
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Genome sequencing has uncovered an array of recurring somatic mutations in different non-Hodgkin lymphoma (NHL) subtypes. If affecting protein-coding regions, such mutations may yield mutation-derived peptides that may be presented by HLA class I proteins and recognized by cytotoxic T cells. A recurring somatic and oncogenic driver mutation of the Toll-like receptor adaptor protein MYD88, Leu265Pro (L265P) was identified in up to 90% of different NHL subtype patients. We therefore screened the potential of MYD88L265P-derived peptides to elicit cytotoxic T cell responses as tumor-specific neoantigens. Based on in silico predictions, we identified potential MYD88L265P-containing HLA ligands for several HLA class I restrictions. A set of HLA class I MYD88L265P-derived ligands elicited specific cytotoxic T cell responses for HLA-B*07 and -B*15. These data highlight the potential of MYD88L265P mutation-specific peptide-based immunotherapy as a novel personalized treatment approach for patients with MYD88L265P+ NHLs that may complement pharmacological approaches targeting oncogenic MyD88 L265P signaling.
Abbreviations
| aAPC | = | artificial antigen-presenting cell |
| APC | = | antigen-presenting cell |
| CFSE | = | carboxyfluorescein diacetate succinimidyl ester |
| CIP | = | cancer immunoguiding program |
| CLL | = | chronic lymphocytic leukemia |
| DLBCL | = | diffuse large B cell lymphoma |
| E/T | = | effector to target ratio |
| FAM | = | fluorescein |
| Fmoc/tBu | = | 9-fluorenylmethyl-oxycarbonyl/tert-butyl |
| HBDs | = | healthy blood donors |
| HIV | = | human immunodeficiency virus |
| IOL | = | intraocular lymphoma |
| L265P | = | Leu265Pro |
| LPL | = | lymphoplasmacytic lymphoma |
| NHL | = | non-Hodgkin lymphoma |
| PBMCs | = | peripheral blood mononuclear cells |
| PCNHL | = | primary cerebral non-Hodgkin's lymphoma |
| PHA | = | phytohaemagglutinin |
| TLR | = | Toll-like receptor |
| WT | = | wild-type (i.e., non-mutated sequence) |
| YAK | = | yakima yellow |
Introduction
The immune system can recognize and to some extent eradicate tumor cells.Citation1,2 Nevertheless, this antitumor response is often inefficient.Citation3 Antigen-specific immunotherapy holds the potential to induce and boost clinically effective anticancer T cell responsesCitation4 and might be used to guide and increase the specificity of cancer immunotherapy in future combination trials,Citation5 especially when combined with newly available immune checkpoint inhibitors.Citation6 For this purpose, the exact knowledge of tumor-associated or tumor-specific immunogenic T cell epitopes is crucial. Tumor-specific neoepitopes, derived from protein-altering mutational events like missense mutations, may be perceived as foreign by the immune system and elicit tumor-specific T cell immunity. Citation7,8
Being tumor-specific, such neoantigens promise high specificity but are largely patient-specific, and therefore, hard to identify and mainly singular events in a patient cohort. Recurrent mutations could overcome this problem of patient-specificity and could be targeted in broadly applicable immunotherapeutic treatments of different types of cancer.
A recurring somatic and oncogenic driver mutation of the Toll-like receptor (TLR) adaptor protein MYD88, Leu265Pro (L265P) was identified in up to 90% of certain non-Hodgkin lymphoma (NHL) subtypes.Citation9-15 NHLs like diffuse large B cell lymphoma (DLBCL) or chronic lymphocytic leukemia (CLL) are the cause of death for more than 199,000 patients annually world-wide.Citation16 The success of established therapies varies strongly between these diseases. Whereas other DLBCL subtypes can be effectively treated, the activated B cell-like lymphoma subtype, for example, is characterized by a less than 40% overall survival following standard therapy.Citation17 For CLL, there is currently no curative treatment and for some patients aggressive chemotherapies or allogenic stem cell transplantations are required.Citation18 Therefore, novel treatments are needed to further improve clinical outcome in these patients. Immunotherapy based on peptide-vaccines or adoptive immune cell transfers might represent ideal treatments, since they are well tolerated and have fewer side effects than chemotherapeutic agents. Growing evidence demonstrates that therapeutic peptide-vaccines can induce specific immune responses, impact clinical outcomeCitation19 and even prolong overall survival in cancer patients.Citation4 Besides, peptide-vaccines experimental cancer immunotherapy using adoptive transfer of antigen-specific or genetically engineered T cellsCitation20,21 as well as antigen-presenting dendritic cellsCitation22 showed also high efficiency in different studies. Furthermore, vaccines may potentially yield a sustained antitumor effect by inducing immunological memory.Citation23 Thus, the identification of cancer-associated and cancer-specific antigens offers a great variety of treatment possibilities in cancer patients.
As MYD88L265P is a widely occurring and tumor-specific mutation, MYD88L265P-based immunotherapy might constitute an ideal alternative or complementation to ongoing attempts to target this mutation pharmacologicallyCitation24 in NHLs. We, therefore, screened the potential of MYD88L265P-containing peptides for CD8+ T cell-mediated immunotherapy and identified potential HLA ligands encompassing the MYD88L265P mutation for several HLA class I restrictions based on in silico predictions. We focused on three HLA-B*07-restricted peptides and one HLA-B*15-restricted peptide to examine the immunogenicity of these tumor-specific neoantigens. The present study shows that MYD88L265P-derived peptides can induce mutation-specific and functional immune responses in vitro, which may pave the way for developing new personalized immunotherapeutic strategies with broad applicability for different NHLs.
Results
In silico prediction yields several likely MYD88L265P-derived HLA class I ligands
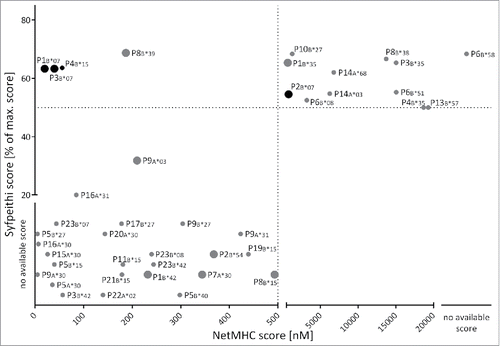
We identified 23 different MYD88L265P-derived peptides predicted by SYFPEITHI and/or NetMHC 3.4 to bind one or more HLA class I allotypes with a score of IC50≤ 500 nM for NetMHC 3.4 or with ≥ 50% of the maximum allotype-specific score for SYFPEITHI. These algorithms predicted 8, 15, and 8 peptides restricted by 5, 17 and 5 different HLA-A, -B, and -C allotypes, respectively (Table S2). Notably, only four ligands were concordantly designated as ligands of the same HLA allotype by both algorithms (). This is mainly due to a different set of available HLA allotypes for the two algorithms (e.g., SYFPEITHI cannot predict 12mer peptides or HLA-B*30:01-restricted peptides).Citation25-29 For effective, on-target immunotherapy the identification of immunogenic tumor-specific antigens that do not elicit cross-reactivity with benign tissue is of paramount importance.Citation30 Therefore, we aimed at identifying mutated peptides with increased binding affinities compared with their non-mutated counterparts. This was realized by predicting the score for every potential mutation-derived ligand, as well as the score for the corresponding WT peptide on the same HLA class I allotype. We highlighted 16 of the 52 predicted mutated peptide:HLA combinations, which have a more than 2-fold higher predicted binding affinity compared with their non-mutated corresponding peptides. Among these, we identified two MYD88L265P-derived gain-of-anchor antigens mutated in their P2-anchor position. These two peptides (P1 and P2) were therefore preferentially selected for further characterization and immunogenicity testing. In total, all 18 8–11mer peptides predicted as potential ligands for HLA class I were synthesized and subjected to functional characterization. For 13/18 predicted HLA class I ligands PBMCs of HLA-matched MYD88L265P-mutated NHL patients were available and could therefore be tested in IFNγ ELISPOT assays for spontaneous memory T cell responses. To verify the ability of the predicted HLA ligands to indeed bind to the respective HLA class I allotypes, 10 of the predicted peptides were analyzed in in vitro monomer refolding assays. The peptides for the refolding experiments were selected according to their predicted binding score as well as available HLA molecules. Four peptide:HLA complexes (P1B*07, P2B*07, P3B*07, and P4B*15) were refolded successfully in vitro (Table S2). The successfully refolded HLA–peptide complexes were all used in experiments.
Figure 1. Prediction of MYD88L265P-derived HLA class I ligands. 50 MYD88L265P-derived 8- to 12-mer peptides were scored by the online prediction tools SYFPEITHI and NetMHC 3.4, as well as an extended in-house database. Peptides with NetMHC 3.4 IC50≤ 500 nM were defined as binders (region left of the dotted line). SYFPEITHI scores are displayed as percent of the maximum score for the respective HLA allotype. The threshold for binders is defined as ≥ 50% of the maximum score (above the dotted line). For some HLA:peptide combinations scoring was possible with only one of the prediction tools due to limited availability of predictors. The figure illustrates the predicted ligands for HLA-A and -B allotypes. Fold-change ratios in binding scores of mutated peptides compared with the corresponding WT peptides are indicated by the size of the respective dot: large dots indicate an at least 2-fold better score, mutated ligands illustrated by small dots exhibit no increased binding score in comparison to their corresponding WT ligand. Out of 50 unique peptide sequences, 23 were scored as potential HLA class I ligands. Four ligands were concordantly designated as ligands of the same HLA allotype by both algorithms with three of them having an at least 2-fold higher score as their corresponding WT peptide. The black dots indicate the peptides which were tested in aAPC-based in vitro primings in HBDs or CLL patients. Abbreviations: max., maximal.
Spontaneous memory T cell responses targeting MYD88L265P-derived peptides are very infrequent in NHL patients
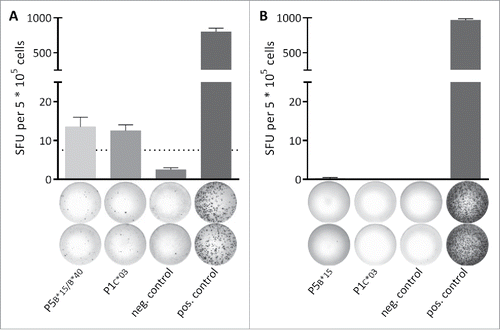
Functional characterization of the predicted candidate HLA class I MYD88L265P-derived NHL-specific ligands was performed by 12-d recall IFNγ ELISPOT assays using PBMCs obtained from MYD88L265P-mutated and MYD88WT patients (Table S1). In one (out of 22 tested) MYD88L265P-mutated NHL patients, memory T cell responses targeting two different MYD88L265P-derived HLA class I ligands were detected by IFNγ ELISPOT (). Importantly, one of the peptides (P5) is a predicted ligand for both, HLA-B*15 and -B*40, which are both expressed by the patient. Therefore, we could not resolve which peptide:HLA combination is responsible for the observed IFNγ secretion. Importantly, no IFNγ secretion was observed for any tested ligand in MYD88WT patients. The frequency of memory T cell responses in 1/22 MYD88L265P-mutated NHL patients appears low, but it is important to be aware of the HLA allotype-specific frequencies. The peptide P1, which is among others analyzed in further immunogenicity experiments, leads to a detectable memory T cell response in 1/3 (33%) tested HLA-matched MYD88L265P-mutated NHL patients. For the other positive peptide P5 memory T cell responses could be detected in 1/4 (25%) HLA-B*15 patients and in 1/3 (33%) HLA-B*40 patients. Furthermore, only two-digit HLA typings were available for the NHL patients. Therefore, it is possible that a negative response is due to a different four-digit HLA restriction of the tested peptide and the HLA type of the patient. Table S3 summarizes all tested MYD88L265P-derived HLA class I ligands.
Figure 2. Spontaneous memory T cell responses are detectable in a leukemia patient. The presence of memory T cell responses in leukemia and lymphoma patients was analyzed using 12-d recall IFNγ ELISPOT assays. (A) In a single (out of 22 tested) MYD88L265P+ NHL patients (CLL-05-R) IFNγ secretion was observed after stimulation with the MYD88L265P-derived peptides P5B*15/B*40 (HQKRPIPI) and P1C*03 (RPIPIKYKAM). (B) Representative example of a MYD88L265P+ patient (CLL-03-R) where no IFNγ secretion was observed after stimulation with the MYD88L265P-derived peptides P5B*15 (HQKRPIPI) and P1C*03 (RPIPIKYKAM). An EBV epitope mix containing the frequently recognized peptides BRLF1 109–117 YVLDHLIVV (HLA-A*02) and EBNA3 247–255 RPPIFIRRL (HLA-B*07) served as positive control. Benign-tissue derived peptide DDX5 YLLPAIVHI (HLA-A*02) served as negative control. The dotted line indicates the 3-fold number of spot forming unit of the negative control. Error bars indicate ± SEM of two independent replicates. Abbreviations: SFU, spot forming unit; neg., negative; pos., positive; SEM, standard error of the mean.
Generation of MYD88L265P-specific T cells in vitro from naive T cells of CLL patients and HBDs
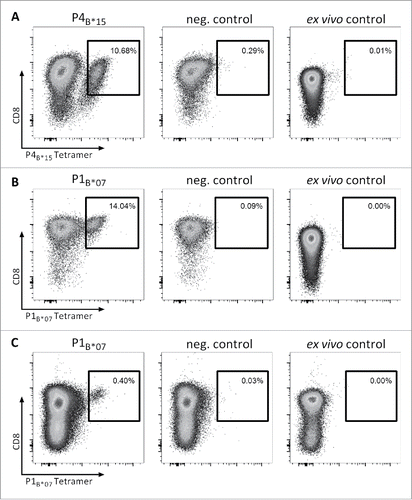
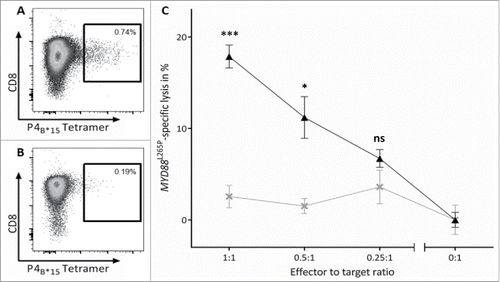
To assess whether MYD88L265P-derived peptide-specific T cell responses can be induced from naive T cells in vitro, we isolated CD8+ T cells from six HLA-B*07+ and three HLA-B*15+ HBDs as well as from two HLA-B*07+ MYD88WT CLL patients. We performed artificial antigen-presenting cell (aAPC)-based in vitro priming using the three HLA-B*07-restricted MYD88L265P-derived peptides RPIPIKYKAM (P1B*07), RPIPIKYKA (P2B*07) and SPGAHQKRPI (P3B*07) as well as the HLA-B*15-restricted peptide HQKRPIPIKY (P4B*15). Using HBD-derived CD8+ T cells, P4B*15-tetramer-positive CD8+ populations with frequencies of 1.50–10.68% of viable cells were detected in 2/3 HBDs after priming (). For the HLA-B*07-restricted peptide P1B*07, we observed tetramer-positive CD8+ populations with frequencies of 0.47–14.04% of viable cells in 6/6 HBDs (). For P2B*07, 1/3 (33%) HBDs showed a tetramer-positive CD8+ population with a frequency of 0.15% of viable cells (Fig. S1A). P3B*07-tetramer positive CD8+ populations with frequencies of 0.20–1.56% of viable cells were detected in 3/4 HBDs (Fig. S1B). Notably, after aAPC-based in vitro priming of CD8+ T cells from MYD88WT B*07+ CLL patients without previous T cell reactivity for P1B*07 (as detected by 12-day recall IFNγ ELISPOT assay and ex vivo tetramer staining), we observed in 1/2 patients a population of 0.40% P1B*07-specific CD8+ T cells within the viable cells (). No tetramer-positive T cell populations >0.10% (>0.50%) were detectable in control stainings with an HLA-B*07 (HLA-B*15)-tetramer containing a control peptide. In ex vivo control stainings no tetramer-positive T cell populations >0.01% were detectable. Furthermore, Peper et al.Citation31 demonstrated that T cell responses observed after three rounds of aAPC-based stimulations were mediated by in vitro primed naive T cells rather than by pre-existing memory T cells, as short-time stimulation of the same PBMC did not result in the detection of specific T cell populations. Collectively, all 4/4 (100%) refolded MYD88L265P-derived peptides thus are able to efficiently prime mutation-specific T cells in certain HLA contexts in HBDs as well as in CLL patients.
Figure 3. Efficient in vitro generation of P4B*15- and P1B*07-specific CD8+ T cells from naive T cells of CLL patients and HBDs. Representative tetramer stainings of CD8+ T cells after three cycles of aAPC-based in vitro priming using CD8+ T cells derived from HLA-matched HBDs primed with (A) the HLA-B*15-restricted peptide HQKRPIPIKY (P4B*15) and (B) the HLA-B*07-restricted peptide RPIPIKYKAM (P1B*07) as well as from HLA-matched MYD88WT CLL patient (CLL-05) primed with (C) the HLA-B*07-restricted peptide RPIPIKYKAM (P1B*07): 1st column: tetramer staining of CD8+ T cells primed with the MYD88L265P-derived peptide; 2nd column: control staining with HLA-matched tetramer containing a non-relevant control peptide on CD8+ T cells derived from the same population as T cells depicted in the 1st column; 3rd column: ex vivo tetramer staining of CD8+ T cells. In vitro primings with HBD-derived PBMCs were performed in six (P1B*07) and three (P4B*15) independent replicates, respectively. For the in vitro priming with PBMCs of CLL patients two independent replicates were conducted. Abbreviations: neg., negative.
MYD88L265P-specific T cells selectively recognize the mutated epitopes and are multi-functional
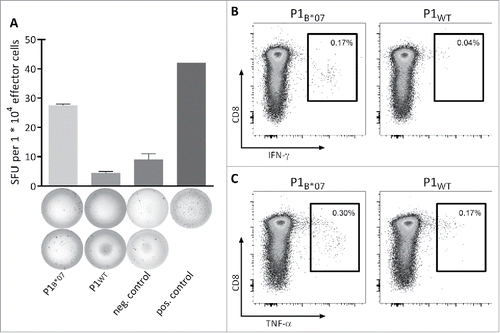
To assess the functional potential of MYD88L265P-specific CD8+ T cells, the peptide-specific secretion of IFNγ and TNFα as well as the expression of the degranulation marker CD107a were analyzed after stimulation with mutation-derived peptides in comparison to the corresponding WT peptides. P1B*07-specific CD8+ T cells of three different HBDs primed in vitro with aAPCs secreted IFNγ after stimulation with the peptide P1B*07 but not after stimulation with the corresponding WT peptide, as detected by IFNγ ELISPOT assay () or intracellular cytokine staining (). P1B*07-specific CD8+ T cells of two tested HBDs also showed an increased TNFα secretion in response to the mutation-derived peptide, but not in response to the corresponding WT peptide (). Moreover, the P1B*07-specific CD8+ T cells of 1/2 donors expressed the degranulation marker CD107a after stimulation with the peptide P1B*07 (data not shown). P4B*15-specific CD8+ T cells showed IFNγ as well as TNFα secretion after stimulation with the peptide P4B*15 but not after stimulation with the respective WT peptide (data not shown).
Figure 4. Functionality and specificity of MYD88L265P-specific T cells. Functionality and specificity of MYD88L265P-specific CD8+ T cells were analyzed by (A) IFNγ ELISPOT assay or (B, C) intracellular cytokine staining. Both assays showed increased production of IFNγ or TNFα after stimulation with the mutation-derived peptide (P1B*07) in comparison with the corresponding WT peptide (P1WT). Representative examples of two different donors are shown. The frequency of P1B*07-specific CD8+ T cell populations was 2.69% (A) and 0.40% (B and C), respectively, as detected by tetramer staining (not shown). Error bars indicate ± SEM of two independent replicates. Abbreviations: SFU, spot forming unit; neg., negative; pos., positive; SEM, standard error of the mean.
To investigate the possibility that the observed T cell responses against the mutated MYD88L265P peptides resulted from molecular mimicry, the MYD88L265P-derived peptides were compared with proteins from microorganisms and viruses. Sequence homology screens against prokaryotic and virus protein sequences (using Smith–Waterman protein searcher), identified no homologous sequences for the peptides P1 (RPIPIKYKAM), P2 (RPIPIKYKA), P3 (SPGAHQKRPI) and P4 (HQKRPIPIKY). Thus indicating that the detected responses are not the result of cross-reacting microorganism- or virus-specific T cells.
MYD88L265P-specific CD8+ T cells elicit mutation-restricted cytotoxicity
To examine peptide recognition and antigen-specific cell lysis of P1B*07- and P4B*15-stimulated CD8+ T cells, cytotoxicity assays (VITAL assays) with in vitro primed effector cells of HBDs were performed. The effector cells were polyclonal cell populations with 0.12% and 0.74% frequencies of P1B*07- and P4B*15-specific CD8+ T cells, respectively (, Fig. S2A). P4B*15-specific CD8+ T cells showed 17.9% (±1.2%) MYD88L265P-peptide-specific significant cell killing at an effector to target ratio (E/T) of 1:1 compared with 2.6% (±1.2%) of non-specific cell lysis of unspecific effector cells against the same targets in three independent replicates, respectively. The specific lysis showed E/T ratio dependent characteristics with specific lysis decreasing with reduced E/T ratios (). P1B*07-specific CD8+ T cells specifically killed 11.4% (±1.7%) of MYD88L265P-loaded targets at an E/T ratio of 0.7:1 in comparison to 2.1% unspecific lysis of unspecific effector cells (Fig. S2C). These results demonstrated clearly MYD88L265P-specificity and cytolytic potential of the in vitro primed CD8+ T cells.
Figure 5. MYD88L265P-selective cytotoxicity of P4B*15-specific effector cells. The MYD88L265P-specific cytotoxicity was analyzed in a VITAL cytotoxicity assay with CD8+ effector cells of in vitro primed cells of HBDs. (A, B) Tetramer staining of polyclonal effector cells one day before the VITAL assay determined the number of P4B*15-specific effector cells in the (A) population of successfully P4B*15-primed CD8+ T cells and in the (B) population of control cells primed with a HLA-matched non-relevant peptide. These control cells were used as unspecific effectors for the determination of the unspecific lysis of target cells. (C) At an effector to target ratio of 1:1 P4B*15-specific effectors (▴) exerted 17.9% (±1.2%) MYD88L265P-specific and significant higher lysis of P4B*15-loaded autologous target cells in comparison to P4WT-loaded cells. P4B*15-unspecific effectors (×) only caused 2.6% (±1.2%) unspecific lysis of the same targets. Results are shown for three independent replicates. Error bars indicate ± SEM. Abbreviations: SEM, standard error of the mean; n.s., not significant; * p > 0.05; ** p > 0.01; *** p > 0.001.
Discussion
T cell based immunotherapy combined with immune checkpoint modulation has enabled new treatment possibilities for a range of solid tumors.Citation32-35 Furthermore, the clinical investigation of T cell based immunotherapy for hematological malignancies has made significant progress over the past years.Citation36,37 Specific anticancer immune responses could be improved and guided further by antigen-specific immunotherapy. To this end, the identification and exact knowledge of immunogenic tumor-specific as well as tumor-associated T cell epitopes is essential.Citation8,38 In NHL, a multitude of studies have examined tumor-associated antigens, and identified an array of promising targets.Citation39-41 Tumor-specific neoantigens, which are derived from protein-altering mutational events, are being viewed as the most attractive targets of T cell based immunotherapy because they may be recognized as foreign by the immune system and can indeed provoke tumor-specific T cell immunity. Citation7,8 Mutations in the nucleophosmin 1 gene, which are among the most common molecular alterations in acute myeloid leukemia, give rise to immunogenic peptides, which elicit spontaneous T cell responses in patients.Citation42 Furthermore, patients with immune responses to these peptides showed an improved overall survival.Citation43 For these reasons, the identification and characterization of further mutation-derived HLA ligands for T cell based immunotherapy is of great interest. The recurrent oncogenic driver mutation of the TLR adaptor protein MYD88, Leu265Pro (L265P) fulfills the criteria of being tumor-specific yet widely occurring with up to 90% frequency of different NHL subtype patients.Citation9-15 In the present study, we therefore screened peptides containing the L265P mutation (MYD88L265P) for immunogenicity and thus as potential tools for tumor-specific immunotherapy.
In a reverse immunological approach we predicted potential MYD88L265P-derived HLA class I ligands. Two of these peptides emerged as gain-of-anchor antigens carrying the mutation in their P2-anchor position. Based on other studies, such peptides have been proposed to be especially suited for immunotherapy since their WT counterpart would not be bound by the same HLA molecule.Citation30
Out of 22 tested MYD88L265P-mutated NHL patients, only one showed a weak preexisting immune response against MYD88L265P-derived epitopes. We analyzed all predicted binders on every HLA-matched MYD88L265P-mutated NHL patient matching only the two digit typing of patient and prediction to maximize the number of possible tests and to account for the inaccuracy of the predictions. Therefore, it is possible that a negative result in the IFNγ ELISPOT could be due to imprecise match with the four digit typing of the predictions and tested patients. However, the allotype-specific frequencies (25–33%) for detectable memory T cell responses in MYD88L265P-mutated NHL patients are comparable to frequencies described in previous studies (CLL, Myeloma). Furthermore, in DLBCL, for example, mutations of genes controlling the immune recognition by T cells have been described.Citation13 However, the overall survival was better in patients carrying the MYD88 mutation suggesting that MYD88-directed immune responses could be involved in tumor rejection.Citation44 Although this awaits formal proof, the observed recall response could be taken as an indication that L265P peptides can be naturally presented. Additionally, in vitro aAPC-based priming in HBDs and CLL patients showed clearly that a T cell response against MYD88L265P-derived peptides could be effectively induced from naïve T cells. The strong immunogenicity of the mutation-derived peptides P1B*07 and P4B*15, as well as the mutation-specificity of the peptide-specific CD8+ T cells was demonstrated by IFNγ and TNFα secretion, expression of the degranulation marker CD107a, and the specific cytotoxic activities of the effector cells. These results demonstrated clearly that the gain-of-function driver mutationCitation12 MYD88L265P could be highly immunogenic. We present evidence that peptide-loaded target cells can be killed by MYD88L265P peptide-primed T cells with an efficiency similar to other published studies.Citation42,45,46 Nevertheless future studies outside the scope of this screening approach should investigate whether killing of cells naturally expressing WT vs. L265P-mutated MYD88 is as effective. Should this indeed be the case, MYD88L265P-based peptide vaccination, alone or in combination with checkpoint inhibitors or additional immunogenic peptides, could maybe be used to induce or boost effective antitumor immune responses; as a complementation to current approaches to target MYD88L265P-mutated NHL pharmacologically at the level of MyD88 dimerizationCitation47 or downstream Interleukin-1 receptor-associated kinase, Bruton's tyrosine kinase or TAK1.Citation48 This could either be done by vaccination of the patient using MYD88L265P-derived peptides or MyD88L265P-encoding RNA;Citation49 alternatively, for example if priming in the patient is suboptimal, naïve T cells could be isolated, primed and expanded ex vivo and re-administered to the patient; third, the transduction of non-specific T cells with TCR α and β chains conferring specificity Citation50 for MYD88L265P peptide:HLA complexes could be envisaged. Further investigations will be necessary to fully explore and harness this potential.
In conclusion, our study shows that MYD88L265P can elicit specific CD8+ T cell responses and therefore may emerge as a promising target for tumor-specific immunotherapy in a variety of different NHL subtypes.
Materials and methods
Patients and blood samples
CLL, DLBCL, lymphoplasmacytic lymphoma (LPL), primary cerebral NHL (PCNHL) or intraocular lymphoma (IOL) patients were recruited at the Erasmus Medical Center Rotterdam, the Department of Hematology, Oncology and Stem Cell Transplantation of the University Medical Center Freiburg, the Department of Hematology and Oncology of the University Hospital Tübingen as well as at the University Eye Hospital Tübingen in collaboration with the Department of Pathology, Tübingen. Informed written consent was obtained in accordance with the Declaration of Helsinki protocol. The study was performed according to the guidelines of and approval by the local ethics committees (373/2011BO2). Patient characteristics are provided in Table S1. Peripheral blood mononuclear cells (PBMCs) from patients as well as PBMCs from healthy blood donors (HBDs) were isolated by density gradient centrifugation (Biocoll, Biochrom GmbH, L 6113). PBMCs from patients were cryopreserved and stored at -80°C until analysis in ELISPOT assays. HBDs were recruited at the University Hospital Tübingen Blood transfusion unit and respective whole blood obtained from blood donations.
MYD88L265P genotyping of cancer cells and HLA typing of patients
For the identification of MYD88L265P-mutated CLL patients, DNA of PBMCs was isolated with the QIAamp DNA Mini Kit (Qiagen, 51304). Each 5 µl reaction consisted of 2.5 µl of TaqMan Universal Master Mix II (life technologies, 4440047), oligonucleotides (0.5 µM of each primer GCAGACAGTGATGAACCTCAGGA and AAGGGCCTGATGCCAGC) from TIB MOLBIOL, 0.25 µM of each probe (YAK-AGCGACCGATCCCCATCA-Q and FAM-AGCGACTGATCCCCATCAAGT-Q) and 10 ng of sample DNA. The probes were labeled with the fluorescent dyes yakima yellow (YAK) and fluorescein (FAM). The PCR was performed in a QuantStudio 7 Flex instrument (life technologies, Carlsbad, California, USA) with the following conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. In a cohort of n = 456 CLL patients, MYD88L265P mutation was detected in 10/456 specimens (2.2% MYD88L265P+ CLL patients). In DLBCL, PCNHL, IOL and LPL the MYD88L265P mutation was determined as described previously.Citation51,52
The HLA typings were performed by the Department of Hematology and Oncology, Tübingen and the German Red Cross blood donation center NSTOB, Institute Dessau (accredited by the European Federation for Immunogenetics). In total, the MYD88L265P+ patient cohort comprised 24 patients covering 11 different HLA-A and 14 HLA-B alleles. HLA typing for HLA-C alleles was performed for 15 patients and revealed seven different HLA-C alleles (Table S1).
Prediction of peptide binding to HLA class I alleles
To discover peptide targets, HLA-binding affinity was predicted across all possible 8- to 12-mer peptides encoded by the MYD88L265P mutation by using the HLA-peptide binding prediction algorithms NetMHC 3.4,Citation26-28,53 SYFPEITHICitation29 and an extended in-house database. This in-house database implements the NetMHC 3.4 prediction server as well as the latest SYFPEITHI matrices which are regularly updated by the newest results generated in our department. For the predictions by SYFPEITHI the threshold for binders was defined as 50% of the maximum score for each HLA allotype. Peptides predicted by NetMHC 3.4 with IC50<50 nM were considered strong binders; peptides with IC50 values of 50–500 nM were considered weak binders. To verify the ability of the identified peptides to indeed bind to the predicted HLA class I molecules HLA:peptide monomers were produced.
Peptide and HLA peptide monomer synthesis
Peptides were synthesized using the automated peptide synthesizer EPS221 (Abimed, Langenfeld, Germany) by applying standard 9-fluorenylmethyl-oxycarbonyl/tert-butyl (Fmoc/tBu) strategy.Citation54,55 Purity was assessed by reversed phase HPLC (e2695; Waters, Eschborn, Germany) and identity affirmed by mass spectrometry. Lyophilized peptides were dissolved at 10 mg/mL in DMSO (WAK Chemie, WAK-DMSO) and further diluted in bidestilled H2O. Biotinylated recombinant HLA molecules, HLA:peptide monomers and fluorescent HLA:peptide tetramers were produced as described previously.Citation56,57
Amplification of peptide-specific T cells for IFNγ ELISPOT assays
PBMCs from patients were cultured as described previously.Citation41,58 In brief, for CD8+ T cell stimulation, PBMCs were pulsed with 1 µg/mL per peptide 24 h after thawing and cultured for 12 days, adding 5 ng/mL human IL-4 (PeproTech, 200–04) and 5 ng/mL human IL-7 (PromoKine, C-61712) on day 0 and 1 as well as 2 ng/mL human IL-2 (R&D Systems, 202-IL) on day 3, 5, 7 and 9, respectively. Peptide-stimulated PBMCs were analyzed by IFNγ ELISPOT assays on day 12.
IFNγ ELISPOT assay
IFNγ ELISPOT assays were performed as described previously.Citation59 In brief, 96-well nitrocellulose plates (Merck Millipore, MSHAN4B50) were coated with 1 mg/mL IFNγ mAb (Mabtech, 3420–3–250) and incubated overnight at 4°C. Plates were blocked with 10% human serum for 2 h at 37°C. 2.5 – 5 × 105 cells/well of pre-stimulated PBMCs were pulsed with 1 µg/mL peptide and incubated for 24 h. For IFNγ ELISPOT assays implementing CD8+ effector cells obtained by aAPC-based in vitro priming, autologous PBMCs depleted of monocytes and CD8+ T cells were used as antigen-presenting cells (APCs). For this purpose, APCs were pulsed before the ELISPOT assay for 2 h with 2 µg/mL of the respective peptide. Secretion of IFNγ was detected using an ELISPOT kit (Mabtech, 3420–2A) according to manufacturer's instructions. Phytohaemagglutinin (PHA; Sigma Life Science, L1668) as well as an EBV epitope mix containing the frequently recognized peptides YVLDHLIVV (BRLF1, HLA-A*02), RLRAEAQVK (EBNA3, HLA-A*03), RPPIFIRRL (EBNA3, HLA-B*07), RAKFKQLL (BZLF1, HLA-B*08), and AEGGVGWRHW (EBNA6, HLA-B*44) served as positive controls. HLA-A*01 (GSEELRSLY, POL_HV1H2), -A*02 (YLLPAIVHI, DDX5_HUMAN), -A*03 (RLRPGGKKK, GAG_HV1BR), -A*24 (AYVHMVTHF, BI1_HUMAN), -B*07 (TPGPGVRYPL, NEF_HV1H2), and -B*08 (DIAARNVL, FAK1_HUMAN) -restricted control peptides derived from the human immunodeficiency virus (HIV) or benign tissues served as negative controls. Spots were counted using an ImmunoSpot S5.0.9.21 analyzer (CTL, Shaker Heights, Ohio, USA). T cell responses were evaluated according to the cancer immunoguiding program (CIP) guidelinesCitation60 and considered to be positive when >10 spots/well were counted and the mean spot count per well was at least 3-fold higher than the mean number of spots in the negative control wells.
Generation of peptide-specific CD8+ T cells by aAPC-based in vitro priming
For generation of aAPC, 5.6-μm-diameter streptavidin-coated polystyrene beads (Bangs Laboratories, CP01N) were re-suspended at 2 × 106 particles per mL, incubated with 200 pM of biotinylated HLA:peptide monomer and 20 nM biotinylated anti-CD28 antibody (produced in-house) for 30 min at room temperature.Citation61 CD8+ T cells from patients and HBDs were enriched from PBMCs by positive selection using magnetic cell sorting (Miltenyi Biotec, 130–045–201). 1 × 106 CD8+ T cells per well were cultured in round bottom 96-well plates (Costar/Corning) and stimulated with 2 × 105 aAPCs and 5 ng/mL human IL-12 (PromoKine, C-62213) three times with a 7 d stimulation interval. 65 U/μL IL-2 (R&D Systems, 202-IL) were added 2 d after stimulation.Citation31 After priming the cells were expanded over 2–6 weeks, fed every 3–4 d with medium containing 150 U/mL IL-2 as well as every 2 weeks with feeder cells (freshly isolated PBMCs of HBDs plus LG2-EBV cells) together with 1 µg/mL PHA-L (Sigma-Aldrich, 11249738001) and 150 U/mL IL-2.
Tetramer staining
The frequency of peptide-specific CD8+ T cells after aAPC priming was determined on a FACS Canto II cytometer (BD Bioscience, Franklin Lakes, New Jersey, USA) by staining with anti-CD8-PerCP (BioLegend, 301030) and HLA:peptide-tetramer-PE as described previously.Citation61 Stainings with tetramers of the same HLA allotype containing irrelevant control peptides served as negative controls. The priming was considered successful if the frequency of peptide-specific CD8+ T cells was ≥ 0.1% of sorted CD8+ T cells and at least 3-fold higher than the frequency of peptide-specific CD8+ T cells in the negative control.
Intracellular cytokine staining
The functionality of peptide-specific CD8+ T cells was analyzed by intracellular IFNγ and TNFα staining as well as staining of the degranulation marker CD107a as described previously.Citation59,62 PBMCs were pulsed with 1 μg/mL of individual peptide and incubated in the presence of 10 μg/mL brefeldin A (Sigma-Aldrich, B6542) and 10 μg/mL GolgiStop (BD Bioscience, 51–2092KZ) for 12–16 h. Cells were labeled using anti-CD107a-FITC (BD Bioscience, 555800), anti-CD8-PerCP (BioLegend, 301030), Cytofix/Cytoperm (BD Bioscience, 554722), anti-TNFα-Pacific Blue (BioLegend, 502920), and anti-IFNγ-PE (BD Bioscience, 559327). Samples were analyzed on a FACS Canto II cytometer (BD Bioscience, Franklin Lakes, New Jersey, USA). Cells stimulated with the corresponding non-mutated (WT) peptide served as negative control.
Cytotoxicity assay (VITAL assay)
The cytolytic capacity of peptide-specific CD8+ T cells was tested using the flow cytometry-based VITAL assay, essentially as described previously.Citation63 Autologous target cells (2 × 106 PBMCs depleted of monocytes and CD8+ T cells) were loaded with peptides by overnight incubation in T cell medium supplemented with 10 μg/mL of the respective peptide. Cells loaded with mutation-derived peptides and the corresponding WT controls were labeled with 0.5 µM of the fluorescent dyes carboxyfluorescein diacetate succinimidyl ester (CFSE) and Far Red (both Invitrogen, C34554, L34973), respectively. The differentially labeled targets were combined in a 1:1 ratio and 6,000 cells of each fluorescent population were plated in a 96-well round-bottomed plate. Effector cells were added in the indicated effector to target ratios (E/T) in triplicates. Following incubation for 24 h at 37°C, all cells were assessed by FACS analysis using a FACS Canto II cytometer (BD Bioscience, Franklin Lakes, New Jersey, USA).
The specific lysis of mutated peptide-loaded target cells was calculated relative to WT peptide-loaded control targets. Effector-independent lysis was assessed in parallel in wells containing no effector cells and the specific lysis was normalized according to the following equation:
Search for homologous peptide sequences
Screening sequence homology of MYD88L265P peptides with prokaryotic and virus proteins was performed for P1 (RPIPIKYKAM), P2 (RPIPIKYKA), P3 (SPGAHQKRPI), and P4 (HQKRPIPIKY), using the Smith–Waterman protein searcher application (SSEARCH) on the European Molecular Biology Laboratory server (http://www.ebi.ac.uk/Tools/sss/fasta/), using default settings.
Software and statistical analysis
Flow cytometric data analysis was performed using FlowJo 10.0.7 (Treestar, Ahland, Oregon, USA). Taqman assay data analysis was performed using the QuantStudio™ Real-Time PCR software (ThermoFisher, Waltham, Massachusetts, USA). GraphPad Prism 6.0 (GraphPad Software, La Jolla, California, USA) was used for the generation of plots and for statistical analysis. Statistical analysis of MYD88L265P-specific lysis was based on unpaired t tests.
Disclosure of potential conflicts of interest
The authors declare no competing financial interests. A.N., J.S.S., O.O.W., D.J.K., H.-G.R., and A.N.R.W. are listed as inventors in a filed patent application.
KONI_A_1219825_s02.pdf
Download PDF (695.8 KB)Acknowledgments
We thank Sabine Dickenhöfer, Patricia Hrstic, Nicole Zuschke and Katharina Graf for excellent technical support. Dr med. Christoph Deuter and Dr med. Daniela Süsskind (Eye Hospital, University of Tübingen) kindly provided us further PBMC samples of lymphoma patients.
Funding
This work was supported by the German Research Foundation (DFG) funded Collaborative Research Center (SFB) 685 “Immunotherapy,” the Else-Übelmesser-Stiftung, the European Union (EU; ERC AdG339842 MUTAEDITING), the German Cancer Consortium (DKTK), the University Hospital and the University of Tübingen.
References
- Boon T, Cerottini J-C, Van den Eynde B, van der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol 1994; 12:337-65; PMID:8011285; http://dx.doi.org/10.1146/annurev.iy.12.040194.002005
- Ostrand-Rosenberg S. Tumor immunotherapy: the tumor cell as an antigen-presenting cell. Curr Opin Immunol 1994; 6:722-7; PMID:7826527; http://dx.doi.org/10.1016/0952-7915(94)90075-2
- Townsend SE, Allison JP. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science 1993; 259:368-70; PMID:7678351; http://dx.doi.org/10.1126/science.7678351
- Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med 2012; 18:1254-61; PMID:22842478; http://dx.doi.org/10.1038/nm.2883
- Perez-Gracia JL, Labiano S, Rodriguez-Ruiz ME, Sanmamed MF, Melero I. Orchestrating immune check-point blockade for cancer immunotherapy in combinations. Curr Opin Immunol 2014; 27:89-97; PMID:24485523; http://dx.doi.org/10.1016/j.coi.2014.01.002
- Ansell SM. Targeting immune checkpoints in lymphoma. Curr Opin Hematol 2015; 22:337-42; PMID:26049755; http://dx.doi.org/10.1097/MOH.0000000000000158
- Coulie PG, Lehmann F, Lethe B, Herman J, Lurquin C, Andrawiss M, Boon T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc Natl Acad Sci U S A 1995; 92:7976-80; PMID:7644523; http://dx.doi.org/10.1073/pnas.92.17.7976
- van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 2013; 31:e439-42; PMID:24043743; http://dx.doi.org/10.1200/JCO.2012.47.7521
- Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, Sheehy P, Manning RJ, Patterson CJ, Tripsas C et al. MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. N Engl J Med 2012; 367:826-33; PMID:22931316; http://dx.doi.org/10.1056/NEJMoa1200710
- Varettoni M, Arcaini L, Zibellini S, Boveri E, Rattotti S, Riboni R, Corso A, Orlandi E, Bonfichi M, Gotti M et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom's macroglobulinemia and related lymphoid neoplasms. Blood 2013; 121:2522-8; PMID:23355535; http://dx.doi.org/10.1182/blood-2012-09-457101
- Xu L, Hunter ZR, Yang G, Zhou Y, Cao Y, Liu X, Morra E, Trojani A, Greco A, Arcaini L et al. MYD88 L265P in Waldenstrom macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood 2013; 121:2051-8; PMID:23321251; http://dx.doi.org/10.1182/blood-2012-09-454355
- Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011; 470:115-9; PMID:21179087; http://dx.doi.org/10.1038/nature09671
- Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet 2011; 43:830-7; PMID:21804550; http://dx.doi.org/10.1038/ng.892
- Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, Escaramis G, Jares P, Bea S, Gonzalez-Diaz M et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011; 475:101-5; PMID:21642962; http://dx.doi.org/10.1038/nature10113
- Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013; 152:714-26; PMID:23415222; http://dx.doi.org/10.1016/j.cell.2013.01.019
- Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. GLOBOCAN 2012 v1.0. Cancer Incidence and Mortality Worldwide: IARC CancerBase No 11, 2013:International Agency for Research on Cancer.
- Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, Xu W, Tan B, Goldschmidt N, Iqbal J et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 2008; 359:2313-23; PMID:19038878; http://dx.doi.org/10.1056/NEJMoa0802885
- Tsimberidou A-M, Keating MJ. Treatment of patients with fludarabine-refractory chronic lymphocytic leukemia: need for new treatment options. Leuk Lymphoma 2010; 51:1188-99; PMID:20545582; http://dx.doi.org/10.3109/10428194.2010.486089
- Schmitt M, Schmitt A, Rojewski MT, Chen J, Giannopoulos K, Fei F, Yu Y, Gotz M, Heyduk M, Ritter G et al. RHAMM-R3 peptide vaccination in patients with acute myeloid leukemia, myelodysplastic syndrome, and multiple myeloma elicits immunologic and clinical responses. Blood 2008; 111:1357-65; PMID:17978170; http://dx.doi.org/10.1182/blood-2007-07-099366
- Porter DL, Levine BL, Bunin N, Stadtmauer EA, Luger SM, Goldstein S, Loren A, Phillips J, Nasta S, Perl A et al. A phase 1 trial of donor lymphocyte infusions expanded and activated ex vivo via CD3/CD28 costimulation. Blood 2006; 107:1325-31; PMID:16269610; http://dx.doi.org/10.1182/blood-2005-08-3373
- Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 2011; 17:4550-7; PMID:21498393; http://dx.doi.org/10.1158/1078-0432.CCR-11-0116
- Timmerman JM, Czerwinski DK, Davis TA, Hsu FJ, Benike C, Hao ZM, Taidi B, Rajapaksa R, Caspar CB, Okada CY. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune responses in 35 patients. Blood 2002; 99:1517-26; PMID:11861263; http://dx.doi.org/10.1182/blood.V99.5.1517
- Park HJ, Neelapu SS. Developing idiotype vaccines for lymphoma: from preclinical studies to phase III clinical trials. Brit J Haematol 2008; 142:179-91; PMID:18422783; http://dx.doi.org/10.1111/j.1365-2141.2008.07143.x
- Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, Lih C-J, Williams PM, Shaffer AL, Gerecitano J. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med 2015; 21:922-6; PMID:26193343; http://dx.doi.org/10.1038/nm.3884
- Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Research 2008; 36:W509-W12; PMID:18463140; http://dx.doi.org/10.1093/nar/gkn202
- Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics 2008; 24:1397-8; PMID:18413329; http://dx.doi.org/10.1093/bioinformatics/btn128
- Nielsen M, Lundegaard C, Worning P, Hvid CS, Lamberth K, Buus S, Brunak S, Lund O. Improved prediction of MHC class I and class II epitopes using a novel Gibbs sampling approach. Bioinformatics 2004; 20:1388-97; PMID:14962912; http://dx.doi.org/10.1093/bioinformatics/bth100
- Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 2003; 12:1007-17; PMID:12717023; http://dx.doi.org/10.1110/ps.0239403
- Schuler MM, Nastke MD, Stevanovikc S. SYFPEITHI: database for searching and T-cell epitope prediction. Methods Mol Biol 2007; 409:75-93; PMID:18449993; http://dx.doi.org/10.1007/978-1-60327-118-9_5
- Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, Pawashe AP, Blanchard T, McMahon D, Sidney J, Sette A et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med 2014; 211:2231-48; PMID:25245761; http://dx.doi.org/10.1084/jem.20141308
- Peper JK, Bösmüller H-C, Schuster H, Gückel B, Hörzer H, Roehle K, Schäfer R, Wagner P, Rammensee H-G, Stevanović S et al. HLA ligandomics identifies Histone Deacetylase 1 as target for Ovarian Cancer immunotherapy. OncoImmunology 2016; 5(5):e1065369; PMID:27467910; http://dx.doi.org/10.1080/2162402X.2015.1065369
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369:122-33; PMID:23724867; http://dx.doi.org/10.1056/NEJMoa1302369
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:22658127; http://dx.doi.org/10.1056/NEJMoa1200690
- Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014; 384:1109-17; PMID:25034862; http://dx.doi.org/10.1016/S0140-6736(14)60958-2
- Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, Sebastian M, Neal J, Lu H, Cuillerot JM et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol 2012; 30:2046-54; PMID:22547592; http://dx.doi.org/10.1200/JCO.2011.38.4032
- Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 2015; 372:311-9; PMID:25482239; http://dx.doi.org/10.1056/NEJMoa1411087
- Martin P, Furman RR, Rutherford S, Ruan J, Ely S, Greenberg J, Coleman M, Goldsmith SJ, Leonard JP. Phase I study of the anti-CD74 monoclonal antibody milatuzumab (hLL1) in patients with previously treated B-cell lymphomas. Leuk Lymphoma 2015; 56(11):3065-70; PMID:25754579; http://dx.doi.org/10.3109/10428194.2015.1028052
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014; 371:2189-99; PMID:25409260; http://dx.doi.org/10.1056/NEJMoa1406498
- Brossart P, Schneider A, Dill P, Schammann T, Grunebach F, Wirths S, Kanz L, Buhring HJ, Brugger W. The epithelial tumor antigen MUC1 is expressed in hematological malignancies and is recognized by MUC1-specific cytotoxic T-lymphocytes. Cancer Res 2001; 61:6846-50; PMID:11559560.
- Walz S, Stickel J, Kowalewski DJ, Schuster H, Weisel K, Backert L, Kahn S, Nelde A, Stroh T, Handel M et al. The antigenic landscape of multiple myeloma: mass spectrometry (re-)defines targets for T-cell based immunotherapy. Blood 2015; 126(10):1203-13; PMID:26138685; http://dx.doi.org/10.1182/blood-2015-04-640532
- Berlin C, Kowalewski DJ, Schuster H, Mirza N, Walz S, Handel M, Schmid-Horch B, Salih HR, Kanz L, Rammensee HG et al. Mapping the HLA ligandome landscape of acute myeloid leukemia: a targeted approach toward peptide-based immunotherapy. Leukemia 2015; 29, 647-59; PMID:25092142; http://dx.doi.org/10.1038/leu.2014.233
- Greiner J, Ono Y, Hofmann S, Schmitt A, Mehring E, Gotz M, Guillaume P, Dohner K, Mytilineos J, Dohner H et al. Mutated regions of nucleophosmin 1 elicit both CD4(+) and CD8(+) T-cell responses in patients with acute myeloid leukemia. Blood 2012; 120:1282-9; PMID:22592607; http://dx.doi.org/10.1182/blood-2011-11-394395
- Greiner J, Schneider V, Schmitt M, Götz M, Döhner K, Wiesneth M, Döhner H, Hofmann S. Immune responses against the mutated region of cytoplasmatic NPM1 might contribute to the favorable clinical outcome of AML patients with NPM1 mutations (NPM1mut). Blood 2013; 122:1087-8; PMID:23929838; http://dx.doi.org/10.1182/blood-2013-04-496844
- Martinez-Trillos A, Pinyol M, Navarro A, Aymerich M, Jares P, Juan M, Rozman M, Colomer D, Delgado J, Gine E et al. Mutations in TLR/MYD88 pathway identify a subset of young chronic lymphocytic leukemia patients with favorable outcome. Blood 2014; 123:3790-6; PMID:24782504; http://dx.doi.org/10.1182/blood-2013-12-543306
- Stanke J, Hoffmann C, Erben U, von Keyserling H, Stevanovic S, Cichon G, Schneider A, Kaufmann AM. A flow cytometry-based assay to assess minute frequencies of CD8+ T cells by their cytolytic function. J Immunological Methods 2010; 360:56-65; PMID:20558172; http://dx.doi.org/10.1016/j.jim.2010.06.005
- Andersen MH, Reker S, Kvistborg P, Becker JC, Straten Pt. Spontaneous Immunity against Bcl-xL in Cancer Patients. J Immunol 2005; 175:2709-14; PMID:16081848; http://dx.doi.org/10.4049/jimmunol.175.4.2709
- Avbelj M, Wolz O-O, Fekonja O, Benčina M, Repič M, Mavri J, Krüger J, Schärfe C, Garcia MD, Panter G. Activation of lymphoma-associated MyD88 mutations via allostery-induced TIR-domain oligomerization. Blood 2014; 124:3896-904; PMID:25359991; http://dx.doi.org/10.1182/blood-2014-05-573188
- Ansell S, Hodge L, Secreto F, Manske M, Braggio E, Price-Troska T, Ziesmer S, Li Y, Johnson S, Hart S. Activation of TAK1 by MYD88 L265P drives malignant B-cell Growth in non-Hodgkin lymphoma. Blood Cancer J 2014; 4:e183; PMID:24531446; http://dx.doi.org/10.1038/bcj.2014.4
- Rittig SM, Haentschel M, Weimer KJ, Heine A, Muller MR, Brugger W, Horger MS, Maksimovic O, Stenzl A, Hoerr I. Intradermal Vaccinations With RNA Coding for TAA Generate CD8+ and CD4+ Immune Responses and Induce Clinical Benefit in Vaccinated Patients. Molecular Therapy 2011; 19:990-9; PMID:21189474; http://dx.doi.org/10.1038/mt.2010.289
- Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, Badros AZ, Garfall A, Weiss B, Finklestein J et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 2015; 21:914-21; PMID:26193344; http://dx.doi.org/10.1038/nm.3910
- Bonzheim I, Giese S, Deuter C, Süsskind D, Zierhut M, Waizel M, Szurman P, Federmann B, Schmidt J, Quintanilla-Martinez L et al. High frequency of MYD88 mutations in vitreoretinal B-cell lymphoma: a valuable tool to improve diagnostic yield of vitreous aspirates. Blood 2015; 126:76-9; PMID:25900979; http://dx.doi.org/10.1182/blood-2015-01-620518
- Schmidt J, Federmann B, Schindler N, Steinhilber J, Bonzheim I, Fend F, Quintanilla-Martinez L. MYD88 L265P and CXCR4 mutations in lymphoplasmacytic lymphoma identify cases with high disease activity. Brit J Haematol 2015; 169:795-803; PMID:25819228; http://dx.doi.org/10.1111/bjh.13361
- Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res 2008; 1;36:W509-12; PMID:18463140; http://dx.doi.org/10.1093/nar/gkn202
- Merrifield B. Solid phase synthesis. Science 1986; 232:341-7; PMID:3961484; http://dx.doi.org/10.1126/science.3961484
- Atherton E, Fox H, Harkiss D, Logan C, Sheppard R, Williams B. A mild procedure for solid phase peptide synthesis: use of fluorenylmethoxycarbonylamino-acids. J Chem Soc Chem Commun 1978; 13:537-9; http://dx.doi.org/10.1039/c39780000537
- Garboczi DN, Hung DT, Wiley DC. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci U S A 1992; 89:3429-33; PMID:1565634; http://dx.doi.org/10.1073/pnas.89.8.3429
- Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science 1996; 274:94-6; PMID:8810254; http://dx.doi.org/10.1126/science.274.5284.94
- Kowalewski DJ, Schuster H, Backert L, Berlin C, Kahn S, Kanz L, Salih HR, Rammensee HG, Stevanovic S, Stickel JS. HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc Natl Acad Sci U S A 2015; 112.2:E166-E75; PMID:25548167; http://dx.doi.org/10.1073/pnas.1416389112
- Widenmeyer M, Griesemann H, Stevanovic S, Feyerabend S, Klein R, Attig S, Hennenlotter J, Wernet D, Kuprash DV, Sazykin AY et al. Promiscuous survivin peptide induces robust CD4+ T-cell responses in the majority of vaccinated cancer patients. Int J Cancer 2012; 131:140-9; PMID:21858810; http://dx.doi.org/10.1002/ijc.26365
- Britten CM, Gouttefangeas C, Welters MJ, Pawelec G, Koch S, Ottensmeier C, Mander A, Walter S, Paschen A, Muller-Berghaus J et al. The CIMT-monitoring panel: a two-step approach to harmonize the enumeration of antigen-specific CD8+ T lymphocytes by structural and functional assays. Cancer Immunol Immunother 2008; 57:289-302; PMID:17721783; http://dx.doi.org/10.1007/s00262-007-0378-0
- Rudolf D, Silberzahn T, Walter S, Maurer D, Engelhard J, Wernet D, Buhring HJ, Jung G, Kwon BS, Rammensee HG et al. Potent costimulation of human CD8 T cells by anti-4-1BB and anti-CD28 on synthetic artificial antigen presenting cells. Cancer Immunol Immunother 2008; 57:175-83; PMID:17657490; http://dx.doi.org/10.1007/s00262-007-0360-x
- Neumann A, Horzer H, Hillen N, Klingel K, Schmid-Horch B, Buhring HJ, Rammensee HG, Aebert H, Stevanovic S. Identification of HLA ligands and T-cell epitopes for immunotherapy of lung cancer. Cancer Immunol Immunother 2013; 62:1485-97; PMID:23817722; http://dx.doi.org/10.1007/s00262-013-1454-2
- Hermans IF, Silk JD, Yang J, Palmowski MJ, Gileadi U, McCarthy C, Salio M, Ronchese F, Cerundolo V. The VITAL assay: a versatile fluorometric technique for assessing CTL- and NKT-mediated cytotoxicity against multiple targets in vitro and in vivo. J Immunol Methods 2004; 285:25-40; PMID:14871532; http://dx.doi.org/10.1016/j.jim.2003.10.017