [Citation17,Citation19]. Activation and accumulation of M1 ATMs in obese WAT can be caused by oxidative stress that increases a number of free fatty acids (FFAs) and LPS [Citation28]. Subsequently, expression of proinflammatory cytokines, such as TNF-α, IL-6, and IL-1β, in ATMs compromises insulin action not only locally in WAT but also systemically as these cytokines are released into circulation (). Thus, inflammation triggered by ATMs constitutes a turning point in the development of obesity-related insulin resistance and T2D.
Kupffer cells (KCs) are resident hepatic macrophages that play central roles in liver injury, such as nonalcoholic steatohepatitis (NASH) [Citation29]. In obesity, excessive fat storage in WAT leads to hepatic ectopic lipid accumulation, resulting in NAFLD and fatty liver diseases. Ectopic fat storage in the liver results in hepatic lipotoxicity, which in turn leads to liver damage and inflammation [Citation30]. The dynamic polarization of KCs determines the pro- or anti-inflammatory conditions in the liver. KCs in normal conditions exhibit an M2-like phenotype and express several receptors such as toll-like receptors (TLRs). In the presence of TLR ligands, KCs become immunogenic and can induce T cell activation and the generation of an efficient cytotoxic T-lymphocytes response [Citation31]. However, obesity-induced proinflammatory cytokines, such as TNF-α, and chemokines, such as monocyte chemoattractant protein-1 and regulated on activation, normal T cell expressed and secreted (RANTES/CCL5), polarize KCs toward the M1 state, which in turn induces insulin resistance in the liver and weakens liver function. Inflammation driven by M1 KCs is counterbalanced by alternatively polarized M2 macrophages that promote resolution of inflammation and tissue repair [Citation32]. The beneficial properties of the alternative M2 KCs have been reported in several inflammatory disorders, including insulin resistance, T2D, and NAFLD [Citation33,Citation34].
Empagliflozin improves insulin resistance by regulating both macrophage recruitment and polarization
Obesity, insulin resistance, and other metabolic disorders are closely associated with chronic inflammation characterized by abnormal cytokine production, increased acute-phase reactants and other mediators, and activation of a network of inflammatory signaling pathways [Citation16]. More than a decade ago, it was reported that TNF-α is overexpressed in the adipose tissue of obese mice; this provided the first clear link between obesity, diabetes, and chronic inflammation [Citation24]. Not only TNF-α but other inflammatory mediators and cytokines are overexpressed in adipose and other tissues in experimental mouse models of obesity and in humans [Citation28]. A lack of TNF-α results in notably improved insulin sensitivity in DIO and ob/ob mice [Citation35], which confirms that inflammation in obesity has a critical role in impairing the physiological response to insulin. SGLT2 inhibitors (dapagliflozin and ipragliflozin) improve inflammation in the kidneys and liver of diabetic mice [Citation36,Citation37]. Empagliflozin also markedly decreases obesity-induced inflammation in the liver and WAT of diet-induced obese (DIO) mice [Citation38]. These findings suggest that SGLT2 inhibitors, particularly empagliflozin, improve insulin resistance partially by attenuating chronic inflammation in obese and diabetic subjects.
Obesity or ectopic fat induces an innate immune response with subsequent recruitment of immune cells, which leads to the development of insulin resistance and NASH. In particular, macrophage recruitment and polarization are pivotal in obesity-induced inflammation and insulin resistance [Citation32]. Thus, strategies to restrain M1 polarization and/or drive the alternative M2 activation of macrophages may have the potential to protect against exacerbated inflammation and insulin resistance and even attenuate progression to NASH. It is noteworthy that we used highly specific gating strategies to determine pure populations of ATMs and M1 and M2 ATMs. A flow cytometry analysis clearly demonstrated a decrease in M1 ATMs that is reciprocal to an increase in M2 ATMs in empagliflozin-treated DIO mice [Citation38]. Moreover, infiltration of Th1 and CD8+ T cells precedes M1-polarized macrophage recruitment, and interactions between T cells and macrophages constitute a maladaptive feed-forward loop, leading to adipocyte inflammation and insulin resistance. Consequently, empagliflozin reduces the accumulation of T cells and M1 macrophages and increases M2 macrophages to alleviate inflammation and insulin resistance in obesity. Thus, empagliflozin attenuates obesity-associated insulin resistance by polarizing M2 ATMs and decreasing inflammation in DIO mice.
Empagliflozin decreases adiposity by shifting fuel selection and promoting fatty acid oxidation
Obesity has been defined as abnormal or excessive fat accumulation in adipocytes that presents a risk to health. Triglycerides are the main form of fat storage in adipose tissue resulting in adiposity. The release of excess FFAs from the lipolysis of visceral adipose tissue into the circulation or portal vein destroys the functions of other organs, such as the liver, heart, and kidneys [Citation39]. Therefore, attenuating the accumulation of triglycerides or enhancing fat utilization in adipose tissue is the main method of treating obesity.
Several studies have shown the protective effects of SGLT2 inhibitors against obesity in rodents. Rats pair-fed with tofogliflozin for 8 weeks show suppressed high-fat diet (HFD)-induced weight gain and hepatic steatosis [Citation40]. Chronic administration of dapagliflozin for 35 days significantly reduces body weight and enhances lipid lipolysis [Citation41]. By contrast, therapeutic treatment with remogliflozin for 4 weeks attenuates hepatic steatosis without affecting weight gain [Citation42]. In addition, luseogliflozin decreases liver weight and ameliorates steatohepatitis in streptozotocin-treated mice fed an HFD without altering weight gain [Citation43]. These observations suggest that the timing of the administration of SGLT2 inhibitors and the mouse model can affect body weight gain. Moreover, in clinical studies, body weight reductions are observed after the administration of SGLT2 inhibitors [Citation9]. Paradoxically, SGLT2 inhibitors can augment energy intake in rodents, which counteracts the beneficial effect on body weight reduction [Citation40,Citation41]. Therefore, we pair-fed an HFD and an HFD with empagliflozin to exclude the influence of increased food intake. We obtained empagliflozin-reduced adiposity despite pair-feeding, which suggests that preventing obesity and its comorbidities is not simply secondary to calorie loss because of glucosuria or reduced calorie intake [Citation38].
Consistent with other SGLT2 inhibitors, administering empagliflozin to HFD-induced obese mice mitigates weight gain and fatty liver. The underlying mechanism for the weight reduction depends partially on increased energy expenditure and enhanced fatty acid oxidation. Empagliflozin increases oxygen consumption and tends to elevate carbon dioxide exhalation, leading to increased sugar and fat utilization [Citation38]. It is important to note that in clinical trials, a small increase in plasma low-density lipoprotein cholesterol (LDL-C) has been reported with SGLT2 inhibitors [Citation9]. Empagliflozin increases the plasma LDL-C level concomitant with higher FFAs and total ketone body levels, which suggests that inhibiting SGLT2 induces ketogenesis and a metabolic switch toward lipid oxidation to counterbalance the carbohydrate restriction [Citation44]. Chronic administration of empagliflozin to patients drives a fuel shift to fat utilization accompanied by decreased tissue glucose disposal and increased lipid use [Citation45]. These findings suggest that empagliflozin suppresses weight gain by shifting energy metabolism toward fat and sugar utilization. A study by Hawley et al. demonstrated that SGLT2 inhibitors promote fatty acid oxidation by activating AMP-activated protein kinase (AMPK)-α in vitro and lowering liver lipid content [Citation46]. Our findings revealed that empagliflozin increases the phosphorylation of AMPK and acetyl-CoA carboxylase (ACC) in the skeletal muscle of DIO mice [Citation38]. These results suggest that empagliflozin enhances fatty acid oxidation partially by activating the AMPK pathway.
Administering empagliflozin increases fatty acid oxidation by altering the expression of adiponectin and leptin in epididymal WAT. The adipose tissue-specific adipokines leptin and adiponectin are involved in the regulation of food intake and energy homeostasis [Citation47]. Plasma leptin levels increase during the development of obesity and decline during weight loss. Leptin stimulates fatty acid esterification to triglycerides and causes an even greater increase in hydrolysis so that there is a net efflux of fatty acids from the cells. By contrast, adiponectin exerts its insulin-sensitizing effects by increasing β-oxidation of fatty acids and reducing serum triglyceride and FFA levels, thus indirectly improving insulin sensitivity. Furthermore, leptin and adiponectin interact with AMPK, which regulates fatty acid and energy metabolism. Administering empagliflozin increases adiponectin mRNA expression and downregulates leptin expression in epididymal WAT and contributes to fat lipolysis and energy expenditure. Taken together, these findings indicate that empagliflozin improves abnormal lipid metabolism and obesity by enhancing fat and sugar utilization and increasing fatty acid oxidation.
Empagliflozin increases energy expenditure by promoting browning in white adipose tissue
Brown adipose tissue (BAT) constitutes a metabolically active tissue responsible for non-shivering thermogenesis and depletion of excess calories. Brown adipocytes produce heat along with increasing the expression of uncoupling proteins (UCPs) by utilizing fatty acids. Among UCPs, UCP1 is the major isoform expressed in BAT, which is regulated by the transcription factor peroxisome proliferator-activated receptor-gamma coactivator 1α [Citation48]. Several studies have revealed that certain depots of WAT take on a BAT phenotype when subjected to certain stimuli: Brown-like adipocytes, also known as beige cells, express UCP1 and contribute to thermogenesis [Citation49,Citation50]. In response to physiological stimuli (such as chronic exposure to cold), hormonal stimuli (such as irisin), pharmacological treatment (such as peroxisome proliferator-activated receptor γ agonist or β-adrenergic stimulation), or a brown fat–like gene expression program (such as UCP1), cell death-inducing DFFA-like effector-a and diodinase 2 are induced in WAT [Citation51,Citation52]. Indeed, brown-like adipocytes have anti-obesity and antidiabetic effects in rodent models [Citation51]. Chronic treatment with empagliflozin increases whole-body energy expenditure, heat production, and the protein levels of UCP1 in both BAT and WAT, which suggests that empagliflozin promotes adipose tissue browning [Citation38]. Some studies have revealed that M2-type macrophages promote browning of WAT by activating type 2 cytokines production during exposure to cold [Citation53,Citation54]. In cold conditions, various type 2 cytokines released from M2 macrophages activate β-adrenergic receptors in adipocytes to turn on the thermogenic program, including induction of UCP1. In addition, adiponectin plays a role in SGLT2 pathway and promoting beige adipocytes [Citation55,Citation56]. Zhao et al. showed elevation of adiponectin downregulates the renal SGLT2 by activating PPARδ, which in turn reduces reabsorption of sodium and glucose [Citation56]. On the other hand, inhibition of SGLT2 by SGLT2 inhibitors increases the expression of adiponectin in diabetic subjects and obese model [Citation38,Citation57]. Thus, empagliflozin promotes browning in WAT, at least in part, by polarizing M2 ATMs and increasing adiponectin expression in WAT. Fibroblast growth factor 21 (FGF21) is a central mediator of fatty acid oxidation and lipid metabolism in WAT and the liver [Citation58,Citation59]. Pharmacological doses of FGF21 improve glucose tolerance, lower serum FFAs, and lead to weight loss in obese mice through increases in energy expenditure [Citation60]. Moreover, FGF21 also activates the β3-adrenergic receptor in WAT and regulates recruitment of beige adipocytes [Citation61], thereby leading to increased energy utilization and browning. Our previous study revealed that chronic treatment of empagliflozin increased the hepatic mRNA expression of FGF21 and plasma levels of FGF21 [Citation38]. These evidence suggest that FGF21 can mediate a shift of energy metabolism toward fat use in response to SGLT2 inhibition. Thus, an increase in FGF21 in the liver and circulation following the administration of empagliflozin may be another factor promoting fat utilization and browning in obesity.
Conclusions and perspectives
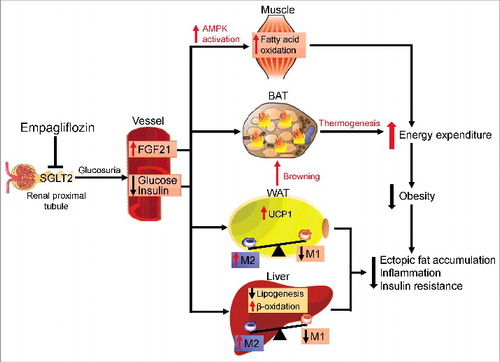
In conclusion, Xu et al. presented compelling evidence that empagliflozin plays a crucial role in obesity-induced adipose tissue inflammation and insulin resistance by regulating macrophage recruitment and M1/M2 status (). Note that Xu et al. demonstrated that empagliflozin acts against adiposity by promoting fat and sugar utilization and enhancing β-oxidation of FFAs. Moreover, they found increased energy expenditure in empagliflozin-treated DIO mice and enhanced expression of UCP1 and browning in WAT, which suggests that empagliflozin regulates the proton influx back into the mitochondrial matrix and dissipates oxidative energy as heat instead of synthesis of adenosine triphosphate (ATP) (). In light of these new data, SGLT2 inhibitors may be a promising treatment for insulin resistance, NAFLD, and T2D. However, the main limitation of this study is that the effects of empagliflozin were evaluated on a preventive, not a therapeutic, treatment schedule, which makes it difficult to translate the results to humans. Therapeutic studies will aid in the translation of experimental results regarding the anti-obesity effects of SGLT2 inhibitors to clinical settings.
Figure 2. Protective effects of empagliflozin in high-fat diet-induced obese mice. Inhibiting SGLT2 with empagliflozin directly decreases blood glucose levels, leading to the following: (1) Empagliflozin promotes fat utilization by enhancing AMPKα and ACC phosphorylation in skeletal muscle and increasing hepatic and plasma levels of FGF21. (2) Empagliflozin enhances browning and thermogenesis in WAT and BAT, which results in increased energy expenditure. (3) Empagliflozin improves insulin sensitivity by polarizing M2 macrophages in fat and liver.
Abbreviations
| ACC | = | Acetyl-CoA carboxylase |
| AMPK | = | AMP-activated protein kinase |
| ATMs | = | Adipose tissue macrophages |
| ATP | = | Adenosine triphosphate |
| BAT | = | Brown adipose tissue |
| DIO | = | Diet-induced obese |
| FFAs | = | Free fatty acids |
| FGF21 | = | Fibroblast growth factor 21 |
| HFD | = | High-fat diet |
| IL-4 | = | Interleukin-4 |
| LPS | = | Lipopolysaccharide |
| NAFLD | = | Non-alcoholic fatty liver disease |
| NASH | = | Non-alcoholic steatohepatitis |
| SGLT | = | Sodium/glucose cotransporter |
| T2D | = | Type 2 diabetes |
| TNF | = | Tumor necrosis factor |
| UCP | = | Uncoupling protein |
| UGE | = | Urinary glucose excretion |
| WAT | = | White adipose tissue |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author Contributions
All authors contributed to the drafting and writing of the present manuscript.
Acknowledgments
This study was supported by a Grant-in-Aid for Scientific Research (B; 25282017) and for Challenging Exploratory Research (15K12698) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and the Japan Diabetes Foundation (TO).
Funding
Ministry of Education, Culture, Sports, Science, and Technology (MEXT), 25282017.
References
- Cangoz S, Chang YY, Chempakaseril SJ, et al. et al. The kidney as a new target for antidiabetic drugs: SGLT2 inhibitors. J Clin Pharm Ther. 2013;38(5):350–9 doi:10.1111/jcpt.12077
- Van den Heuvel LP, Assink K, Willemsen M, et al. Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2). Hum Genet. 2002;111(6):544–7 doi:10.1007/s00439-002-0820-5
- Santer R, Calado J. Familial renal glucosuria and SGLT2: from a mendelian trait to a therapeutic target. Clin J Am Soc Nephrol. 2010;5(1):133–41 doi:10.2215/CJN.04010609
- Vallon V, Rose M, Gerasimova M, et al. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol. 2013;304(2):F156–67 doi:10.1152/ajprenal.00409.2012
- Ferrannini E, Seman L, Seewaldt-Becker E, et al. A Phase IIb, randomized, placebo-controlled study of the SGLT2 inhibitor empagliflozin in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15(8):721–8 doi:10.1111/dom.12081
- Rosenwasser RF, Rosenwasser JN, Sutton D, et al. Tofogliflozin: a highly selective SGLT2 inhibitor for the treatment of type 2 diabetes. Drugs Today (Barc). 2014;50(11):739–45 doi:10.1358/dot.2014.50112232267
- Polidori D, Mari A, Ferrannini E. Canagliflozin, a sodium glucose co-transporter 2 inhibitor, improves model-based indices of beta cell function in patients with type 2 diabetes. Diabetologia. 2014;57(5):891–901 doi:10.1007/s00125-014-3196-x
- Vivian EM. Sodium-glucose co-transporter 2 (SGLT2) inhibitors: a growing class of antidiabetic agents. Drugs Context. 2014;3:212264. doi:10.7573/dic.212264
- Nauck MA. Update on developments with SGLT2 inhibitors in the management of type 2 diabetes. Drug Des Devel Ther. 2014;8:1335–80 doi:10.2147/DDDT.S50773
- White JR, Jr.. Empagliflozin, an SGLT2 inhibitor for the treatment of type 2 diabetes mellitus: A review of the evidence. Ann Pharmacother. 2015; doi:10.1177/1060028015573564
- Roden M, Weng J, Eilbracht J, et al. Empagliflozin monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Diabetes & Endocrinology. 2013;1(3):208–219 doi:10.1016/s2213-8587(13)70084-6
- Grempler R, Thomas L, Eckhardt M, et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012;14(1):83–90 doi:10.1111/j.1463-1326.2011.01517.x
- Sarashina A, Koiwai K, Seman LJ, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of single doses of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in healthy Japanese subjects. Drug Metabolism and Pharmacokinetics. 2013;28(3):213–219 doi:10.2133/dmpk.DMPK-12-RG-082
- Scheen AJ. Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin Pharmacokinet. 2014;53(3):213–25 doi:10.1007/s40262-013-0126-x
- Seman L, Macha S, Nehmiz G, et al. Empagliflozin (BI 10773), a potent and selective SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clinical Pharmacology in Drug Development. 2013;2(2):152–161 doi:10.1002/cpdd.16
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–7 doi:10.1038/nature05485
- Kitade H, Sawamoto K, Nagashimada M, et al. et al. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes. 2012;61(7):1680–90 doi:10.2337/db11-1506/-/DC1
- Huh JY, Park YJ, Ham M, et al. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cells. 2014;37(5):365–71 doi:10.14348/molcells.2014.0074
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–84 doi:10.1172/JCI29881
- Dey A, Allen J, Hankey-Giblin PA. Ontogeny and polarization of macrophages in inflammation: blood monocytes versus tissue macrophages. Front Immunol. 2014;5:683. doi:10.3389/fimmu.2014.00683
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35 doi:10.1038/nri978
- Brown BN, Ratner BD, Goodman SB, et al. Macrophage polarization: an opportunity for improved outcomes in biomaterials and regenerative medicine. Biomaterials. 2012;33(15).3792–802 doi:10.1016/j.biomaterials.2012.02034
- Kraakman MJ, Murphy AJ, Jandeleit-Dahm K, et al. Macrophage polarization in obesity and type 2 diabetes: weighing down our understanding of macrophage function? Front Immunol. 2014;5:470. doi:10.3389/fimmu.2014.00470
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha_ direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91 doi:10.1126/science.7678183
- Xu H, Barnes GT, Yang Q, et al. et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–30 doi:10.1172/JCI19451
- Feuerer M, Herrero L, Cipolletta D, et al. et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–9 doi:10.1038/nm.2002
- Molofsky AB, Nussbaum JC, Liang HE, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210(3):535–49 doi:10.1084/jem.20121964
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–45 doi:10.1146/annurev-immunol-031210-101322
- Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. 2014;59(5):2034–42 doi:10.1002/hep.26754
- Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–46 doi:10.1146/annurev-physiol-021909-135846
- Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science. 2013;342(6161):1242974. doi:10.1126/science.1242974
- Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–95 doi:10.1172/JCI59643
- Jager J, Aparicio-Vergara M, Aouadi M. Liver innate immune cells and insulin resistance: the multiple facets of Kupffer cells. J Intern Med. 2016;280(2):209–20 doi:10.1111/joim.12483
- Xu L, Kitade H, Ni Y, et al. Roles of chemokines and chemokine receptors in obesity-associated insulin resistance and nonalcoholic fatty liver disease. Biomolecules. 2015;5(3):1563–79 doi:10.3390/biom5031563
- Uysal KT, Wiesbrock SM, Marino MW, et al. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389(6651):610–4 doi:10.1038/39335
- Tahara A, Kurosaki E, Yokono M, et al. et al. Effects of SGLT2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur J Pharmacol. 2013;715(1-3):246–55 doi:10.1016/j.ejphar.2013.05.014
- Terami N, Ogawa D, Tachibana H, et al. et al. Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS One. 2014;9(6):e100777. doi:10.1371/journal.pone.0100777
- Xu L, Nagata N, Nagashimada M, et al. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine. 2017;20:137–149 doi:10.1016/j.ebiom.2017.05.028
- Haslam DW, James WPT. Obesity. The Lancet. 2005;366(9492):1197–1209 doi:10.1016/s0140-6736(05)67483-1
- Obata A, Kubota N, Kubota T, et al. et al. Tofogliflozin improves insulin resistance in skeletal muscle and accelerates lipolysis in adipose tissue in male mice. Endocrinology. 2016;157(3):1029–42 doi:10.1210/en.2015-1588
- Devenny JJ, Godonis HE, Harvey SJ, et al. Weight loss induced by chronic dapagliflozin treatment is attenuated by compensatory hyperphagia in diet-induced obese (DIO) rats. Obesity (Silver Spring). 2012;20(8):1645–52 doi:10.1038/oby.2012.59
- Nakano S, Katsuno K, Isaji M, et al. Remogliflozin etabonate improves fatty liver disease in diet-induced obese male mice. J Clin Exp Hepatol. 2015;5(3):190–8 doi:10.1016/j.jceh.2015.02.005
- Qiang S, Nakatsu Y, Seno Y, et al. et al. Treatment with the SGLT2 inhibitor luseogliflozin improves nonalcoholic steatohepatitis in a rodent model with diabetes mellitus. Diabetol Metab Syndr. 2015;7:104. doi:10.1186/s13098-015-0102-8
- Briand F, Mayoux E, Brousseau E, et al. Empagliflozin, via switching metabolism toward lipid utilization, moderately increases LDL cholesterol levels through reduced LDL catabolism. Diabetes. 2016;65(7):2032–8 doi:10.2337/db16-0049
- Ferrannini E, Baldi S, Frascerra S, et al. Shift to fatty substrate utilization in response to sodium-glucose votransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes. 2016;65(5):1190–1195 doi:10.2337/db15-1356
- Hawley SA, Ford RJ, Smith BK, et al. The Na+/glucose cotransporter inhibitor canagliflozin activates AMPK by inhibiting mitochondrial function and increasing cellular AMP levels. Diabetes. 2016;65(9):2784–94 doi:10.2337/db16-0058
- Adya R, Tan BK, Randeva HS. Differential effects of leptin and adiponectin in endothelial angiogenesis. J Diabetes Res. 2015;2015:648239. doi:10.1155/2015/648239
- Lowell BB, Spiegelman BM. Towards a molecular understanding of adaptive thermogenesis. Nature. 2000;404(6778):652–60 doi:10.1038/35007527
- Cohen P, Levy JD, Zhang Y, et al. et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell. 2014;156(1-2):304–16 doi:10.1016/j.cell.2013.12.021
- Seale P, Bjork B, Yang W, et al. et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454(7207):961–7 doi:10.1038/nature07182
- Wu J, Cohen P, Spiegelman BM. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev. 2013;27(3):234–50 doi:10.1101/gad.211649.112
- Lo KA, Sun L. Turning WAT into BAT: a review on regulators controlling the browning of white adipocytes. Biosci Rep. 2013;33(5): doi:10.1042/BSR20130046
- Nguyen KD, Qiu Y, Cui X, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011;480(7375):104–8 doi:10.1038/nature10653
- Qiu Y, Nguyen KD, Odegaard JI, et al. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell. 2014;157(6):1292–308 doi:10.1016/j.cell.2014.03.066
- Hui X, Gu P, Zhang J, et al. et al. Adiponectin enhances cold-induced browning of subcutaneous adipose tissue via promoting M2 macrophage proliferation. Cell Metab. 2015;22(2):279–90 doi:10.1016/j.cmet.2015.06.004
- Zhao Y, Gao P, Sun F, et al. et al. Sodium intake regulates glucose homeostasis through the PPARdelta/adiponectin-mediated SGLT2 pathway. Cell Metab. 2016;23(4):699–711 doi:10.1016/j.cmet.2016.02.019
- Tanizawa Y, Araki E, Tobe K, et al. Efficacy and safety of tofogliflozin administered for 52 weeks as monotherapy or combined with other oral hypoglycaemic agents in Japanese patients with type 2 diabetes. Diabetologia. 2013;56:S82–S83
- Inagaki T, Dutchak P, Zhao G, et al. et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5(6):415–25 doi:10.1016/j.cmet.2007.05.003
- Badman MK, Pissios P, Kennedy AR, et al. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5(6):426–37 doi:10.1016/j.cmet.2007.05.002
- Kharitonenkov A, Shiyanova TL, Koester A, et al. et al. FGF21 as a novel metabolic regulator. J Clin Invest. 2005;115(6):1627–35 doi:10.1172/JCI23606
- Fisher FM, Kleiner S, Douris N, et al. et al. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012;26(3):271–81 doi:10.1101/gad.177857.111