
ABSTRACT
Exploiting gut mucosal immunity to design new antitumor vaccination strategy remains unexplored. Tumor cell-derived microparticles (T-MP) are natural biomaterials that are capable of delivering tumor antigens and innate signals to dendritic cells (DC) for tumor-specific T cell immunity. Here, we show that T-MPs by oral vaccination route effectively access and activate mucosal epithelium, leading to subsequent antitumor T cell responses. Oral vaccination of T-MPs generated potent inhibitory effect against the growth of B16 melanoma and CT26 colon cancer in mice, which required both T cell and DC activation. T-MPs, once entering intestinal lumen, were mainly taken up by ileac intestinal epithelial cells (IEC), where T-MPs activated NOD2 and its downstream MAPK and NF-κB, leading to chemokine releasing, including CCL2, from IECs to attract CD103+ CD11c+ DCs. Furthermore, ileac IECs could transcytose T-MPs to the basolateral site, where T-MPs were captured by those DCs for cross-presentation of loaded antigen contents. Elucidating these molecular and cellular mechanisms highlights T-MPs as a novel antitumor oral vaccination strategy with great potential of clinical applications.
Introduction
Design of potent and cost-effective prophylactic vaccines against cancer is highly desirable for cancer prevention. Although this goal has been facilitated by the development of new technologies such as transcriptome sequencing,Citation1 computational modeling,Citation2 and material engineering,Citation3 the foundation for generating more efficient vaccines lies in a better understanding of the immunological principle. For instance, despite the subcutaneous or intravenous injection as the common means to deliver tumor vaccines,Citation4,5 recent advances in mucosal immunityCitation6,7 provide new opportunities to explore the oral route for prophylactic and therapeutic tumor vaccines. Oral vaccination has been practiced for hundreds years and successfully prevented serious viral and bacterial infections.Citation8,9 However, their utility seems to be limited to mucosal pathogens and application of oral vaccines to control cancer has not yet been appreciated. Compared to the systemic delivery, oral route has significant advantages by its simplicity, safety, and induction of both mucosal and systemic immune responses.Citation10 Nevertheless, oral vaccination has to overcome several gastrointestinal barriers, including the extremely low pH value in the stomach, the presence of proteolytic enzymes and bile salts and the low intestinal permeability, which limit conventional DNA, peptide and tumor cell-based vaccines for oral delivery. Thus, new strategies that can overcome the barriers of oral delivery are urgently needed for developing high efficacious and efficient tumor vaccines.
Despite the delivery barriers, other issues are also challenging for eliciting effective antitumor T cell immunity by oral tumor vaccination. For example, even if tumor antigens can cross intestinal epithelium and reach the site where they are captured by local resident antigen-presenting cells (APC), the induction of tumor-specific T cells is still not ensured. This is because appropriate innate signals and cytokine microenvironment are necessary for inducing antitumor T cell immunity. Innate signals are required to stimulate DCs to upregulate co-stimulatory molecules such as CD80 and CD86.Citation11,12 Meanwhile, the presence of Th1 cytokines such as IL-12 and type I interferon are critical for priming tumor-cytotoxic T cells.Citation13 Currently, synthetic biomaterials such as nanoparticles are used to deliver multifunctional and spatiotemporally controllable innate signals to immune cells for a better antitumor immunity.Citation14,15 Notwithstanding these advances, whether these artificial biomaterials, especially for their natural counterparts, are useful for oral vaccination remains elusive.
Tumor cells as natural biomaterials may potentially become ideal tumor vaccines. Theoretically, tumor cells harbor the tumor antigen repertoire that can generate epitopes for both CD8+ and CD4+ T cells, leading to optimal antitumor immunity and greatly diminishing the chance of tumor escape. At the same time, tumor cells also contain various danger signals such as HMGB1, HSPs, RNAs and DNAs, which can stimulate the maturation of DCs and facilitate the generation of a suitable cytokine milieu.Citation16,17 On the other hand, tumor cells also carry various immunosuppressive signals, which may diminish the efficaciousness of tumor cell-based vaccines.Citation18,19 How to enhance tumor cell-carried danger stimuli while minimize their immunosuppressive signals is one of the most critical aspects for developing desirable tumor cell-based vaccines.
Upon stimulation or apoptosis, cells may change their cytoskeletons, leading to encapsulation of cytosolic contents by cellular membrane to form vesicles that are subsequently released into the extracellular space. These specialized subcellular vesicles with a diameter of 100 to 1,000 nm are called microparticles (MP). Previously, we showed that MPs released by Listeria monocytogenes-infected macrophages can transfer L. monocytogenes antigens to DCs, leading to the induction of protective T cell immunity,Citation20 indicating a certain link of MPs to vaccines. Furthermore, we found that tumor cell-derived MPs could deliver both tumor antigens and innate signals to DCs and triggered strong antitumor immunity, thus representing a great potential to act as tumor vaccines.Citation21,22 In the present study, we further provide evidence that T-MPs can activate NOD2 signaling in ileac epithelium through oral delivery leading to strong vaccination outcome of antitumor immunity.
Results
Oral administration of T-MPs inhibits tumor growth in vivo
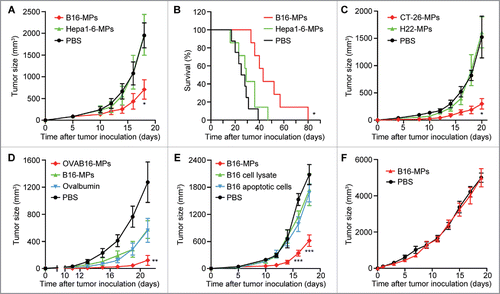
To explore whether oral vaccination of T-MPs generates antitumor effect, C57BL/6 mice pretreated with anti-acidic agent were immunized with B16 melanoma cell-derived MPs (B16-MPs) by intragastric administration (i.g.) three times ahead of a s.c. inoculation of B16 tumor cells. As shown in , compared to PBS control, the oral vaccination of B16-MPs significantly inhibited B16 tumor growth and prolonged the survival of mice (). Intriguingly, MPs derived from Hepa1-6 hepatocarcinoma cells, which have the same genetic background as B16-MPs, did not generate the anti-B16 melanoma effect (). In parallel, we found that the oral administration of MPs from CT-26 colon cancer cells rather than those from H22 hepatocarcinoma cells (both are BALB/c background) led to inhibition of CT-26 tumor growth in BALB/c mice (). Therefore, oral T-MPs seem to be capable of inducing immune protection against the growth of their parental tumors. To further verify the specificity of the above immune protection, mice were i.g. administered with OVAB16-MPs, B16-MPs, or ovalbumin, and then challenged with OVAB16 tumor cells. We found that compared with B16-MPs or ovalbumin alone, OVAB16-MPs resulted in much stronger protection outcome (). Previously, we have demonstrated that subcutaneous vaccination of T-MPs is superior to either tumor lysates or tumor exosomes.Citation21 Here, we further compared the immune protective effect of oral administration of B16-MPs, tumor lysates, and apoptotic B16 cells. To make fair comparison, these three agents were prepared from the same number of B16 cells. However, unlike B16-MPs, oral administration of B16 lysates or apoptotic B16 cells did not generate the immune protective effect, compared to the PBS control (). In addition, in T-cell-deficient nude mice, we found that B16-MPs failed to elicit the aforementioned antitumor effect (). Collectively, these data suggested that T-MPs may elicit potent antitumor T cell immunity through oral delivery.
Figure 1. Oral administration of T-MPs inhibits tumor growth in vivo. (A,B) C57BL/6 mice were immunized i.g. with B16-MPs, Hepa1-6-MPs or PBS on days -13, -11, and -7, followed by s.c. injection with 1×105 B16 tumor cells on day 0 (n = 6 per group). Tumor volumes were measured and calculated. Error bars represent mean ± SEM; *p < 0.05, B16-MPs group compared with Hepa1-6-MPs or PBS group (A). The long-term survival was analyzed. *p < 0.05, B16-MPs group compared with Hepa1-6-MPs or PBS group (B). (C) BALB/c mice were immunized i.g. with CT-26-MPs, H22-MPs or PBS on days -13, -11, and -7, followed by s.c. injection with 1×105 CT-26 tumor cells on day 0 (n = 6 per group). Tumor volumes were measured and calculated. Error bars represent mean ± SEM; *p < 0.05, CT-26-MPs group compared with H22-MPs or PBS group. (D) C57BL/6 mice were immunized i.g. with OVAB16-MPs, B16-MPs, ovalbumin or PBS on days -13, -11, and -7, followed by s.c. injection with 1×105 OVAB16 tumor cells on day 0 (n = 6 per group). Tumor volumes were measured and calculated. Error bars represent mean ± SEM; **p < 0.01, OVAB16-MPs group compared with B16-MPs, ovalbumin, or PBS group. (E) C57BL/6 mice were immunized i.g. with B16-MPs, lysate, apoptotic cells, or PBS on days -13, -11, and -7, followed by s.c. injection with 1×105 B16 tumor cells on day 0 (n = 6 per group). Tumor volumes were measured and calculated. Error bars represent mean ± SEM; ***p < 0.001, B16-MPs group compared with lysate, apoptotic cells, or PBS group. (F) Nude mice were immunized i.g. with B16-MPs or PBS on days -13, -11, and -7, followed by s.c. injection with 1×105 B16 tumor cells on day 0 (n = 6 per group). Tumor volumes were measured and calculated. Error bars represent mean ± SEM.
Oral administration of T-MPs induces systemic tumor-specific T cell immunity
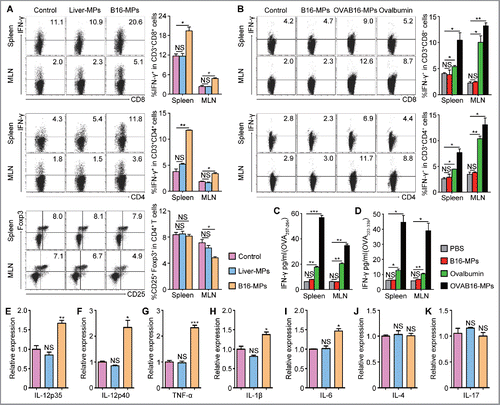
The above data indicated that the antitumor effect of oral T-MPs is T cell dependent. To further dissect the influence of oral administration of T-MPs on T cells, mice were orally administrated with B16-MPs for three times. Seven days later, cells from mesenteric lymph nodes (MLN) and spleen were isolated and stimulated with PMA and ionomycin in vitro. The results showed that the percentage of IFNγ+ CD8+ and IFNγ+ CD4+ T cells both were significantly increased while Treg cells were decreased in MLN (), however, the frequency of Th2, Th17, and Th22 cells was not changed (Figs. S1A and B), indicating that T-MPs may induce the activation and differentiation of T cells toward Th1 program. Intriguingly, as a comparison, the oral administration of normal liver cell-derived MPs could not induce T cell activation ( and Figs. S1A and B), suggesting that the activation of T cells by oral administration of T-MPs seems to be tumor-specific. To further confirm that oral administration of T-MPs indeed induces tumor-specific T cell immune response, we immunized i.g. mice with OVAB16-MPs, B16-MPs, and ovalbumin three times. Two weeks after the last vaccination, cells isolated from spleen and MLN were stimulated with OVA peptides (OVA257-264 or OVA323-339). The flow cytometric results showed that IFNγ expression of CD8+ and CD4+ T cells was strikingly upregulated in the OVAB16-MPs group and moderately upregulated in the ovalbumin group, while remained unchanged in the B16-MPs group (), which was consistent with IFNγ levels in the supernatants detected by ELISA kit (). In addition to IFNγ, we found most Th1-related inflammatory cytokines such as IL-12, TNF-α, IL-1β, and IL-6 but not IL-4 and IL-17 were also upregulated in the spleen, as evaluated by qPCR (). Together, these data suggested that oral administration of T-MPs induces systemic tumor-specific T cell immunity.
Figure 2. Oral administration of T-MPs induces systemic tumor-specific T cell immunity. (A) C57BL/6 mice were immunized i.g. with B16-MPs, Liver-MPs, or PBS control on days 1, 3, and 7 (n = 3 per group). On day 14, lymphocytes from spleen and MLN were isolated and cultured in vitro for 5 h in presence of PMA (80 nM), ionomycin (1.3 μM), and Brefeldin A (5 μg/mL), followed by flow cytometric analysis. The percentages of IFNγ+ cells in both CD8+ and CD4+ T cells were shown, as well as the percentages of Treg cells. Error bars represent mean ± SEM; *p < 0.05; **p < 0.01. (B–D) C57BL/6 mice were immunized i.g. with OVAB16-MPs, B16-MPs, ovalbumin or PBS control on days 1, 3, and 7 (n = 3 per group). On day 21, lymphocytes isolated from spleen and MLN were restimulated with OVA257-264 and OVA323-339 in vitro, respectively, for 72 h. The percentages of IFNγ+ cells in both CD8+ and CD4+ T cells were analyzed by flow cytometry (B), and the IFNγ levels in the culture supernatant were measured by ELISA (C, D). Error bars represent mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001. (E–K) C57BL/6 mice were immunized i.g. with B16-MPs, Liver-MPs, or PBS control on days 1, 3, and 7 (n = 3 per group). On day 14, lymphocytes from spleen were evaluated for the expression of inflammatory cytokines IL-12p35 (E), IL-12p40 (F), TNF-α (G), IL-1β (H), IL-6 (I), IL-4 (J), and IL-17 (K) by qPCR. Error bars represent mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001; all experimental groups compared with control group.
Dendritic cells are required for oral T-MP-induced antitumor T cell immunity
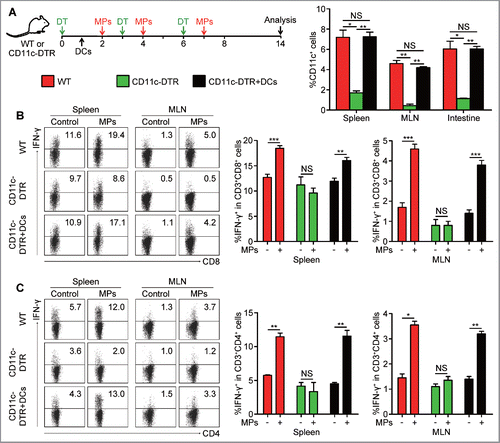
Next, we tried to explore how tumor-specific T cell immunity was initiated by oral administration of T-MPs. We previously found that T-MPs alone were not sufficient to stimulate T cell proliferation,Citation21 suggesting that uptake of T-MPs by APC is critical for antigen presentation. Although there are different types of APCs at intestinal site, DCs are generally considered as the professional APCs that are indispensable for the initiation of adaptive immune responses. Thereby, we tested whether the above oral T-MP-induced T cell immunity was mediated through DCs presenting antigens. We used diphtheria toxin (DT) to deplete DCs in CD11c-DTR mice (). A high depleting efficiency was observed at the sites of the spleen, MLN, and intestine (). Due to the depletion of DCs, T-MPs failed to stimulate the generation of IFNγ-secreting CD8+ and CD4+ T cells (). To further validate this result, we carried out gain-of-function experiment. 1×107 CD11c+ cells isolated from the spleen of WT mice were adoptively transferred to CD11c-DTR mice after the initial DT injection (). Such adoptive transfer led to the recovery of DCs in the spleen, MLN, and intestine (). More significantly, this restoration could rescue T-MP-induced T cell responses in CD11c-DTR mice, as evaluated by the increased number of IFNγ-secreting CD8+ and CD4+ T cells (). Together, these data suggested that DCs are required to present antigens of oral T-MPs for the subsequent antitumor T cell immunity.
Figure 3. Dendritic cells are required for T-MP-induced antitumor T cell immunity. (A) CD11c-DTR mice were treated with DT at 4 ng/g every three days, while 1×107 CD11c+ cells isolated from the spleen of mice were adoptively transferred to CD11c-DTR mice at day 1 for reconstitution of DCs. Mice were immunized i.g. with B16-MPs or PBS on days 2, 4, and 7 followed by analysis on day 14. The percentages of CD11c+ cells in spleen, MLN and intestine were tested by flow cytometry after the last dose of DT in mice of WT, CD11c-DTR, and CD11c-DTR with DC recovery (n = 6 per group). Error bars represent mean ± SEM; *p < 0.05; **p < 0.01. (B, C) On day 14, lymphocytes in spleen and MLN were isolated from mice of each group and analyzed for the percentages of IFNγ+ cells in both CD8+ and CD4+ T cells by flow cytometry. Error bars represent mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001.
Oral T-MPs are mainly taken up by ileac IECs
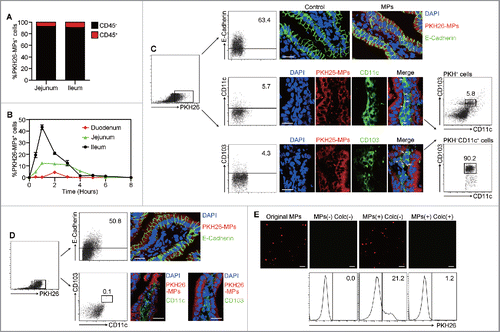
Although DCs are capable of taking up T-MPs effectively,Citation21 the intestinal epithelium may act as a barrier to prevent the direct contact of DCs with oral T-MPs. Here, we wondered how T-MPs or their antigens crossed the epithelial barrier and then how they could be captured by DCs. To this end, PKH26-labeled T-MPs were i.g. administered to mice. One hour later, the analysis of cells in intestinal epithelium compartment showed that more than 95% PKH26+ cells were CD45− cells and less than 5% PKH26+ cells were CD45+ cells (), suggesting that oral T-MPs are taken up by non-immune cells at the intestinal site. To further dissect this process in detail, we isolated the cells from duodenum, jejunum, and ileum, respectively, at different time point (0, 0.5, 1 to 8 h). The flow cytometric analysis showed that the majority of T-MPs were taken up in the ileum, and the uptaking peak came only 1 h after oral administration ( and Fig. S2A). To clarify that such MP-containing non-immune cells are IECs, we stained E-cadherin, a typical surface marker for IECs. As expected, we found that 63.4% of PKH26+ cells showed E-cadherin positive (), which was in accordance with the result of fluorescent immunostaining (). In addition, we also observed that among PKH26+ cells, majority of CD45+ cells exhibited a phenotype of CD103+ CD11c+ DCs, confirmed by flow cytometry and fluorescent immunostaining (). Intriguingly, after treated with microtubule inhibitor, colchicine, to block transcytosis, these PKH26+ DCs disappeared (), suggesting that T-MPs are transferred through transcytosis of IECs to DCs. To further support that IECs transfer T-MPs to DCs via transcytosis, we isolated ileac IECs one hour after oral administration of PKH26-labeled T-MPs and cultured them for 1 h in vitro. Using confocal microscope, we observed that T-MPs were released from IECs to the supernatants, however, the addition of colchicine blocked the release of T-MPs by IECs (). As a support, these released fluorescent T-MPs could be taken up by DCs (). In addition, we found that although capable of taking up the orally administered T-MPs, IECs appeared not to have antigen-presenting function to present T-MP antigens. The ileac IECs were isolated at different time point (4, 6, and 8 h) after i.g. administration of OVAB16-MPs. It was found that the antibody that recognizes H-2Kb-OVA257-264 peptide complex could only bind to CD11c+ cells but not E-cadherin+ cells (Fig. S2B). Altogether, these data suggested that T-MPs in the intestinal lumen are mainly taken up by ileac IECs after being orally administered, then transferred to a small population of CD103+ CD11c+ DCs for antigen presentation.
Figure 4. Oral T-MPs are mainly taken up by ileac IECs. (A) BALB/c mice were given i.g. PKH26-labeled H22-MPs followed by cell isolation from jejunum and ileum at 1 h, and the percentages of CD45+ and CD45− cells in PKH26+ cells were analyzed by flow cytometry (n = 3 per group). Data are representative of three independent experiments. (B) BALB/c mice were given i.g. PKH26-labeled H22-MPs, and cells in IE compartment were isolated from duodenum, jejunum, and ileum, respectively, at different time points (0 h, 0.5 h, 1 h to 8 h). The percentages of PKH26+ cells were analyzed by flow cytometry. Error bars represent mean ± SEM. (C) PKH26+ cells from ileum at 1 h were stained with E-Cadherin, CD11c, and CD103, and analyzed by flow cytometry (n = 3 per group). At the same time, the ileum tissues were removed for frozen sections and immunofluorescent staining, and observed by confocal fluorescent microscopy. Scale bars, 20 μm. Data are representative of three independent experiments. (D) BALB/c mice were given i.g. PKH26-labeled H22-MPs 30 min ahead of a colchicine i.v. treatment at 5 μM/g. One hour later, the percentages of E-Cadherin+ and CD103+ CD11c+ cells in PKH26+ cells from ileum were analyzed by flow cytometry. The ileum tissues were also removed for frozen sections and immunofluorescent staining. Scale bars, 20 μm. Data are representative of three independent experiments. (E) One hour after BALB/c mice were given i.g. PKH26-labeled H22-MPs, isolated ileac cells were sorted by MACS for CD45− IECs. The PKH26+ IECs were cultured with or without 10 μM colchicine for one hour, and the supernatant was collected for fluorescent microscopy or co-incubated with DCs for another 2 h followed by flow cytometric analysis. Scale bars, 5 μm. Data are representative of three independent experiments.
IECs release CCL2 to attract CD103+ CD11c+ DCs after taking up T-MPs
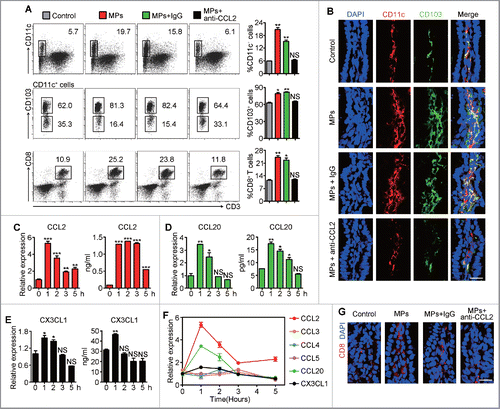
Next, we investigated whether and how CD103+ CD11c+ DCs migrated to the subepithelium of intestine where they met and captured IEC-transferred T-MPs for antigen presentation. In line with this idea, oral delivery of T-MPs caused the recruitment of CD103+ CD11c+ DCs at the intestinal epithelial site, as evaluated by flow cytometry and immunostaining (). Also, these recruited DCs upregulated the expression of CD80, CD86, and CCR7, the surface markers of mature DCs (Fig. S3A). We thereby speculated that oral administration of T-MPs could result in the release of chemokines that recruited DCs to the intestinal subepithelium. To test speculation, we analyzed a panel of chemokines that are related to DC recruitment. It was observed that only CD45− cells but not CD45+ cells, which were isolated from the ileum at different time point after the oral administration of T-MPs, significantly upregulated the expression of CCL2, CCL20, and CX3CL1 evaluated by qPCR (). Consistently, the protein levels of CCL2, CCL20, and CX3CL1 were increased in the intestinal tissue after the oral administration of T-MPs (). However, the expression of CCL3, CCL4, and CCL5 remained unaltered ( and Figs. S3B–K). Notably, among CCL2, CCL20, and CX3CL1, the increase of CCL2 was the most striking and maintained for the longest, while the expression of CCL20 and CX3CL1 dropped to the basal levels after a short increase (), suggesting that CCL2 might be the major contributor for DC migration to the ileac site. Indeed, the migration of CD103+ CD11c+ DCs cells to the ileac site was blocked by CCL2 neutralizing antibody (). In addition, at the ileac site, we also observed the increased percentage of CD8+ T cells after oral administration of T-MPs, which could also be blocked by CCL2 neutralizing antibody treatment (). Together, these data suggested that uptake of T-MPs by ileac IECs leads to releasing CCL2 for CD11c+ CD103+ DC attraction.
Figure 5. IECs release CCL2 to attract CD103+ CD11c+ DCs after taking up T-MPs. (A, B, G) 2 h after BALB/c mice were given i.g. H22-MPs, PBS control or pretreatment i.p. with anti-CCL2 neutralizing antibody (n = 3 per group), cells in IE compartment were isolated from ileum and analyzed for the percentages of CD11c+, CD103+ and CD3+ CD8+ cells by flow cytometry (gating all live cells). Error bars represent mean ± SEM; *p < 0.05; **p < 0.01; all experimental groups compared with control group (A). The ileum tissues were removed for frozen sections and immunofluorescent staining. Scale bars, 20 μm. Data are representative of three independent experiments (B, G). (C–F) The isolated ileac cells at different time point (0 h to 5 h) after H22-MPs treatment were sorted by MACS for both CD45+ and CD45− cells. The expression of CCL2 (C), CCL20 (D), and CX3CL1 (E) together with other chemokines (F) in CD45− cells was measured by qPCR, and the protein levels were evaluated by ELISA (C–E). Error bars represent mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001; 1 h to 5 h groups compared with 0 h group.
T-MPs activate NOD2 signaling of IECs for CCL2 production
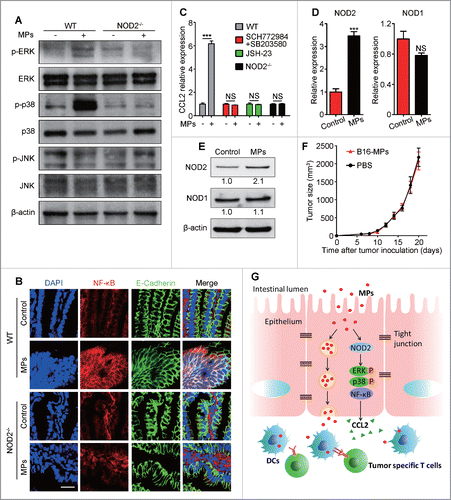
Finally, we investigated the signaling pathway through which T-MP-engaged IECs upregulated the expression of CCL2. Existing evidence indicates that CCL2 as an inflammation effector molecule is generally induced through inflammatory signaling pathway such as MAPK and NF-κB.Citation23 As shown in , although JNK was not altered, ERK and p38 were remarkably phosphorylated one hour after oral administration of T-MPs. In parallel with MAPK activation, the activation and nuclear translocation of NF-κB in ileac IECs were also observed (). Then, we used MAPK and NF-κB inhibitor to block their corresponding signaling pathway in mice treated with oral T-MPs. As expected, the T-MP-mediated increase in CCL2 expression in IECs could be canceled by blocking either MAPK or NF-κB (). NOD1 and NOD2, which are members of nucleotide-binding oligomerization domain (NOD)-like receptors, act as cytosolic sensors of various danger signals in IECs.Citation24 It has been reported that MAPK and NF-κB could be activated by NOD2 and serve as the downstream effectors of NOD2 signaling.Citation25 Here, we observed that the expression of NOD2 but not NOD1 was upregulated in ileac IECs isolated from oral T-MP-treated mice (). Then, NOD2−/− mice were used to test whether the increased NOD2 was involved in the activation of MAPK and NF-κB and the subsequent CCL2 production. It was shown that ERK and p38 in IECs could not be phosphorylated and activated by oral T-MP treatment in NOD2 deficient mice (). Consistently, the translocation of NF-κB from the cytoplasm to the nucleus was also blocked in NOD2 deficient mice (). Moreover, NOD2−/− mice did not show increased CCL2 levels in ileac epithelium after oral administration of T-MPs (). Also, in NOD2-deficient mice, T-MP-triggered recruitment of CD103+ CD11c+ DCs and CD8+ T cells to the ileac epithelial site was inhibited (Fig. S4). In line with the role of NOD2 in CCL2 production, we found that oral vaccination of B16-MPs could not generate a protective effect against B16 cell challenge in NOD2−/− mice (). Together, these data suggested that T-MPs could activate NOD2 signaling in IECs to enhance CCL2 production.
Figure 6. T-MPs within IECs activate NOD2 signaling for CCL2 production. (A) Phosphorylation of ERK, p38, and JNK in CD45− IECs from WT and NOD2−/− mice one hour after H22-MPs i.g. treatment was detected by Western blot. Data are representative of three independent experiments. (B) One hour after H22-MPs i.g. treatment, the ileum tissues from WT and NOD2−/− mice were removed for frozen sections and immunofluorescent staining. Scale bars, 20 μm. Data are representative of three independent experiments. (C) One hour after H22-MPs i.g. treatment, the expression of CCL2 in ileac IECs from mice of WT, NOD2−/−, pretreatment with ERK inhibitor SCH772984 (50mg/kg, i.p.) and p38 inhibitor SB203580 (20 μM, p.o.) for one week, or pre-treatment with NF-κB inhibitor JSH-23 (120 μg per day, p.o.) for one week, was measured by qPCR (n = 3 per group). Error bars represent mean ± SEM; ***p < 0.001. (D, E) The expression of NOD2 and NOD1 in ileac IECs one hour after H22-MPs i.g. treatment was measured by qPCR (n = 3 per group). Error bars represent mean ± SEM; ***p < 0.001, H22-MPs group compared with PBS control group (D). The protein levels were evaluated by Western blot and the relative intensity was further analyzed. Data are representative of three independent experiments (E). (F) NOD2−/− mice were immunized i.g. with B16-MPs or PBS on days -13, -11, and -7, followed by s.c. injection with 1×105 B16 tumor cells on day 0 (n = 6 per group). Tumor volumes were measured and calculated. Error bars represent mean ± SEM.
Discussion
Microparticles are enriched with a selective repertoire of proteins and nucleic acids from parental cells and are thought to play an active role in conferring intercellular signals. Previously, we showed that MPs as a carrier could deliver chemotherapeutic drugs,Citation26,27 oncolytic virus,Citation28 microRNAs,Citation29 DNA,Citation21 adhesion molecules,Citation30 and enzymesCitation31 to tumor cells or immune cells. We further revealed that T-MPs contain tumor antigens and innate signals (DNA and others) to stimulate DCs for highly efficient induction of antitumor T cell immunity.Citation21 Given this immunogenic properties of T-MPs as well as their lipid raft-based structure and the desirable stability, in the present study, we examined whether T-MPs could be employed for oral vaccination. It was shown that orally administered T-MPs could interact with ileac intestinal epithelial cells. They could not only activate the NOD2 signaling of IECs for CCL2 production and CD103+ CD11c+ DCs recruitment, but also be transported to the basolateral site by IECs and captured by DCs, inducing both mucosal and systemic antitumor T cell immunity (). To our knowledge, this represents the first report to reveal a novel property of T-MPs for oral antitumor vaccination that can deliver both antigens and the adjuvant activity to gut mucosal DCs.
A successful oral vaccination has to overcome a series of gastrointestinal barriers. In this study, we found that T-MPs are unable to resist gastric acid. Without the pretreatment of anti-acidic agent, oral T-MPs could not generate tumor-specific T cell immunity against tumor growth, as evidenced by striking decrease of uptake of T-MPs by IECs (Fig. S5). Though vulnerable to extreme low pH value, T-MPs are very stable under other conditions. It was evidently indicated that they are resistant to shaking and sunlight and can remain stable at the room temperature for days.Citation26 This might be ascribed to their lipid raft-based membrane structure.Citation32 Such lipid rafts probably also contribute to the resistance of T-MPs to proteolytic enzymes and bile salts in the intestinal lumen, or other unknown properties of T-MPs confer this resistance. In addition to the above barriers, a key issue for oral vaccination of T-MPs lies in their high intestinal permeability. A layer of mucus on the surface of intestinal epithelium prevents the access of bacteria to the epithelium.Citation33,34 M cells located in the Peyer's patches have a thinner unanchored glycocalyx, which allows them to easily take up and transport antigens,Citation35,36 but M cells are far less abundant than columnar enterocytes. In our study, we found that T-MPs are taken up by most IECs, which did not support that M cells mediate T-MPs crossing the epithelium. Thus, T-MPs may inherently have the ability to cross the layer of mucus. This could be explained by the possibilities that (1) enough gap sizes exist in the structure of mucus that guarantee the migration of T-MPs within, and (2) T-MPs possess well biological compatibility with mucus and cannot be stuck by mucus. In addition, in this study, we found that a majority of T-MPs are taken up at the ileac site rather than the jejunal site. This is probably due to the anatomical specificity of ileum. However, the precise mechanism is worthy of further investigation.
Oral administration of antigens such as food antigens commonly induces tolerance rather than immune responses.Citation37 Such unresponsiveness might be a physiological requirement of avoidance of unnecessary inflammation, which, however, impedes the efficacy and efficiency of oral tumor vaccination.Citation38 Oral tolerance might be due to several reasons. IECs as non-conventional APCs express low or even no co-stimulatory molecules on their surface. As a result, antigens captured and presented by IECs results in T cell anergy and tolerance.Citation39,40 On the other hand, DCs as professional APCs in mucosal system are able to induce efficient T cell responses, which, however, depends on DC maturation and its expressing high levels of co-stimulatory molecules such as CD80 and CD86.Citation41 In the inflammatory microenvironment, pathogen- or damage-associated molecular patterns (PAMP or DAMP) interact with pattern recognition receptors (PRR) expressed by DCs, leading to DC activation and maturation.Citation42,43 Under physiological condition, the intestinal environment is absent of inflammation, leading to failure of DC maturation. Subsequently, their presenting antigen causes anergic T cells or Treg cells. Concerning the issue of oral tolerance, current oral vaccination regimens are supplemented with mucosal adjuvants, including derivatives of cholera toxin, recombinant cholera toxin B-subunit, and CpG oligodeoxynucleotides, to enhance the required innate signals.Citation44-46 Unlike these adjuvants, T-MPs themselves include various innate stimulatory activities. T-MPs contains both genomic and mitochondrial DNA fragments that can activate cGAS-STING signaling pathway for induction of type I interferon expression.Citation21 In addition, DCs seem to possess strong ability to take up MPs and are induced to upregulate CD80 and CD86.Citation21 In our unpublished study, we found that T-MPs entered the lysosomes of DCs and increased the lysosomal pH value, leading to an increase of cellular ROS levels. Thus, T-MPs as oral vaccines not only provide tumor antigens but also deliver innate signals to stimulate mucosal DCs, leading to immunity rather than oral tolerance.
Notwithstanding T-MPs transferring tumor antigens and innate signals to mucosal DCs, an important issue lies in how T-MPs cross intestinal epithelium so to access DCs. In this study, we found that IECs effectively take up T-MPs. Intriguingly, IECs can sense the innate signals of T-MPs to activate the NOD2 signaling pathway, followed by the activation of downstream signal molecules MAPK and NF-κB and the resultant production of chemokines including CCL2. We found that CCL2 plays a major role in attraction of CD103+ CD11c+ DCs. Physiologically, IECs have the ability to transcytose nutrients or non-nutrients such as IgA.Citation47,48 In addition to responding to innate signals carried by T-MPs, we have provided evidence that IECs can transcytose T-MPs to the basolateral site where T-MPs meet the attracted DCs. Colchicine inhibits transcytosis by binding tubulin and subsequently inhibiting microtubule polymerization.Citation49,50 We found that the colchicine treatment resulted in inaccessibility of T-MPs for DCs at the intestinal site. More convincingly, we show that IECs isolated from T-MP-orally administered mice could release T-MPs in vitro. Thus, T-MPs, once escaped from gastric acid-mediated destruction and arrived at the intestinal lumen, are taken up and transferred by IECs to CD103+ CD11c+ DCs, leading to effective cross-presentation of the tumor antigens by the DCs to specific T cells for antitumor immune responses.
In summary, our data show that T-MPs, by virtue of their physical and chemical properties, are able to cross the layer of epithelial mucosa to activate IECs and mucosal DCs. Our findings suggest that systemic tumor protection can be achieved by vaccination of T-MPs via the gastrointestinal route. This T-MP-based novel oral vaccination strategy is not only suitable for combating tumors, but also potent for the prevention of various infectious diseases.
Materials and methods
Animals and cell lines
BALB/c, BALB/c-nude, and C57BL/6 mice (6–8 weeks old) were purchased from Vital River. CD11c-DTR mice and NOD2−/− mice were friendly provided by Drs Yangxin Fu and Zhihua Liu laboratories, respectively, at the Institute of Biophysics of Chinese Academy of Science. All animal experiments were approved by the Institutional Committee for the Ethics of Animal Care and Treatment. Mouse B16 and OVAB16 melanoma, Hepa1-6 hepatocarcinoma, CT-26 colon carcinoma, and H22 hepatocarcinoma tumor cell lines were purchased from Cell Resource Center of Peking Union Medical College (Beijing, China), and cultured according to the guidelines given. Cells were tested for mycoplasma detection, interspecies cross contamination and authenticated by isoenzyme and short tandem repeat (STR) analyses in Cell Resource Centre of Peking Union Medical College before the beginning of the study and spontaneously during the research. All cell lines were maintained in culture for a maximum of 20 passages (two months).
Preparation of T-MPs, tumor-cell lysates, and apoptotic tumor cells
Tumor cells were exposed to ultraviolet irradiation (300 J/m2) for 1 h, and 12 h later, supernatants were used for microparticles isolation as described previously.Citation26 In brief, supernatants were centrifuged at 1,000 g for 10 min to remove whole cells and then centrifuged for 2 min at 14,000 g to get rid of debris. The supernatants were further centrifuged for 60 min at 14,000 g to pellet microparticles. The pellets were washed three times and resuspended in PBS for the subsequent experiments. The ultracentrifuge tubes were purchased from JET BIOFIL (Guangzhou Jet Bio-Filtration Co., Ltd). Tumor-cell lysates were prepared by subjecting tumor cells in PBS to three cycles of rapid freezing in liquid nitrogen and thawing at 37°C. The lysates were centrifuged at 2,000 g for 10 min to remove cellular debris. Apoptotic tumor cells were obtained by exposing tumor cells to UV irradiation (300 J/m2) in culture medium for 1 h, and apoptotic cells were suspended in PBS after 5 min 400 g centrifugation.
The endotoxin concentration of microparticles, cell lysates, and apoptotic cells derived from all cell lines was less than 0.05 EU/mL, which was assessed by ToxinSensor Chromogenic LAL Endotoxin Assay Kit (Genscript).
Prophylactic immunization and tumor challenge
In B16 tumor model, C57BL/6 mice were immunized i.g. three times with T-MPs or control materials on days -13, -11, and -7. On day 0, mice were challenged s.c. with B16 tumor cells 1×105. Palpable tumors were measured by calipers, and tumor volume was calculated according to the formula: volume = (length×widthCitation2)/2, where length and width are measured in millimeters. For CT-26 tumor model, BALB/c mice received s.c. injection of 1×105 CT-26 tumor cells after immunization with T-MPs as described above. For the OVAB16 tumor model, C57BL/6 mice received s.c. injection of 1×105 OVAB16 tumor cells after immunization with T-MPs or control materials as described above. For the immune-deficient model, nude mice were treated i.g. with B16-MPs or PBS as described above ahead of s.c. injection of B16 cells 1×105. For the NOD2-deficient model, NOD2−/− mice were vaccinated i.g. with B16-MPs or PBS as described above followed by a challenge of B16 cells 1×105. All kinds of mice from experiments in vivo should go through a night of fasting to guarantee the gastrointestinal tract emptying and receive i.v. omeprazole (OME) at 5 μg/g as an anti-acidic agent 30 min ahead of vaccination orally.
Flow cytometric analysis
The surface staining antibodies were as follows: anti-CD3 (17A2), anti-CD4 (GK1.5), anti-CD8a (53-6.7), anti-CD25 (PC61.5), anti-CD103 (2E7), anti-CD11c (N418), anti-CD45 (30-F11), anti-CD80 (16-10A1), anti-CD86 (GL1), anti-CCR7 (4B12), anti-OVA257-264 peptide bound to H-2Kb (25-D1.16). For intracellular cytokine staining, lymphocytes prepared from spleen were cultured for 5 h in the presence of PMA (80 nM), ionomycin (1.3 μM), and Brefeldin A (5 μg/mL). Cells were treated with anti-IFNγ (XMG1.2) and anti-Foxp3 (FJK-16s) on the basis of the cytoplasmic and nuclear proteins staining protocols (Biolegend). All antibodies were purchased from Biolegend, and Flow cytometric analysis was performed with Accuri C6.
Cell isolation from intestine
The small intestine was separated and Peyer's patches were dissected out. After longitudinally cut and washed in PBS to remove fecal contents and food debris, the intestine was sliced into approximately 1 cm pieces and transferred to Ca2+/Mg2+-free HBSS supplemented with 5% FBS and 2 mM EDTA. Cells were obtained by horizontally shaking the tissue twice at 250 rpm for 20 min at 37°C followed each time by filtering the supernatants through 70-μm cell strainer. The cells, a mixture of IECs and CD45+ immune cells, were washed in PBS and used for subsequent experiments.
Immunofluorescence microscopy
4% paraformaldehyde-fixed frozen sections of intestine tissues were permeabilized in 0.1% Triton X-100 for 10 min. After blocking with 5% BSA for 1 h, the sections were incubated with the primary antibodies overnight at 4°C, followed by fluorescent secondary antibodies for 1 h at room temperature, and DAPI was stained at last. The unlabeled primary antibodies included hamster anti-mouse CD11c (N418) mAb (Abcam), rat anti-mouse CD103 (AP-MAB0828) mAb (Abcam), and rabbit anti-mouse NF-κB p65 (L8F6) mAb (Cell Signaling Technology). The secondary antibodies included TRITC-conjugated anti-hamster IgG (ImmunoReagent, Inc.), DyLight 488-conjugated anti-hamster IgG (Biolegend), Alexa Fluor 488-conjugated anti-rat IgG (Jackson ImmunoResearch Laboratories), and Alexa Fluor 594-conjugated anti-rabbit IgG (LiankeBio, China). In some cases, the sections were incubated with the fluorescent primary antibodies overnight. The IECs were labeled with Alexa Fluor 488-conjugated anti-mouse E-Cadherin (DECMA-1, eBioscience), and intestinal CD8+ T cells were labeled with eFluor 615-conjugated anti-mouse CD8a (53-6.7, eBioscience). All sections were visualized by OLYMPUS Confocal Microscopy, FV1000MPE.
Enzyme linked immunosorbent assay (ELISA)
The small intestine (100 mg) was cut into pieces and immersed in 500μL PBS with protease inhibitor. Then tissue homogenate was made by electric homogenizer. After centrifugation at 15,000g for 15 min, the supernatants were gathered and assessed by CCL2 ELISA Kit (eBioscience), CCL20 ELISA Kit (R&D Systems), and CX3CL1 ELISA Kit (R&D Systems) according to the manufacturer's protocols. In some cases, cell culture supernatants were collected and analyzed by IFNγ ELISA Kit (DAKEWE, China).
Western blot analysis
Lysate proteins of IECs, the CD45− cells sorted by MACS in cells isolated from IE compartment, were extracted and analyzed by Western blot antibodies including anti-mouse Phospho-p44/42 MAPK (ERK1/2), anti-mouse p44/42 MAPK (ERK1/2), anti-mouse Phospho-p38 MAPK, anti-mouse p38 MAPK, anti-mouse Phospho-p46/54 MAPK (JNK1/2), anti-mouse p46/54 MAPK (JNK1/2), anti-mouse NOD1, anti-mouse NOD2, and anti-mouse β-actin at recommended dilutions followed by secondary horseradish peroxidase-conjugated antibodies and visualized by enhanced chemiluminescence according to the manufacturer's protocol (ECL kit; Pierce). All the antibodies were purchased from Cell Signaling Technology except anti-mouse NOD2 (Abcam).
Labeling of microparticles
Isolated MPs were labeled with PKH26 (Sigma) according to the manufacturer's protocol.
Other reagents
The ERK inhibitor SCH772984, p38 inhibitor SB203580, and NF-κB inhibitor JSH-23 were purchased from Selleck, and administrated according to the instructions given. The anti-CCL2 neutralizing antibody was purchased from Bio X Cell, and administrated according to the instructions given.
Real-time quantitative PCR analysis
Total RNA was extracted from tissues and cells with TRIzol reagent (Invitrogen). Real-time quantitative PCR was performed with THUNDERBIRD SYBR qPCR mix (TOYOBO) on ABI applied biosystems (StepOnePlus). mRNA levels were normalized to β-actin. The primer sequences were as follows: mouse β-actin, sense 5′-CCTTCTTGGGTATGGAATCCTG-3′, antisense 5′-CAATGCCTGGGTACATG GTG-3′; mouse CCL2, sense 5′-TTAAAAACCTGGATCGGAACCAA-3′, antisense 5′-GCATTAGCTTCAGATTTACGGGT-3′; mouse CCL20, sense 5′-GCCGAT GAAGCTTGTGACAT-3′, antisense 5′-GCTGTGTCCAATTCCATCCC-3′; mouse CX3CL1, sense 5′-GATGACCTCACGAATCCCAG-3′, antisense 5′-GCGTCTTG GACCCATTTCTC-3′; mouse CCL3, sense 5′-TTCTCTGTACCATGACACT CTGC-3′, antisense 5′-CGTGGAATCTTCCGGCTGTAG-3′; mouse CCL4, sense 5′- TTCCTGCTGTTTCTCTTACACCT-3′, antisense 5′-CTGTCTGCCTCTTTTGGTC AG-3′; mouse CCL5, sense 5′-GAGTATTTCTACACCAGCAGCA-3′, antisense 5′- TCTTGAACCCACTTCTTCTCTG-3′; mouse IL-12p35, sense 5′-GGAACTACACA AGAACGAGAG-3′, antisense 5′-AAGTCCTCATAGATGCTACCA-3′; mouse IL-12p40, sense 5′-TGGTTTGCCATCGTTTTGCTG-3′, antisense 5′-ACAGGTG AGGTTCACTGTTTCT-3′; mouse TNF-α, sense 5′-CCACGTCGTAGCAAA CCAC-3′, antisense 5′- TTGTCCCTTGAAGAGAACCTG-3′; mouse IL-1β, sense 5′-CCTGTTCTTTGAAGTTGACGG-3′, antisense 5′-AATGAGTGATACTGCCTG CC-3′; mouse IL-6, sense 5′- AACAAGAAAGACAAAGCCAGAG-3′, antisense 5′- GTTAGGAGAGCATTGGAAATTGG-3′; mouse IL-4, sense 5′-GGTCTCAACCCC CAGCTAGT-3′, antisense 5′-GCCGATGATCTCTCTCAAGTGAT-3′; mouse IL-17, sense 5′-CGATCATCCCTCAAAGCTCAG-3′, antisense 5′-TATCAGGGTCTTCA TTGCGG-3′.
Statistical analysis
Results were expressed as mean values ± SEM and interpreted by repeated-measure analysis of variance or Kaplan–Meier analysis. Differences were considered to be statistically significant when p < 0.05.
Disclosure of potential conflicts of interest
B. Huang was supported by Soundny (Sheng-Qi-An) Biotech. The other authors declare no competing financial interests.
Author contributions
WD, HZ, XY, YL, DC, XL, XJ, and JL designed and performed the experiments and analyzed the data. KT, JM, ZH, and XQ analyzed the data. WD and BH designed the experiments, interpreted the results, and wrote the manuscript.
Supplementary_materials.docx
Download MS Word (1 MB)Funding
This work was supported by National Basic Research Program of China (2014CB542103), National Natural Science Foundation of China (81661128007, 81472653, 81530080), National Natural Science Fund for Young Scholars of China (81502473), and CAMS Initiative for Innovative Medicine (2016-I2M-1-007).
References
- Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov 2011; 1:391-407; PMID:22096659; http://dx.doi.org/10.1158/2159-8290.cd-11-0209
- Hanash S.. Integrated global profiling of cancer. Nat Rev Cancer 2004; 4:638-44; PMID:15286743; http://dx.doi.org/10.1038/nrc1414
- Hubbell JA, Thomas SN, Swartz MA. Materials engineering for immunomodulation. Nature 2009; 462:449-60; PMID:19940915; http://dx.doi.org/10.1038/nature08604
- Best SR, Peng S, Juang CM, Hung CF, Hannaman D, Saunders JR, Wu TC, Pai SI. Administration of HPV DNA vaccine via electroporation elicits the strongest CD8+ T cell immune responses compared to intramuscular injection and intradermal gene gun delivery. Vaccine 2009; 27:5450-9; PMID:19622402; http://dx.doi.org/10.1016/j.vaccine.2009.07.005
- Buchan S, Gronevik E, Mathiesen I, King CA, Stevenson FK, Rice J. Electroporation as a “prime/boost” strategy for naked DNA vaccination against a tumor antigen. J Immunol 2005; 174:6292-8; PMID:15879128; http://dx.doi.org/10.4049/jimmunol.174.10.6292
- Perez-Lopez A, Behnsen J, Nuccio SP, Raffatellu M. Mucosal immunity to pathogenic intestinal bacteria. Nat Rev Immunol 2016; 16:135-48; PMID:26898110; http://dx.doi.org/10.1038/nri.2015.17
- Lycke N.. Recent progress in mucosal vaccine development: potential and limitations. Nat Rev Immunol 2012; 12:592-605; PMID:22828912; http://dx.doi.org/10.1038/nri3251
- Holmgren J, Svennerholm AM. Vaccines against mucosal infections. Curr Opin Immunol 2012; 24:343-53; PMID:22580196; http://dx.doi.org/10.1016/j.coi.2012.03.014
- Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nat Med 2005; 11:S45-53; PMID:15812489; http://dx.doi.org/10.1038/nm1213
- Mitragotri S.. Immunization without needles. Nat Rev Immunol 2005; 5:905-16; PMID:16239901; http://dx.doi.org/10.1038/nri1728
- Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 2014; 54:289-96; PMID:24766893; http://dx.doi.org/10.1016/j.molcel.2014.03.040
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol 2013; 31:51-72; PMID:23157435; http://dx.doi.org/10.1146/annurev-immunol-032712-100008
- Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 2011; 208:1989-2003; PMID:21930769; http://dx.doi.org/10.1084/jem.20101158
- Koshy ST, Mooney DJ. Biomaterials for enhancing anti-cancer immunity. Curr Opin Biotech 2016; 40:1-8; PMID:26896596; http://dx.doi.org/10.1016/j.copbio.2016.02.001
- Irvine DJ, Swartz MA, Szeto GL. Engineering synthetic vaccines using cues from natural immunity. Nat Mater 2013; 12:978-90; PMID:24150416; http://dx.doi.org/10.1038/nmat3775
- Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT. LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the toll adapters TRAM and TRIF. J Exp Med 2003; 198:1043-55; PMID:14517278; http://dx.doi.org/10.1084/jem.20031023
- Hornung V, Latz E. Intracellular DNA recognition. Nat Rev Immunol 2010; 10:123-30; PMID:20098460; http://dx.doi.org/10.1038/nri2690
- Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, Corzo A, Cho HI, Celis E, Lennox B et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med 2010; 16:880-6; PMID:20622859; http://dx.doi.org/10.1038/nm.2172
- Dhodapkar MV, Dhodapkar KM, Palucka AK. Interactions of tumor cells with dendritic cells: balancing immunity and tolerance. Cell Death Differ 2008; 15:39-50; PMID:17948027; http://dx.doi.org/10.1038/sj.cdd.4402247
- Zhang Y, Zhang R, Zhang H, Liu J, Yang Z, Xu P, Cai W, Lu G, Cui M, Schwendener RA et al. Microparticles released by Listeria monocytogenes-infected macrophages are required for dendritic cell-elicited protective immunity. Cell Mol Immunol 2012; 9:489-96; PMID:23064105; http://dx.doi.org/10.1038/cmi.2012.33
- Zhang H, Tang K, Zhang Y, Ma R, Ma J, Li Y, Luo S, Liang X, Ji T, Gu Z et al. Cell-free tumor microparticle vaccines stimulate dendritic cells via cGAS/STING signaling. Cancer Immunol Res 2015; 3:196-205; PMID:25477253; http://dx.doi.org/10.1158/2326-6066.cir-14-0177
- Zhang H, Huang B. Tumor cell-derived microparticles: a new form of cancer vaccine. Oncoimmunology 2015; 4:e1017704; PMID:26405568; http://dx.doi.org/10.1080/2162402x.2015.1017704
- Chatterjee D, Chaudhuri K. Vibrio cholerae O395 outer membrane vesicles modulate intestinal epithelial cells in a NOD1 protein-dependent manner and induce dendritic cell-mediated Th2/Th17 cell responses. J Biol Chem 2013; 288:4299-309; PMID:23275338; http://dx.doi.org/10.1074/jbc.M112.408302
- Motta V, Soares F, Sun T, Philpott DJ. NOD-like receptors: versatile cytosolic sentinels. Physiol Rev 2015; 95:149-78; PMID:25540141; http://dx.doi.org/10.1152/physrev.00009.2014
- Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol 2006; 6:9-20; PMID:16493424; http://dx.doi.org/10.1038/nri1747
- Tang K, Zhang Y, Zhang H, Xu P, Liu J, Ma J, Lv M, Li D, Katirai F, Shen GX et al. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat Commun 2012; 3:1282; PMID:23250412; http://dx.doi.org/10.1038/ncomms2282
- Ma J, Zhang Y, Tang K, Zhang H, Yin X, Li Y, Xu P, Sun Y, Ma R, Ji T et al. Reversing drug resistance of soft tumor-repopulating cells by tumor cell-derived chemotherapeutic microparticles. Cell Res 2016; 26:713-27; PMID:27167569; http://dx.doi.org/10.1038/cr.2016.53
- Ran L, Tan X, Li Y, Zhang H, Ma R, Ji T, Dong W, Tong T, Liu Y, Chen D et al. Delivery of oncolytic adenovirus into the nucleus of tumorigenic cells by tumor microparticles for virotherapy. Biomaterials 2016; 89:56-66; PMID:26950165; http://dx.doi.org/10.1016/j.biomaterials.2016.02.025
- Li D, Jia H, Zhang H, Lv M, Liu J, Zhang Y, Huang T, Huang B. TLR4 signaling induces the release of microparticles by tumor cells that regulate inflammatory cytokine IL-6 of macrophages via microRNA let-7b. Oncoimmunology 2012; 1:687-93; PMID:22934260; http://dx.doi.org/10.4161/onci.19854
- Ma J, Cai W, Zhang Y, Huang C, Zhang H, Liu J, Tang K, Xu P, Katirai F, Zhang J et al. Innate immune cell-derived microparticles facilitate hepatocarcinoma metastasis by transferring integrin αMβ2 to tumor cells. J Immunol 2013; 191:3453-61; PMID:23956429; http://dx.doi.org/10.4049/jimmunol.1300171
- Tang K, Liu J, Yang Z, Zhang B, Zhang H, Huang C, Ma J, Shen GX, Ye D, Huang B. Microparticles mediate enzyme transfer from platelets to mast cells: a new pathway for lipoxin A4 biosynthesis. Biochem Bioph Res Co 2010; 400:432-6; PMID:20801099; http://dx.doi.org/10.1016/j.bbrc.2010.08.095
- Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005; 106:1604-11; PMID:15741221; http://dx.doi.org/10.1182/blood-2004-03-1095
- Johansson ME, Hansson GC. Microbiology. Keeping bacteria at a distance. Science 2011; 334:182-3; PMID:21998374; http://dx.doi.org/10.1126/science.1213909
- Johansson ME, Sjovall H, Hansson GC. The gastrointestinal mucus system in health and disease. Nat Rev Gastro Hepa 2013; 10:352-61; PMID:23478383; http://dx.doi.org/10.1038/nrgastro.2013.35
- Pelaseyed T, Bergstrom JH, Gustafsson JK, Ermund A, Birchenough GM, Schutte A, van der Post S, Svensson F, Rodríguez-Piñeiro AM, Nyström EE et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol Rev 2014; 260:8-20; PMID:24942678; http://dx.doi.org/10.1111/imr.12182
- Ermund A, Schutte A, Johansson ME, Gustafsson JK, Hansson GC. Studies of mucus in mouse stomach, small intestine, and colon. I. Gastrointestinal mucus layers have different properties depending on location as well as over the Peyer's patches. Am J Physiol-Gastr L 2013; 305:G341-7; PMID:23832518; http://dx.doi.org/10.1152/ajpgi.00046.2013
- Pabst O, Mowat AM. Oral tolerance to food protein. Mucosal Immunol 2012; 5:232-9; PMID:22318493; http://dx.doi.org/10.1038/mi.2012.4
- Poonam P.. The biology of oral tolerance and issues related to oral vaccine design. Curr Pharm Design 2007; 13:2001-7; PMID:17627533; http://dx.doi.org/10.2174/138161207781039814
- Goto Y, Ivanov II. Intestinal epithelial cells as mediators of the commensal-host immune crosstalk. Immunol Cell Biol 2013; 91:204-14; PMID:23318659; http://dx.doi.org/10.1038/icb.2012.80
- Hershberg RM, Mayer LF. Antigen processing and presentation by intestinal epithelial cells - polarity and complexity. Immunol Today 2000; 21:123-8; PMID:10689299; http://dx.doi.org/10.1016/S0167-5699(99)01575-3
- Shakhar G, Lindquist RL, Skokos D, Dudziak D, Huang JH, Nussenzweig MC, Dustin ML. Stable T cell-dendritic cell interactions precede the development of both tolerance and immunity in vivo. Nat Immunol 2005; 6:707-14; PMID:15924144; http://dx.doi.org/10.1038/ni1210
- Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature 2007; 449:419-26; PMID:17898760; http://dx.doi.org/10.1038/nature06175
- Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012; 12:265-77; PMID:22437871; http://dx.doi.org/10.1038/nrc3258
- Kim D, Kim YG, Seo SU, Kim DJ, Kamada N, Prescott D, Philpott DJ, Rosenstiel P, Inohara N, Núñez G. Nod2-mediated recognition of the microbiota is critical for mucosal adjuvant activity of cholera toxin. Nat Med 2016; 22:524-30; PMID:27064448; http://dx.doi.org/10.1038/nm.4075
- Yin Y, Qin T, Wang X, Lin J, Yu Q, Yang Q. CpG DNA assists the whole inactivated H9N2 influenza virus in crossing the intestinal epithelial barriers via transepithelial uptake of dendritic cell dendrites. Mucosal Immunol 2015; 8:799-814; PMID:25492476; http://dx.doi.org/10.1038/mi.2014.110
- Holmgren J, Adamsson J, Anjuere F, Clemens J, Czerkinsky C, Eriksson K, Flach CF, George-Chandy A, Harandi AM, Lebens M et al. Mucosal adjuvants and anti-infection and anti-immunopathology vaccines based on cholera toxin, cholera toxin B subunit and CpG DNA. Immunol Lett 2005; 97:181-8; PMID:15752556; http://dx.doi.org/10.1016/j.imlet.2004.11.009
- Gutzeit C, Magri G, Cerutti A. Intestinal IgA production and its role in host-microbe interaction. Immunol Rev 2014; 260:76-85; PMID:24942683; http://dx.doi.org/10.1111/imr.12189
- Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 2014; 14:141-53; PMID:24566914; http://dx.doi.org/10.1038/nri3608
- Ferreira VH, Dizzell S, Nazli A, Kafka JK, Mueller K, Nguyen PV, Tremblay MJ, Cochrane A, Kaushic C. Medroxyprogesterone acetate regulates HIV-1 uptake and transcytosis but not replication in primarygenital epithelial cells, resulting in enhanced T-cell infection. J Infect Dis 2015; 211:1745-56; PMID:25538276; http://dx.doi.org/10.1093/infdis/jiu832
- Skoufias DA, Wilson L. Mechanism of inhibition of microtubule polymerization by colchicine: inhibitory potencies of unliganded colchicine and tubulin-colchicine complexes. Biochemistry 1992; 31:738-46; PMID:1731931; http://dx.doi.org/10.1021/bi00118a015