
ABSTRACT
Toll-like receptor (TLR) 4 agonists have emerged as a new group of molecules used for cancer therapy. They have been exploited to enhance the immunogenicity of current chemotherapeutic regimens. However, their effects on cancer cells remain elusive. Here, we showed that a TLR4 agonist, namely a synthetic lipid A analog (ALA), OM-174, exhibits antitumor effects in several mammary tumor mouse models. We also showed that immune components are involved in such effects, as attested to by the failure of ALA to induce tumor regression or an increase of animal survival in mice knocked-out for interferon γ (IFNγ) or TLR4. TLR4 and IFNγ receptor (INFR2) expressed by cancer cells are involved in the antitumor efficacy of ALA since this last did not inhibit tumor growth in mice bearing a tumor but lacking TLR4 or IFNγ receptor 2 (IFNR2). Mechanistic investigations revealed that nitric oxide (NO), superoxide and peroxynitrite produced by uncoupling of inducible NO synthase (NOS II) in cancer cells are key mediators of ALA and IFNγ-mediated tumor growth inhibition. We present here a comprehensive picture of tumor cell death induction, in vivo and in vitro, by immunotherapy and for the first time the involvement of the TLR4/IFNγ/NOS II pathway in immunotherapy was investigated.
Introduction
TLRs are a family of evolutionarily conserved pattern recognition receptors. Originally identified as controllers of the dorsal–ventral embryonic polarity of Drosophila melanogaster,Citation1 orthologs have been characterized in humans Citation2 and implicated in innate immune responses.Citation3 To date, 10 TLRs have been identified in humans (TLR1-TLR10) and 13 in mice (Tlr1-tlr13).Citation4,5 These protein receptors are characterized by their ability to respond to invading pathogens by recognizing conserved molecular structures known as microbe-associated molecular patterns (MAMPs), including flagellin, bacterial lipopolysaccharide (LPS), nucleic acids derived from viruses and zymosan of fungi. Moreover, several TLRs recognize the so-called “damage-associated molecular patterns” (DAMPS), including heat shock proteins,Citation6 and the high-mobility group box 1 (HMGB1),Citation7 which both operate as TLR2/TLR4 agonists. TLRs are mainly expressed by immune cells including monocytes and macrophages, mast cells and dendritic cells (DCs).Citation8 Recently, elevated TLR expression was observed in cancer cells and this was linked to oncogenesis and cancer progression.Citation9 However, TLR and cancer interrelation remains controversial. On the one hand, TLRs appear able to suppress cancer progression,Citation10-12 but on the other hand, they have been reported to enhance cancer progression.Citation13,14 Enhanced TLR expression within tumors has made these molecules attractive therapeutic targets. The most efficacious TLR ligand in cancer therapy is the Bacillus Calmette and Guérin (BCG) TLR2/TLR4 agonist, which was used successfully for the treatment of bladder cancer for more than three decades.Citation15 During the following 10 y, other TLR agonists besides BCG were the subject of an intense wave of preclinical investigations.Citation16 The safety in patients and ability to induce therapeutic effects against a variety of tumors was confirmed for some of these, for example, monophosphoryl lipid A (MPL),Citation17 imiqimod,Citation18 and OM-174.Citation19
Even if the molecular mechanisms of TLR agonist-mediated tumor regression are not well understood, several mechanisms have been characterized. For instance, dying tumor cells that release specific DAMPS elicited by precise chemotherapeutic agents exert immune-stimulator functions by activating TLR4 on DCs. Hence, the cross-priming of antigen-specific T lymphocytes is promoted.Citation20 However, how tumor cells are killed to provide DCs with antigen has not been determined. It is known that the controlled activation of TLR4 by ALA induces the production of several pro-inflammatory cytokines such as IFNγ, tumor necrosis factor α (TNFα), as well as NO produced by NO synthases (NOS). These mediators induce apoptosis of chemotherapy-resistant tumor cells,Citation12,21 or tumor cells sensitized to ionizing radiationCitation22 and to chemotherapeutic agents such as oxaliplatin.Citation23 However, the precise mechanisms at the origin of ALA-mediated toxicity remain incompletely understood.
Here, we report the investigation of the antitumor efficacy of an agonist of TLR4, ALA OM-174, in mouse models of mammary cancer. We also studied mechanisms leading to this antitumor activity. Our results indicated that ALA inhibited in vivo mammary tumor growth. The antitumor efficacy of ALA involved host TLR4 and IFNγ, but not NOS II, and TLR4, IFNR2 and NOS II of mammary cancer cells. NOS II-derived NO and ROS, and peroxynitrite produced within tumor cells were found to be key mediators in ALA-induced tumor regression.
Results
The analog lipid A, OM-174 had an in vivo antitumor effect, depending on cancer, host TLR4+ cells and host IFNγ
The synthetic derivative lipid A OM-174 (ALA) exerts an antitumor effect in different models of cancers.Citation11,24 However, the molecular mechanisms involved remain unclear. Here, we showed that ALA was also effective in two models of breast cancer in mice. In NT2 () or EMT-6H () tumor-bearing mice, ALA induced tumor regression and raised mouse survival, respectively. Seventy percent of EMT-6H tumor-bearing mice were still alive 35 d after i.v. tumor cell injection when treated with ALA, while all the mice treated with saline solution died before day 35 (). Similarly, 100% of mice with s.c. NT2 tumors became tumor-free after 35 d upon ALA treatment (). Mechanistically, ALA was ineffective in TLR4−/− mice, since all such mice died before day 35, whether or not they were treated with ALA (). This indicates that host TLR4-expressing cells are involved in ALA antitumor activity.
Figure 1. ALA-induced tumor growth inhibition depends on both cancer and host TLR4. (A) Tumor growth curve of FvB mice bearing subcutaneous NT2 cells after treatment or not with ALA (8 mg/kg) (8/group). Statistical test: Mann–Whitney U-test. **p < 0.01, ***p < 0.001. (B–C) mice survival curves of (B) Balb/c mice bearing EMT-6H cells, in wild-type (wt) mice or mice with the TLR4 gene knocked-out (TLR4−/−) after treatment or not with ALA (8 mg/kg) (20/group) or (C) mice bearing EMT-6H cells knocked-down for TLR4 (clone A and B) after treatment or not with ALA (8 mg/kg) (10/group). Survival curves were statistically different (***p <0.001 Log–Rank (Mantel–Cox) test). (D) Quantification of ALA, as measured by β-hydroxymyristic acid analysis, in different mice tissues 24 h after the 3rd ALA injection. Organs and lung nodules were taken from 27 treated and 12 control mice. Data are representative of at least two experiments. Student's t test *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
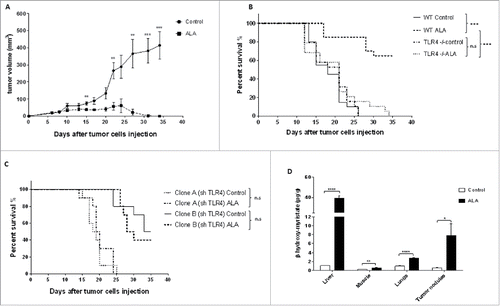
To determine whether a direct effect of ALA on tumor cells was involved in its antitumor effect in vivo, we down regulated the expression of TLR4 in EMT-6H cells using sh-RNA. This leads to a membrane reduction of 76% and 57% of TLR4 in clone A and B, respectively, as analyzed by flow cytometry. The survival of mice injected with either clone A or clone B, was not significantly different between vehicle and ALA-treated mice (). We observed an increase in mice survival with clone B, which could be explained by a potential immune-driven effect. We also demonstrated that ALA could reach tumors and interact with cancer cells. The concentration of β-hydroxymyristic acid, a specific component of ALA,Citation25 was measured in various organs of mice and shown to be higher in tumor nodules and lung tissues of ALA-treated mice than in their non-treated counterparts. In treated mice, β-hydroxymyristic acid was also found in the liver and at very low concentration in muscles (). Together, all these results suggested that the antitumor effect of ALA required TLR4 expression in tumor cells.
Considering that the activation of immune cell TLR4 induced the production of pro-inflammatory cytokines including IFNγ, that increased tumor immunogenicity,Citation22 we investigated whether the antitumor effect of ALA requires IFNγ.
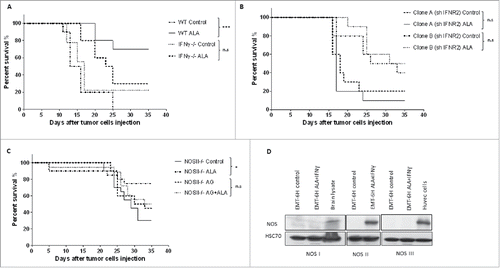
We tested this hypothesis using IFNγ knock-out (IFNγ−/−) mice. As expected, the ALA-treated wild-type mice (wt ALA) survived longer than mice in the other groups, but there was no difference between IFNγ−/− mice treated with ALA or not (). This indicates that the IFNγ produced by the host was involved in the antitumor effect of ALA. In an attempt to discriminate between a direct effect of IFNγ on tumor cells and an indirect one, we evaluated the antitumor efficacy of ALA in mice bearing clone A or B tumors in which the expression of the IFNR2 was reduced by sh-RNA. This leads to a membrane reduction of 37% and 38% of IFNγR in clone A and B, respectively, as analyzed by flow cytometry. When the two IFNR2 defective clones were injected into mice, no survival difference was found within clone A or clone B groups in ALA treated or untreated mice (). Suppression of the ALA effect when tumor cells were less receptive to IFNγ suggested that IFNγ was necessary for the effectiveness of ALA in this model by acting directly on tumor cells.
Figure 2. ALA-induced tumor growth inhibition depends on both IFNγ and NOS II. (A) Wild-type (wt) Balb/c mice or mice with the IFNγ gene knocked-out (IFNγ−/−) bearing EMT-6H tumors were treated with ALA (8 mg/kg) or not (control) and mice survival was monitored for 35 d (10/group) (***p < 0.001, Log–Rank (Mantel–Cox) test). (B) Clones A and B of shIFNR2 EMT-6H cells were injected into wild-type mice before treatment or not with ALA (8 mg/kg). Mice survival was monitored for 35 d (10/group). Survival curves were statistically non-significant (p > 0.05 Log–Rank (Mantel–Cox) test). Data are representative of two experiments. (C) Balb/c mice with the NOS II gene knocked-out (NOS II−/−) and bearing EMT-6H tumors were treated as in (B), in the presence or absence of the NOS II inhibitor aminoguanidine (AG, 50 mg/kg). Mice survival was monitored for 35 d (20/group).*p < 0.05 Log–Rank (Mantel–Cox) test)). (D) Western blot analysis of the protein expression of NOS isoforms in EMT-6H cells treated or not with ALA (500 ng/mL) and IFNγ (33 ng/mL). Brain lysate and human umbilical vein endothelial cells (HUVEC) were used as positive controls. HSC70 served as a loading control.
Inducible NOS II is involved in the ALA-mediated antitumor effect
We further investigated the molecular mechanisms mediated by TLR4 and INFγ at the origin of the antitumor effect of ALA. It is well known that IFNγ interacts synergistically with LPS to induce various pro-inflammatory mediators including transcription of the inducible NOS II.Citation26 To test the involvement of NOS II in the antitumor efficacy of ALA, we analyzed the effect of ALA in mice lacking NOS II. EMT-6H tumor-bearing NOS II−/− mice treated with ALA survived significantly longer than NOS II−/− control mice (), indicating that ALA efficacy did not involve host cell NOS II. To test whether this enzyme, which was expressed by tumor cells stimulated in vivo with ALA and IFNγ, could explain the antitumor effect, NOS II−/− mice with tumors were treated with ALA and aminoguanidine (AG), known to inhibit NOS II of cancer cells that did not express NOS I or NOS III (). ALA increased the number of mice alive on day 35 in knockout mice (NOS II−/− with ALA versus control NOS II−/−) but not in knock-out mice treated with AG (NOS II−/− with ALA + AG vs. control NOS II−/− with AG) (). These results indicated that NOS II of cancer cells but not host NOS II was indispensable for the ALA-mediated antitumor effect.
In vitro induction of cancer cell death by ALA plus IFNγ was dependent on TLR4, IFNγ and NOS II
To further investigate the molecular mechanisms involved in the ALA-inducing antitumor effect, we explored the cytotoxic potential of ALA and IFNγ in vitro. To this end, EMT-6H, shTLR4-EMT-6H and shIFNγR2-EMT-6H cells were treated with OM-147, IFNγ or both these compounds and viable cells (adherent cells) were measured by the methylene blue colorimetric test. ALA alone or IFNγ alone had no cytotoxic effect on EMT-6H cells (Fig. S1), but the combination of the two compounds had a strong cytotoxic effect on EMT-6H tumor cells (, Fig. S1). It should be noted that another pro-inflammatory cytokine, TNFα, failed to synergize with ALA to induce tumor cell death (Fig. S1). However, the ALA plus IFNγ combination failed to induce cell death in TLR4-depleted cells (shTLR4-EMT-6H) (), IFNγR2-depleted cells (shIFNγR2-EMT-6H) () or in cells in which NOS II activity was inhibited by two specific inhibitors, AG and 1400W (). It should be noted that there is some cell death in shINFR2 treated with ALA+IFNγ. This could be a consequence of depletion efficacy. Indeed, more than 60% of IFNγR remain expressed in shIFNγR clones as showed before. These results confirmed those obtained in vivo that demonstrated the involvement of TLR4, IFNγ and NOS II in ALA-induced tumor regression. We also showed that ALA-dependent TLR4 activation and IFNγ are at the origin of NOS II induction, as observed in vivo. Indeed, treatment of EMT-6H cells with ALA and IFNγ induced NOS II expression, as detected by Western blot (). NOS II expression was significantly inhibited in EMT-6H cells depleted for either TLR4 or IFNγR2 () as evaluated by Western blot and NO production attested by nitrite accumulation (Fig. S2). We concluded that ALA plus IFNγ-mediated induction of NOS II and NO represents a crucial pathway in mammary cancer cell death.
Figure 3. ALA and IFNγ induce cell death in EMT-6H cells in vitro through TLR4, IFNR and NOS II. (A) Cell viability analysis of clones A and B of TLR4 deficient EMT-6H cells or not after 48 h of cell incubation with ALA (500 ng/mL) and IFNγ (33 ng/mL). (B) Cell viability analysis of clones A and B of EMT-6H cells deleted or not for IFNR2 after 48 h treatment with ALA plus IFNγ as in (A). (C) Effects of NOS II inhibitors (aminoguanidine [AG; 0.5 mM] and 1400 W; 10 µM) on cell viability induced by ALA plus IFNγ. Data (A–C) are the mean of at least three independent experiments. Student's t test *p < 0.05. (D–F) Western blot analysis of NOS II expression induced by ALA and/or IFNγ as in (A) in WT EMT-6H cells (D) or those deleted for TLR4 (sh TLR4) or INFR2 (sh IFNR2) (E). Data are representative of three independent experiments. HSC70 served as a loading control.
![Figure 3. ALA and IFNγ induce cell death in EMT-6H cells in vitro through TLR4, IFNR and NOS II. (A) Cell viability analysis of clones A and B of TLR4 deficient EMT-6H cells or not after 48 h of cell incubation with ALA (500 ng/mL) and IFNγ (33 ng/mL). (B) Cell viability analysis of clones A and B of EMT-6H cells deleted or not for IFNR2 after 48 h treatment with ALA plus IFNγ as in (A). (C) Effects of NOS II inhibitors (aminoguanidine [AG; 0.5 mM] and 1400 W; 10 µM) on cell viability induced by ALA plus IFNγ. Data (A–C) are the mean of at least three independent experiments. Student's t test *p < 0.05. (D–F) Western blot analysis of NOS II expression induced by ALA and/or IFNγ as in (A) in WT EMT-6H cells (D) or those deleted for TLR4 (sh TLR4) or INFR2 (sh IFNR2) (E). Data are representative of three independent experiments. HSC70 served as a loading control.](/cms/asset/75cac8dc-1e49-4770-9579-8c784c237b4e/koni_a_1123369_f0003_b.gif)
NO alone was not sufficient to induce the antitumor effect of ALA plus IFNγ but required reactive oxygen species (ROS)
To test the role of NO in ALA and IFNγ-induced cell death, EMT-6H cells were pretreated with CPTIO (2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide), an NO scavenger, before adding ALA + IFNγ. Under these conditions, cytotoxicity decreased 3-fold (). The intracellular NO donor GTN at 50–60 µM produced the same amount of nitrite as NOS II, but was not toxic to EMT-6H cells (). Furthermore, different NO donors, such as GTN and SNP (sodium nitroprusside), when delivered at concentrations that produced the same amount of nitrite as NOS II did, were not cytotoxic even in the presence of IFNγ (Fig. S3). Therefore NO produced by NOS II seemed necessary but not sufficient to induce cytotoxicity, implying that another mediator was required to act with NO.
Figure 4. ALA plus IFNγ induce NO and ROS production necessary but not sufficient by themselves to induce tumor cell toxicity. (A) Analysis of EMT-6H cell death after treatment with ALA (500 ng/mL), IFNγ(33 ng/mL) or cPTIO(2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide; 50 µM) for 48 h. The data of one experiment representative of five independent ones are shown. Student's t test ****p < 0.0001. (B) Cell death analysis (in black) of EMT-6H cells and NO production (in white) as attested by nitrite accumulation after treatment for 48 h with different concentrations of GTN (Glyceryltrinitrate). (C) EMT-6H cells were treated with ALA (500 ng/mL) and IFNγ (33 ng/mL) in the presence or absence of 10 µM 1400 W for 24 h. ROS production was detected by flow cytometry after adding the DCFH-2-DA probe for 30 min. (D) EMT-6H cells were treated as in (C). Production of superoxide was detected by Electronic paramagnetic resonance after adding CMH for 15 min. Data show the mean light intensity emitted and are representative of two independent experiments. (E) Mice were treated as described in . Lung tissues were harvested after the 3rd ALA injection and cryo-sections were incubated with DHE (dihydroethidium), which fluoresces red in the presence of ROS. Cell nuclei were stained with DAPI (blue). Scale bars: 20 μm. (F) Survival curve of Balb/c mice bearing EMT-6H tumors were given drinking water ad libitum containing 3.5 mg/mL N-acetyl cysteine (NAC) or not throughout the treatment with ALA (8/group) (8 mg/kg) (*p < 0.05 Log–Rank (Mantel–Cox) test). The data from one experiment representative of two are shown.
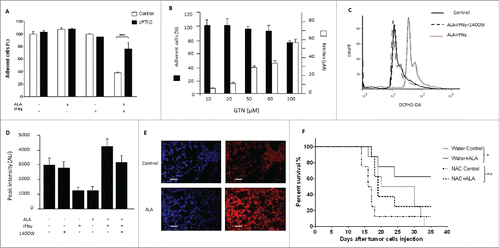
ROS were candidate cytotoxic agents. As shown in , the combination of ALA and IFNγ induced ROS production in EMT-6H cells as detected by flow cytometry with the DCFH2-DA ROS-sensitive probe. Addition of the NOS inhibitor 1400 W to the cells abolished the increase in ROS production (). Superoxide was also detected by electron paramagnetic resonance (EPR) spectroscopy (). The detection of ROS in cryo-sections of lungs and tumor nodules with DHE (dihydroethidium), a second ROS-sensitive probe, showed an increase of ROS in ALA-treated mice (). Strikingly, the inhibition of NOS II activity reduced ROS production irrespective of the inhibitor used, whether it was 1400 W () AG, NMMA (N(G)-monomethyl-L-arginine) or NAME (NG-Nitro-L-arginine methyl ester) (data not shown). Our results suggested that ALA and IFNγ together induced NO and ROS production in EMT-6H cells via NOS II activity. To confirm the involvement of ROS in the antitumor effect of ALA, EMT-6H tumor-bearing mice were given N-acetyl cysteine (NAC), a ROS inhibitor, in their drinking water during ALA treatment. shows a group effect: mice treated with ALA survived significantly longer than mice from other test groups. As NAC reduced the antitumor activity of ALA, ROS production likely has an antitumor effect in vivo.
The toxic effect of ALA plus IFNγ on EMT-6H cells was due to peroxynitrite
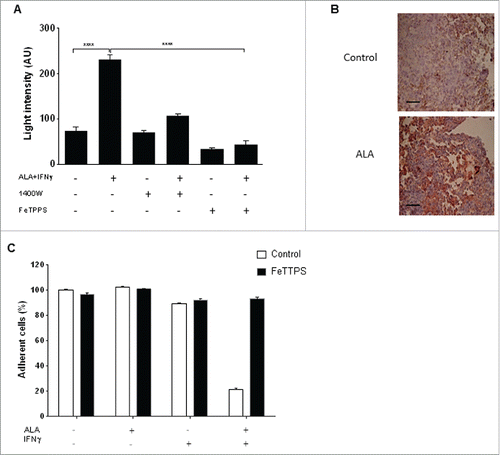
NO and superoxide are known to react together to give rise to highly cytotoxic peroxynitrite. We thus searched for evidence of peroxynitrite formation in the tumor cells, as detected by luminescence in the presence of luminol, a non-specific compound to measure peroxynitrite. The association of ALA and IFNγ had a potent effect on the amount of luminescence produced in EMT-6H cells (), whereas IFNγ alone increased the luminescence only slightly and ALA alone had no effect (not shown). The production of peroxynitrite in tumors from ALA-treated mice was assessed in vivo using tumor sections and anti-NO-tyrosine antibodies (). When 1400 W or FeTPPS (5,10,15,20-tetrakis (4-sulfonatophenyl) porphyrinato iron (III) chloride), a catalyst for peroxynitrite degradation,Citation27 was added to EMT-6H cells treated with ALA and IFNγ, less luminescence was detected than in the absence of these former molecules () and the cytotoxicity of ALA plus IFNγ was inhibited ().
Figure 5. The toxic effect of ALA plus IFNγ on EMT-6H cells is due to peroxynitrite. (A) EMT-6H cells were treated with or without ALA (500 ng/mL) plus IFNγ (33 ng/mL) and 10 µM 1400 W or FeTPPS (5,10,15,20-tetrakis (4-sulfonatophenyl) porphyrinato iron (III) chloride; 150 µM), for 48 h. Peroxynitrite production was detected by adding luminol to cells and immediately measuring luminescence. Student's t test ****p < 0.0001. (B) Lung tissues were taken after the 3rd injection of ALA or saline solution as . The sections were stained with an anti-NO-tyrosine antibody (red). Scale bars: 20 μm. (C) EMT-6H cells were treated with or without ALA, IFNγ or FeTPPS for 48 h and cell viability was measured as described in Materials and Methods.
ALA plus IFNγ induced uncoupling of NOS II activity to produce NO plus superoxide anion
NADPH oxidases (NOX) are the main enzymes that catalyze the production of superoxide by cells. However, the inhibition of NOX with various inhibitors (apocynin, R6G, oxypurinol) did not decrease ALA plus IFNγ cytotoxicity (), showing that NOX were not involved in the ALA plus IFNγ antitumor effect.
Figure 6. NOS II uncoupling was observed when EMT-6H cells were activated with ALA plus IFNγ. (A) EMT-6H cells were treated with or without NOX inhibitors: 300 µM apocynin, 0.3 µM rhodamine 6G, or 150 µM oxypurinol, for 1 h before adding ALA (500 ng/mL) plus IFNγ (33 ng/mL) for 48 h. Cell viability was detected as described in Materials and methods. Data from one representative of three independent experiments in triplicate are shown. (B) EMT-6H cells were treated for 1 h with 0.5 mM AG or 10 µM 1400 W, then for 48 h with or without ALA and IFNγ. Nitrite accumulation was measured by the Griess assay. (C) EMT-6H cells were treated with or without ALA or IFNγ. L-arginine was measured in cell extracts by HPLC using homo-L-arginine as the internal standard. Data from one experiment representative of two are shown.
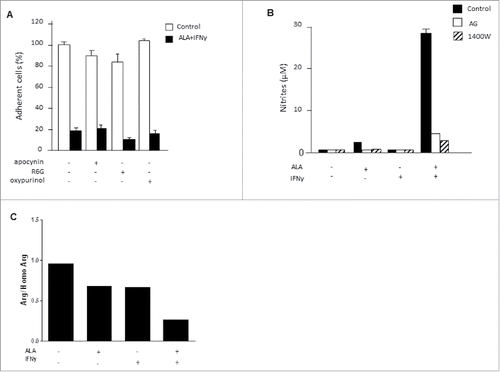
EMT-6H cells did not produce nitrites when treated with ALA plus IFNγ and NOS II inhibitors such as AG and 1400 W (). Interestingly, 1400 W (), like AG (data not shown), also suppressed ROS and superoxide accumulation in tumor cells. These data led us to examine whether NOS II itself was both an NO and ROS producer, in a so-called “uncoupling process.” NOS II uncoupling is regulated by the availability of its substrate, L-arginine (L-Arg) and the co-factor tetrahydrobiopterin (H4B). Modifying the amounts of H4Band L-Arg, particularly decreasing the L-Arg concentration, can lead to the generation of superoxide anions and hydrogen peroxide by NOS II.Citation28 We therefore measured the concentrations of H4B and L-Arg in EMT-6H cells treated with ALA and IFNγ. The amount of H4B in treated EMT-6H cells was not significantly different from that in control cells (data not shown). In contrast, the amounts of L-Arg decreased as much as 3-fold in treated cells compared to untreated cells (). We thus concluded that a reduced availability of L-Arg caused NOS II uncoupling in EMT-6H cells treated with ALA and IFNγ and that under these conditions NOS II of EMT-6H cells produced both NO and ROS.
Altogether, our results clearly demonstrated for the first time that the antitumor effect of two immune-stimulators in a model of mammary cancer are mediated by NOS II via its capacity to produce both NO and ROS in cancer cells, a so-called “NOS uncoupling process,” infrequently observed in physiology.
Discussion
Cancer immunotherapy was revealed in the late 19th century, when Coley observed that bacterial extracts exhibit antitumor activity. Subsequent work demonstrated that several TLR agonists induce anticancer responses in different types of cancer.Citation29 Here, in two different mammary cancer models we showed that treatment with a TRL4 agonist, a synthetic lipid A OM-174 (ALA), controlled tumor growth. This effect involved TLR4 and IFNγ of immune cells and TLR4 and NOS II of cancer cells. Although TLR4 signaling in immune cells is critical for the reduction of tumor growth,Citation20 the role it plays in cancer cells remains controversial. Stimulation of TLR4 expression on tumor cell surfaces has been demonstrated to have a positive role in tumorigenesis,Citation30,31 but other data have shown that the TLR4 pathway activation on tumor cells contributes positively to inhibition of tumor growth.Citation32,33 These discrepancies are not fully understood. Our results indicated that the simple activation of TLR4 in cancer cells failed to induce cell death, but its combination with IFNγ triggered cell demise and tumor growth inhibition. This suggested that the sensitivity of cancer cells to TLR4-mediated cell death needs the presence of a second signal; in the case examined activation of the IFNγ receptor by IFNγ. Previously, along with Ridder et al., we reported that this cytokine could be produced by immune cells when they were activated by OM-174.Citation22,34 Nunez et al.Citation33 evoked such a scenario. They demonstrated that a TLR-activated melanoma produces IFNβ, which contributes positively to the antitumor immune response. We anticipate that systematic application of the TLR4 ligand will engage TLR4 to produce IFNγ on both host immune cells and cancer cells, where IFNγ cooperates with activated TLR4 to induce cell death and tumor growth inhibition. Based on our findings, we hypothesize that those tumors such as IFNγ-secreting immune cell infiltration, i.e. with a certain immune environment, are sensitive, in terms of cell death, to treatment with exogenous or endogenous TLR4 analogs such as lipid A and HMGB1 or HSP60, respectively. In the current study, we also showed, both in vitro and in vivo, that NO and ROS produced by tumor cell NOS II, but not by the host enzyme, mediate the cytotoxic effect of the ALA–IFNγ combination. The capacity of NOS II to produce both NO and ROS rarely occurs except when substrate and cofactor availability are reduced, referred to as NOS uncoupling.Citation35 In our investigation, we showed that ALA plus IFNγ induced a decrease in the intracellular L-Arginine concentration, which could explain NOS II uncoupling. Alternatively, the drop in intracellular arginine could be due to NOS II-dependent consumption and to some enzymes involved in the arginine recycling or degradation pathways; such as arginine decarboxylase, arginine:glycine amidinotransferase, or arginase, respectively.Citation36 We did not find appreciable changes in arginase activity between control and ALA plus IFNγ-treated cells (data not shown). Proving an involvement of the arginine recycling pathway enzymes in arginine depletion needs further investigation. Considering the key role of arginine in survival and proliferation of various malignancies, L-arginine depletion has been considered a promising approach for the treatment of these diseases. Accordingly, recent clinical trials have been performed or are ongoing to examine the use of arginine deprivation as a treatment for advanced malignancies.Citation37 Our findings have also uncovered a new tool for depleting arginine using lipid A and IFNγ. Further studies are needed to confirm the impact of arginine depletion in lipid A plus IFNγ-mediated cancer cell death.
Collectively, our data imply that combining the activation of TLR4 and IFNγ pathways in cancer cells can trigger an anticancer response in mammary cancer cells. Mechanistically, our results highlight the major role that NOS II plays in cancer cell death. Whatever the methods, any therapeutic strategy that permits the activation of TLR4 and IFNγ in a tumor microenvironment should be deleterious for cancer cell survival.
Materials and methods
Cells, animals and treatment
Murine mammary EMT-6H cells were obtained after cloning of the EMT-6 cell lineCitation34 and the mammary NT2 cancer cell line was purchased from the ATCC. Balb/c mice, Fvb mice, IFNγ, TLR4, and NOS II knockout mice were purchased from Charles River Laboratory International. All experiments involving animals were performed in accordance with institutional guidelines and approved by the University of Burgundy's Ethics Committee.
On day 0, EMT-6H cells (105 cells) were injected intravenously (i.v.) into syngenic Balb/c mice giving rise to lung tumor nodules that killed the mice within 5 weeks. NT2 cells (5 × 105 cells) were injected subcutaneously (s.c.) into syngenic Fvb mice on day 0. For mice bearing EMT-6H tumors, treatment consisted of five i.v. injections of ALA OM-174 (OM PHARMA) at 5 d intervals from day 5 until day 25 at the dose of 8 mg/kg. For mice bearing NT2 tumors, treatment consisted of three i.v. injections of 8 mg/kg ALA at day 8 and every week thereafter. AG, an inhibitor of NOS II, was injected i.p. at the dose of 50 mg per kg, two times per week, whereas N-acetyl cysteine (NAC) (3.5 mg/mL), a ROS inhibitor, was given orally, in drinking water. On day 35, all surviving mice were euthanized and autopsied.
Measurement of live cell density
EMT-6H cells (104 cells) were seeded into a 96-well plate for 24 h, treated with or without 500 ng/mL ALA, 33 ng/mL IFNγ, or 10 µM Glyceryltrinitrate (GTN) and incubated for a further 48 h. The density of live cells was determined by a methylene blue colorimetric test.
Determination of NO and ROS production
NO production was estimated using the Griess microassay to determine nitrite accumulation in the cell culture medium. The intracellular ROS production was measured using the 2′,7′-dichlorodihydrofluorescein diacetate (DCFH2-DA) probe. Tumor cells were incubated in the dark with the DCFH2-DA probe. Fluorescence was measured with a FACScan flow cytometer.
Lipid A quantitation
Organs from mice were collected and immediately stored in liquid nitrogen. Samples were ground up and the amounts of ALA were determined by direct quantitation of β-hydroxymyristic acid in the extracts by gas chromatography-mass spectrometry (GC-MS).Citation38
Statistics
Unless otherwise stated in the figure legends data from one experiment representative of three experiments are shown. In vitro data are presented as the mean ± SEM and paired groups were compared with the Student's t test. Tumor sizes between two groups were compared using Mann–Whitney U-test. Survival analysis was assessed by Mante–Cox log-rank test; p <0.05 was considered significant compared with controls.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
Myriam Lamrani, Nejia Sassi, Jean-Luc Boucher, Jérôme Labbé, Cédric Seignez, Cindy Racoeur, Anne Athias, Catherine Vergely conceived and carried out experiments and analyzed data, Romain Guerreiro analyzed data, Luc Rochette conceived experiments and discussed data. Catherine Paul conceived and carried out experiments, analyzed and discussed data and edited the manuscript; Ali Bettaieb conceived experiments, analyzed and discussed data and edited the manuscript; Jean-François Jeannin conceived experiments, analyzed and discussed data, coordinated the experiments, prepared and edited the manuscript.
KONI_A_1123369_supplementary_material.zip
Download Zip (174.3 KB)Funding
This work was supported by a grant FEDER 2009-0431SGO003S01173 from European Union and no. 32403 from Région de Bourgogne and grants from Ligue Contre le Cancer (CCIR-GE, 90X.2009), Fondation de France (2011-00020354) and INCA (686/08).
References
- Anderson KV, Bokla L, Nüsslein-Volhard C. Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the Toll gene product. Cell 1985; 42:791-8; PMID:3931919; http://dx.doi.org/10.1016/0092-8674(85)90275-2
- Taguchi T, Mitcham JL, Dower SK, Sims JE, Testa JR. Chromosomal localization of TIL, a gene encoding a protein related to the Drosophila transmembrane receptor Toll, to human chromosome 4p14. Genomics 1996; 32:486-8; PMID:8838819; http://dx.doi.org/10.1006/geno.1996.0150
- Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997; 388:394-7; PMID:9237759; http://dx.doi.org/10.1038/41131
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011; 34:637-50; PMID:21616434; http://dx.doi.org/10.1016/j.immuni.2011.05.006
- Hennessy EJ, Parker AE, O'Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov 2010; 9:293-307; PMID:20380038; http://dx.doi.org/10.1038/nrd3203
- Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem 2002; 277:15028-34; PMID:11836257; http://dx.doi.org/10.1074/jbc.M200497200
- Yamazaki T, Hannani D, Poirier-Colame V, Ladoire S, Locher C, Sistigu A, Prada N, Adjemian S, Catani JP, Freudenberg M et al. Defective immunogenic cell death of HMGB1-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ 2014; 21:69-78; PMID:23811849; http://dx.doi.org/10.1038/cdd.2013.72
- Ozato K, Tsujimura H, Tamura T. Toll-like receptor signaling and regulation of cytokine gene expression in the immune system. Biotechniques 2002; Suppl:66-8, 70, 72 passim; PMID:12395929
- Sato Y, Goto Y, Narita N, Hoon DS. Cancer Cells Expressing Toll-like Receptors and the Tumor Microenvironment. Cancer Microenviron 2009; 2 Suppl 1:205-14; PMID:19685283; http://dx.doi.org/10.1007/s12307-009-0022-y
- Coley WB. II. Hawkins on tubercular peritonitis. Ann Surg 1893; 17:462-4; PMID:17859910; http://dx.doi.org/10.1097/00000658-189301000-00101
- Onier N, Hilpert S, Arnould L, Saint-Giorgio V, Davies JG, Jeannin JF. Cure of colon cancer metastasis in rats with the new lipid A OM 174. Apoptosis of tumor cells and immunization of rats. Clin Exp Metastasis 1999; 17:299-306; PMID:10545016; http://dx.doi.org/10.1023/A:1006663017149
- Garay RP, Viens P, Bauer J, Normier G, Bardou M, Jeannin JF, Chiavaroli C. Cancer relapse under chemotherapy: why TLR2/4 receptor agonists can help. Eur J Pharmacol 2007; 563:1-17; PMID:17383632; http://dx.doi.org/10.1016/j.ejphar.2007.02.018
- Zhang YB, He FL, Fang M, Hua TF, Hu BD, Zhang ZH, Cao Q, Liu RY. Increased expression of Toll-like receptors 4 and 9 in human lung cancer. Mol Biol Rep 2009; 36:1475-81; PMID:18763053; http://dx.doi.org/10.1007/s11033-008-9338-9
- Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007; 317:121-4; PMID:17615358; http://dx.doi.org/10.1126/science.1140485
- Yoo KH, Lim TJ, Chang SG. Monthly intravesical bacillus Calmette-Guérin maintenance therapy for non-muscle-invasive bladder cancer: 10-year experience in a single institute. Exp Ther Med 2012; 3:221-5; PMID:22969872; http://dx.doi.org/10.3892/etm.2011.400
- Vacchelli E, Vitale I, Tartour E, Eggermont A, Sautès-Fridman C, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Anticancer radioimmunotherapy. Oncoimmunology 2013; 2:e25595; PMID:24319634; http://dx.doi.org/10.4161/onci.25595
- Pedersen C, Breindahl M, Aggarwal N, Berglund J, Oroszlán G, Silfverdal SA, Szüts P, O'Mahony M, David MP, Dobbelaere K et al. Randomized trial: immunogenicity and safety of coadministered human papillomavirus-16/18 AS04-adjuvanted vaccine and combined hepatitis A and B vaccine in girls. J Adolesc Health 2012; 50:38-46; PMID:22188832; http://dx.doi.org/10.1016/j.jadohealth.2011.10.009
- Holcmann M, Drobits B, Sibilia M. How imiquimod licenses plasmacytoid dendritic cells to kill tumors. Oncoimmunology 2012; 1:1661-3; PMID:23264929; http://dx.doi.org/10.4161/onci.22033
- Isambert N, Fumoleau P, Paul C, Ferrand C, Zanetta S, Bauer J, Ragot K, Lizard G, Jeannin JF, Bardou M. Phase I study of OM-174, a lipid A analogue, with assessment of immunological response, in patients with refractory solid tumors. BMC Cancer 2013; 13:172; PMID:23547558; http://dx.doi.org/10.1186/1471-2407-13-172
- Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007; 13:1050-9; PMID:17704786; http://dx.doi.org/10.1038/nm1622
- D'Agostini C, Pica F, Febbraro G, Grelli S, Chiavaroli C, Garaci E. Antitumour effect of OM-174 and cyclophosphamide on murine B16 melanoma in different experimental conditions. Int Immunopharmacol 2005; 5:1205-12; PMID:15914325; http://dx.doi.org/10.1016/j.intimp.2005.02.013
- De Ridder M, Verovski VN, Chiavaroli C, Van den Berge DL, Monsaert C, Law K, Storme GA. The radiosensitizing effect of immunoadjuvant OM-174 requires cooperation between immune and tumor cells through interferon-gamma and inducible nitric oxide synthase. Int J Radiat Oncol Biol Phys 2006; 66:1473-80; PMID:17056198; http://dx.doi.org/10.1016/j.ijrobp.2006.07.1381
- Seignez C, Martin A, Rollet CE, Racoeur C, Scagliarini A, Jeannin JF, Bettaieb A, Paul C. Senescence of tumor cells induced by oxaliplatin increases the efficiency of a lipid A immunotherapy via the recruitment of neutrophils. Oncotarget 2014; 5:11442-51; PMID:25347345; http://dx.doi.org/10.18632/oncotarget.2556
- Gautier T, Paul C, Deckert V, Desrumaux C, Klein A, Labbe J, Le Guern N, Athias A, Monier S, Hammann A et al. Innate immune response triggered by triacyl lipid A is dependent on phospholipid transfer protein (PLTP) gene expression. FASEB J 2010; 24:3544-54; PMID:20418497; http://dx.doi.org/10.1096/fj.09-152876
- Maitra SK, Schotz MC, Yoshikawa TT, Guze LB. Determination of lipid A and endotoxin in serum by mass spectroscopy. Proc Natl Acad Sci U S A 1978; 75:3993-7; PMID:16592555; http://dx.doi.org/10.1073/pnas.75.8.3993
- Martin E, Nathan C, Xie QW. Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J Exp Med 1994; 180:977-84; PMID:7520478; http://dx.doi.org/10.1084/jem.180.3.977
- Misko TP, Highkin MK, Veenhuizen AW, Manning PT, Stern MK, Currie MG, Salvemini D. Characterization of the cytoprotective action of peroxynitrite decomposition catalysts. J Biol Chem 1998; 273:15646-53; PMID:9624158; http://dx.doi.org/10.1074/jbc.273.25.15646
- Xia Y, Roman LJ, Masters BS, Zweier JL. Inducible nitric-oxide synthase generates superoxide from the reductase domain. J Biol Chem 1998; 273:22635-9; PMID:9712892; http://dx.doi.org/10.1074/jbc.273.35.22635
- Galluzzi L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautès-Fridman C, Tartour E, Zitvogel L, Kroemer G. Trial Watch: Experimental Toll-like receptor agonists for cancer therapy. Oncoimmunology 2012; 1:699-716; PMID:22934262; http://dx.doi.org/10.4161/onci.20696
- Huang B, Zhao J, Li H, He KL, Chen Y, Chen SH, Mayer L, Unkeless JC, Xiong H. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res 2005; 65:5009-14; PMID:15958541; http://dx.doi.org/10.1158/0008-5472.CAN-05-0784
- Szczepanski MJ, Czystowska M, Szajnik M, Harasymczuk M, Boyiadzis M, Kruk-Zagajewska A, Szyfter W, Zeromski J, Whiteside TL. Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res 2009; 69:3105-13; PMID:19318560; http://dx.doi.org/10.1158/0008-5472.CAN-08-3838
- Andreani V, Gatti G, Simonella L, Rivero V, Maccioni M. Activation of Toll-like receptor 4 on tumor cells in vitro inhibits subsequent tumor growth in vivo. Cancer Res 2007; 67:10519-27; PMID:17974996; http://dx.doi.org/10.1158/0008-5472.CAN-07-0079
- Núñez NG, Andreani V, Crespo MI, Nocera DA, Breser ML, Morón G, Dejager L, Libert C, Rivero V, Maccioni M. IFNβ produced by TLR4-activated tumor cells is involved in improving the antitumoral immune response. Cancer Res 2012; 72:592-603; http://dx.doi.org/10.1158/0008-5472.CAN-11-0534
- Gauthier N, Lohm S, Touzery C, Chantôme A, Perette B, Reveneau S, Brunotte F, Juillerat-Jeanneret L, Jeannin JF. Tumour-derived and host-derived nitric oxide differentially regulate breast carcinoma metastasis to the lungs. Carcinogenesis 2004; 25:1559-65; PMID:15059928; http://dx.doi.org/10.1093/carcin/bgh158
- Sun J, Druhan LJ, Zweier JL. Dose dependent effects of reactive oxygen and nitrogen species on the function of neuronal nitric oxide synthase. Archives of biochemistry and biophysics 2008; 471:126-33; PMID:18201545; http://dx.doi.org/10.1016/j.abb.2008.01.003
- Wu G, Morris SM, Jr. Arginine metabolism: nitric oxide and beyond. Biochem J 1998; 336 (Pt 1):1-17; PMID:9806879; http://dx.doi.org/10.1042/bj3360001
- Feun LG, Kuo MT, Savaraj N. Arginine deprivation in cancer therapy. Curr Opin Clin Nutr Metab Care 2015; 18:78-82; PMID:25474015; http://dx.doi.org/10.1097/MCO.000000000000-0122
- Gautier T, Klein A, Deckert V, Desrumaux C, Ogier N, Sberna AL, Paul C, Le Guern N, Athias A, Montange T et al. Effect of plasma phospholipid transfer protein deficiency on lethal endotoxemia in mice. J Biol Chem 2008; 283:18702-10; PMID:18458077; http://dx.doi.org/10.1074/jbc.M802802200