
ABSTRACT
In recent years, immunotherapies, such as those involving chimeric antigen receptor (CAR) T cells, have become increasingly promising approaches to non-small-cell lung cancer (NSCLC) treatment. In this study, we explored the antitumor potential of prostate stem cell antigen (PSCA)-redirected CAR T and mucin 1 (MUC1)-redirected CAR T cells in tumor models of NSCLC. First, we generated patient-derived xenograft (PDX) mouse models of human NSCLC that maintained the antigenic profiles of primary tumors. Next, we demonstrated the expression of PSCA and MUC1 in NSCLC, followed by the generation and confirmation of the specificity and efficacy of PSCA- and MUC1-targeting CAR T cells against NSCLC cell lines in vitro. Finally, we demonstrated that PSCA-targeting CAR T cells could efficiently suppress NSCLC tumor growth in PDX mice and synergistically eliminate PSCA+MUC1+ tumors when combined with MUC1-targeting CAR T cells. Taken together, our studies demonstrate that PSCA and MUC1 are both promising CAR T cell targets in NSCLC and that the combinatorial targeting of these antigens could further enhance the antitumor efficacy of CAR T cells.
Introduction
Globally, lung cancer is the greatest killer among all cancers,Citation1,2 and non-small-cell lung cancer (NSCLC) accounts for approximately 85% of all cases of lung cancer.Citation3,4 Current therapeutic strategies, including surgery, radiation and chemotherapy, have not yielded significant survival benefits. Tyrosine kinase inhibitors (TKIs) targeting EGFR and ALK have been widely used to treat NSCLC, but frequent resistance to these drugs develops due to acquired mutations of EGFRCitation5-7 and of ALK.Citation8 Furthermore, recently introduced CTLA4, PD-1 and PD-L1 immune checkpoint inhibitors have had noCitation9 or only moderate effects on NSCLC.Citation10-13 Therefore, novel treatment regimens are still needed.
Chimeric antigen receptor (CAR) T cells that target CD19 have generated exciting results in leukemia and lymphoma.Citation14-16 However, the broad applicability of these cells for solid cancer is limited by the paucity of truly tumor-specific target antigens. Additionally, the heterogeneity of tumor-associated antigens (TAAs) in solid cancers complicates CAR T cell therapies, as the targets may differ among various cancers and even patients of a same cancer. Therefore, it is important to define the TAA profile of a solid cancer before using TAA-oriented personalized CAR T cell immunotherapies.
A few CAR T cell antigens have been targeted to treat NSCLC. Glypican-3 was recently reported as a promising target for lung squamous cell carcinoma.Citation17 In a phase I clinical trial of anti-epidermal growth factor receptor (EGFR) CAR T cells for lung cancer, only 2 of 11 patients achieved partial responses.Citation18 The expression of MUC1, a transmembrane glycoprotein, is aberrantly upregulated in many types of cancer, including NSCLC.Citation19 A trial of MUC1-targeting CAR T cells is currently recruiting patients with four types of solid cancers, including NSCLC (ClinicalTrials.gov Identifier: NCT02587689). Hence, MUC1 is a promising CAR T cell target in NSCLC.
Prostate stem cell antigen (PSCA) is a glycosylphosphoinositol-anchored cell surface antigenCitation20 that is overexpressed mainly in prostate cancer,Citation21 although its expression has also been reported in other tumors such as gallbladder adenocarcinomaCitation22 and gastric cancer.Citation23 Surprisingly, PSCA is frequently overexpressed in NSCLC,Citation20 although this requires confirmation. Antibody-based, PSCA-targeted therapies, as well as a peptide vaccine, have been explored for the treatment of prostate cancer.Citation24-28 Furthermore, PSCA-targeting CAR T cells have been used to treat pancreatic cancer in humanized mice,Citation29 and clinical trials of anti-PSCA CAR T cells for the treatment of prostate, bladder and pancreatic cancers are ongoing (ClinicalTrials.gov Identifier, NCT02092948 and NCT02744287). It remains unknown, however, whether anti-PSCA CAR T cells could be used to treat NSCLC.
Patient-derived xenograft (PDX) models have been widely used in translational cancer research,Citation30 which faithfully resemble the original tumors from which they were developed and this similarity is maintained across passages.Citation31 In this study, we generated a PDX model of NSCLC in which we detected strong histological expression of PSCA and weak expression of MUC1. We subsequently proved the capacities of PSCA- and MUC1-targeted CAR T cells to recognize and kill NSCLC cells expressing the respective target antigens. Finally, we observed enhanced efficacy of a combination of both PSCA- and MUC1-targeted CAR T cells against double-positive NSCLC samples. Our results suggest that PSCA and MUC1 are NSCLC-specific targets of CAR T cells and indicate that combinatorial antigen targeting could enhance the antitumor efficacy of these cells.
Results
PDX models retained molecular phenotype of NSCLC cells
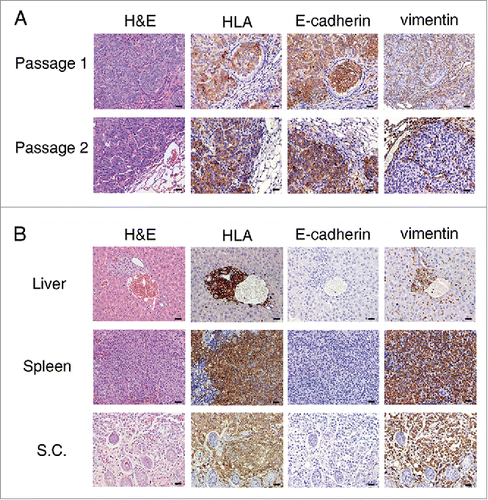
Using TALEN-mediated gene targeting, we previously generated a NOD-SCID-IL2Rγ−/− strain (NSI) of mice capable of engrafting and modeling human hematopoietic cells.Citation32 Here, we generated human NSCLC PDX mice by subcutaneously or intravenously implanting dissected primary tumor masses or cell suspensions that could be serially transplanted and engrafted in NSI mice. Immunohistochemistry results showed that HLA+NSCLC cells from a patient (patient P2) that had been engrafted in the lungs of NSI mice expressed E-cadherin, but not vimentin, during both the first and second passages after transplantation (). Moreover, NSCLC cells from third passage of another patient (patient P1) metastasized from the initial subcutaneous implants to the livers and spleens of NSI mice, and tumor cells from all locations unanimously expressed vimentin but not E-cadherin (). Therefore, PDX models of NSCLC retained the molecular phenotypes of NSCLC cells across different passages, as well as in different organs of metastasis.
Figure 1. Generation and molecular characterization of patient-derived xenograft (PDX) models of non-small-cell lung cancer. (A) Hematoxylin and eosin (H&E) staining and immunohistochemistry detection of human leukocyte antigen (HLA), E-cadherin and vimentin in tumor sections from both the first and second passages of PDX mice for patient P2 (see from ). Although HLA+ cells from PDX mice also expressed E-cadherin, cells from both passages were negative for vimentin. (B) H&E staining and immunohistochemistry detection of HLA, E-cadherin and vimentin in sections from liver, spleen and subcutaneous (s.c.) tissue. All sections were from a single mouse among the third passage of PDX mice for patient P1 (see from ). Scale bar = 20 μm.
Frequent expression of PSCA in NSCLC
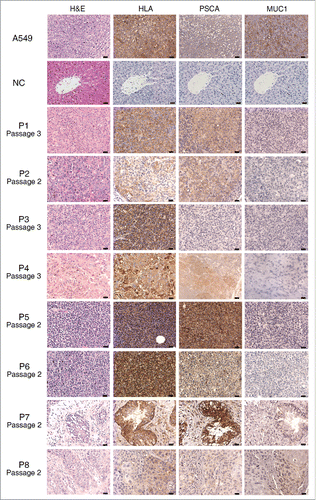
NSCLC samples from eight patients were successfully engrafted in NSI mice to generate PDX models (). We subsequently harvested the tumors and evaluated the expression of PSCA and MUC1. Although the latter is frequently overexpressed in NSCLC,Citation19 only two of the eight patients in our study expressed this antigen; in contrast, seven patients, including two patients who expressed MUC1, expressed PSCA (). Collectively, our results demonstrate the frequent expression of PSCA in human NSCLC cells, consistent with a previous report.Citation20 The co-expression of PSCA and MUC1 in patients with NSCLC prompted an attempt to evaluate the efficacy of combination CAR T cells for dual antigen targeting in our PDX models.
Table 1. Clinical information of the patients and characteristics of the corresponding PDX models.
Figure 2. Sections of tumors from eight patient-derived xenograft (PDX) models. Representative images correspond to tumors derived from eight patients; all sections were stained with hematoxylin and eosin (H&E) and antibodies against human leukocyte antigen (HLA), PSCA and MUC1. The passage numbers of each PDX for the patients were indicated. The negative controls (NC) are the liver tissues from a same mouse of third passage of PDX for patient P3. Scale bar = 20 μm. PSCA and MUC1 detection results are also shown in .
Generation of CAR T cells targeting PSCA and MUC1
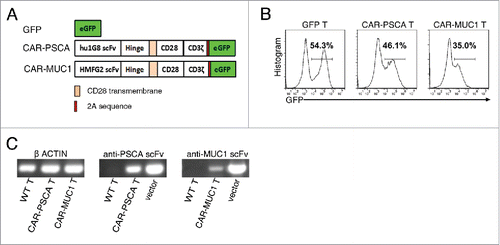
To redirect T lymphocytes to PSCA and MUC1, we used a second-generation PSCA-specific CAR and MUC1-specific CAR, which respectively consisted of the single-chain variable fragments (scFvs) derived from the humanized 1G8 anti-PSCA antibodyCitation33 and anti-MUC1 HMFG2 monoclonal antibody,Citation34 and signaling domains from the co-stimulatory molecule CD28 and the CD3ζ(). Lentiviral vectors encoding green fluorescent protein (GFP; negative control), CAR-PSCA and CAR-MUC1 were transfected into pre-activated human T cells to generate GFP T, CAR-PSCA T and CAR-MUC1 T cells, respectively. Transduction efficiencies were measured as the percentages of GFP+ cells (). We used reverse transcription polymerase chain reaction (PCR) analyses of the scFv sequences to further confirm the expression of anti-PSCA CAR and anti-MUC1 CAR in T cells ().
Figure 3. Construction of anti-prostate stem cell antigen (PSCA) and anti-mucin 1 (MUC1) chimeric antigen receptor (CAR) T cells. (A) Structures of the genes used for lentiviral transfection. GFP, control without CAR; CAR-PSCA, anti-PSCA CAR; CAR-MUC1, anti-MUC1 CAR. (B) Representative flow cytometric analysis of transfected T cells. (C) Reverse transcription-PCR detection of the following: β-ACTIN in wild type (WT), CAR-PSCA and CAR-MUC1 T cells (left); anti-PSCA scFv in WT and CAR-PSCA T cells and CAR-PSCA vector as a positive control (middle) and anti-MUC1 scFv in WT and CAR-MUC1 T cells and CAR-MUC1 vector as a positive control (right).
CAR-PSCA T cells and CAR-MUC1 T cells specifically targeted PSCA+ and MUC1+ lung cancer cells, respectively, in vitro
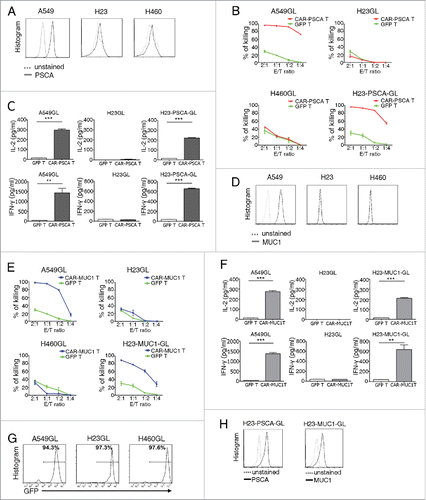
We evaluated the specificity and efficacy of CAR-PSCA T cells against lung cancer cell lines in vitro. First, in a PSCA expression analysis of three lung cancer cell lines, A549, H23 and H460, only A549 cells were found to strongly express PSCA (). Immunohistochemistry analyses also consistently detected PSCA in A549 cells (). A luciferase-based in vitro killing assay demonstrated that CAR-PSCA T cells specifically killed A549GL and H23-PSCA-GL cells (). Enzyme-linked immunosorbent assay (ELISA) results showed the PSCA-specific induction of IL-2 and IFNγ production in supernatants from the killing assay (). Taken together, these findings indicate that CAR-PSCA T cells can recognize and kill PSCA+ cells in vitro. We also confirmed the specificity and efficacy of CAR-MUC1 T cells. Like PSCA, MUC1 was only detected on A549 cells, but not H23 and H460 cells (). CAR-MUC1 T cells killed A549GL and H23-MUC1-GL cells, but not H460GL or H23GL cells, in vitro (). We additionally observed MUC1-specific induction of IL-2 and IFNγ production in culture supernatants (). Next, A549, H460, H23 and H23-PSCA cells were transduced with a lentiviral vector expressing GFP and luciferase (), and H23-PSCA-GL and H23-MUC1-GL cells were generated by transducing lentiviral vectors encoding PSCA and MUC1 into H23GL cells ().
Figure 4. T cells expressing the prostate stem cell antigen (PSCA) or mucin 1 (MUC1) chimeric antigen receptor (CAR) specifically killed PSCA+ or MUC1+ lung cancer cell lines, respectively, in vitro. (A) Flow cytometric analysis of PSCA expression on A549, H23 and H460 cell lines. (B) Percentages of lung cancer line cells killed by GFP T cells and CAR-PSCA T cells at the indicated effector (E): target (T) ratios. The ratios were of the absolute number of CAR T cells vs target cells (corrected for transduction efficiency). T cells were co-cultured with A549GL, H460GL, H23GL or H23-PSCA-GL cells for 18 h, and luciferase activities were measured using a D-luciferin substrate. % of killing = % (total activities without T cells − activities with T cells)/ total activities without T cells. Data were representative of killing assays using T cells from three different donors. (C) Results of enzyme-linked immunosorbent assays (ELISAs) to detect IL-2 and IFNγ in the supernatants of co-cultures at a E:T ratio of 1:1. (D) Flow cytometric analysis of MUC1 expression on A549, H23 and H460 cell lines. (E) Percentages of lung cancer line cells killed by GFP T cells and CAR-MUC1 T cells at the indicated E:T ratios. (F) Results of ELISAs to detect IL-2 and IFNγ in the supernatants of cocultures at an E:T ratio of 1:1. Data were representative of killing assays using T cells from three different donors. (G) Post-transfection GFP-luciferase (GL) expression was detected in A549GL, H23GL and H460GL cell lines by flow cytometry. GFP served as a marker of luciferase expression. (H) Flow cytometric detection of PSCA (left) and MUC1 (right) in H23GL cells after lentiviral transduction. Error bars denote standard errors of the means, and groups were compared using the unpaired t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
CAR-PSCA T cells were efficacious against PSCA+NSCLC in PDX mice
We used a PDX model generated from the PSCA+, MUC1− tumor of one patient (patient P2) to further confirm the efficacy of CAR-PSCA T cells against NSCLC (). Briefly, dissected tumor masses (∼2 mm × 2 mm) were subcutaneously transplanted in NSI mice to generate PDX mice, which subsequently received two infusions of T cells (); the tumors were calibrated until day 40. NSCLC tumor mass growth was significantly suppressed by CAR-PSCA T cells, but not by CAR-MUC1 T cells (). On day 40, the smallest tumors were those in mice treated with CAR-PSCA T cells (), and tumors treated with CAR-PSCA T cells had much lower weights than those left untreated or treated with GFP T cells (); however, no significant difference was found when CAR-MUC1 T cells were used, further suggesting that our CAR T cells recognized and killed NSCLC PDX tumors in an antigen-dependent manner. These results prove the efficacy of CAR-PSCA T cells against PSCA+ NSCLC in PDX mice.
Figure 5. Prostate stem cell antigen chimeric antigen receptor (CAR-PSCA) expressing T cells inhibit the growth of non-small-cell lung cancer (NSCLC) and exhibit synergistic efficacy with mucin 1 CAR (CAR-MUC1) expressing T cells against NSCLC in patient-derived xenograft (PDX) models. (A) Diagram of the experiment with primary NSCLC tumors from patient P2 or P8 in NSI mice. Mice were inoculated subcutaneously with dissected tumor masses from patient P2 or P8 (2 mm × 2 mm), infused with 5 × 106 total T cells on days 7 and 10, and culled on day 40 for tumor analysis. (B–E) Results from the PDX model of patient P2. (B) T cells were analyzed for transfection efficiency before infusion into PDX mice of patient P2. (C) Tumor growth curves in groups treated with no T (n = 3), GFP T (n = 3), CAR-PSCA T (n = 4) or CAR-MUC1 T (n = 4) cells. (D) Tumors from mice treated with no T, GFP T, CAR-PSCA T or CAR-MUC1 T cells on day 40 are shown. One mouse from both the no T and GFP T groups died when tumors were small, which was not shown. (E) Comparison of the weights of tumors described in D. (F, J) Results from the PDX model of patient P8. (F) T cells were analyzed for transfection efficiency before infusion into PDX mice of patient P8. (G) Tumor growth curves in groups treated with no T (n = 3), GFP T (n = 3), CAR-PSCA T (n = 4), CAR-MUC1 T (n = 4), and combinatorial CAR T cells (n = 4). (H) Tumors from different groups in (G) on day 40 were shown. Also, both the no T and GFP T groups had one mouse died when tumors were small. (I) Comparison of the weights of tumors in H. (J) Tumors from CAR-PSCA T, CAR-MUC1 T and combinatorial groups were singled out for comparison. Error bars denote standard errors of the means, and groups were compared using the unpaired t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
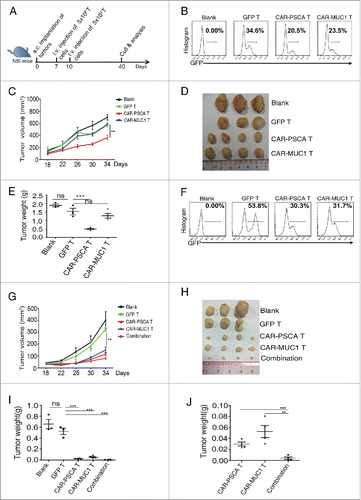
CAR-PSCA T and CAR-MUC1 T cells synergistically inhibited NSCLC growth in PDX mice
We next evaluated the efficacy of a combination of CAR-PSCA T and CAR-MUC1 T cells in a NSCLC PDX model generated from another patient (patient P8) whose tumor expressed both PSCA and MUC1 (). PDX mice were untreated (blank) or treated with identical numbers of GFP T, CAR-PSCA T, CAR-MUC1 T, or a 1:1 mix of CAR-PSCA T and CAR-MUC1 T cells (). Tumor growth was dramatically inhibited by CAR-PSCA T cells, CAR-MUC1 T cells and combined T cells ( and ). Furthermore, the tumor weights in mice treated with combined CAR T cells were significantly less than the weights in mice treated with a single type of CAR T cells ( and ). Collectively, the combination of CAR-PSCA and CAR-MUC1 T cells exhibited superior efficacy against NSCLC, compared with each cell type alone.
Discussion
The treatment of most patients with solid cancers would require the development of genetically redirected T cells that target private somatic mutations, or neoantigens.Citation35 However, this is an arduous task, given the heterogeneity of the mutational landscape within a tumor mass and between metastases. Despite the paucity of tumor-specific antigens shared across various types of solid cancers and among patients within one type of cancer, the TAAs of a single specific cancer are much less heterogeneous than neoantigens. Therefore, CAR T cell immunotherapy remains important during the development of neoantigen targeting techniques.
Although numerous TAAs have been detected in NSCLC,Citation36 a few have been targeted by CAR T cells.Citation37,38 The use of these few non-cancer-specific antigens, which include FAP,Citation39 EGFR,Citation40 mesothelinCitation41 and glypican-3,Citation17 has led to poor or undefined therapeutic outcomes in patients. It is therefore important to broaden the NSCLC-specific targets of CAR T cells. PSCA-targeting CAR T cells has been developed,Citation29,42,43 and is ready for use in clinical trials of safety in patients with prostate, bladder and pancreatic cancers (ClinicalTrials.gov Identifier: NCT02092948 and NCT02744287). Additionally, a trial of MUC1-targeting CAR T cells is currently recruiting patients for NSCLC (ClinicalTrials.gov Identifier: NCT02587689). In this study, we frequently detected PSCA and MUC1 expression in NSCLC cells and thereby demonstrated the usefulness of anti-PSCA and anti-MUC1 CAR T cells.
Dual targeting of erbB2 and MUC1 by T cells expressing both CARs has been reported to deliver complimentary signals, enhance CAR T cell proliferation, but reduce IL-2 production.Citation44 Combinatorial antigen recognition of PSCA and PSMA by a CAR that provides suboptimal activation and a chimeric co-stimulatory receptor (CCR) respectively has been reported to improve specificity and reduce off-target effects.Citation42 This strategy requires two types of chimeric receptors expressing on T cells together. In this study, we tested dual targeting of PSCA and MUC1 by mixed CAR T cells targeting either PSCA or MUC1 in NSCLC PDX models. Cancerous cells within a tumor mass may not unanimously express a single specific antigen, and even the PSCA+MUC1+ samples in our study might contain single positive cancerous cells; therefore, a CAR T cell-based combinatorial targeting strategy may broaden the populations of targeted cancerous cells.
Although the CAR-PSCA and CAR-MUC1 T cells used in our study caused considerable reductions in tumor sizes, they could not completely eradicate NSCLC in PDX mice. This result could be attributed to the poor survival of CAR T cells in the immunosuppressive PDX tumor microenvironment. Strategies to improve the survival and infiltrating capacities of CAR T cells, such as optimized co-stimulationCitation45-47 and cytokine co-expression,Citation47-51 are worthy of exploration for all CAR T cells in solid tumors.
Overall, we have demonstrated that because PSCA and MUC1 are both rational targets in NSCLC, PSCA- and MUC1-targeting CAR T cells might comprise novel therapeutic agents for patients with NSCLC. Our results also suggest that a mixture of CAR T cells with different specificities could target simultaneously expressed tumor antigens and lead to better therapeutic outcomes.
Materials and methods
Vector design
To generate CAR-PSCA and CAR-MUC1 lentiviral vectors, scFvs derived from the humanized 1G8 anti-PSCA antibodyCitation33 and anti-MUC1 HMFG2 monoclonal antibodyCitation34 were codon optimized and synthesized by Genscript (Piscataway, NJ, USA). These scFvs were cloned into a CAR-encoding vector backbone comprising the CD8a leader sequence, human IgD hinge, portions of the CD28 transmembrane domains and the CD28 and CD3ζendodomains within the second-generation lentiviral vector pWPXLd. The amino acid (aa) sequences of the CARs were CD8a leader (aa 1–21), scFv, IgD hinge (aa 187–289), CD28 (aa 153–220) and CD3ζ (aa 52–163).
Lentivirus production and transduction of primary human T cells
Lentivirus particles were produced in HEK-293T cells via polyethyleneimine (Sigma-Aldrich, St. Louis, MO, USA) transfection. The pWPXLd-based lentiviral plasmid and two packaging plasmids, psPAX2 and pMD.2G, were co-transduced into HEK-293T cells. Lentivirus-containing supernatants were harvested at 48 and 72 h post-transduction and filtered through a 0.45-μm filter. Peripheral blood mononuclear cells (PBMCs) were isolated from the buffy coats of healthy donors using Lymphoprep (Fresenius Kabi Norge, AS, Berg i Østfold, Norway). T cells were negatively selected from PBMCs using a MACS Pan T Cell Isolation Kit (Miltenyi Biotec, Bergish Gladbach, Germany) and activated using microbeads coated with anti-human CD3, anti-human CD2 and anti-human CD28 antibodies (Miltenyi Biotec) at a 3:1 bead:cell ratio for 3 d in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, 2 mM glutamine and 1% penicillin/streptomycin. On day 3 post-activation, T cells were transfected with CAR lentiviral supernatants in the presence of 8μg /mL polybrene (Sigma). Twelve hours post-transfection, T cells were cultured in fresh media containing IL-2 (300 U/mL); subsequently, fresh media was added every 2–3 d to maintain cell density within the range of 0.5–1 × 106/mL. Healthy PBMC donors and all patients who provided primary patient specimens gave informed consent to the use of their samples for research purposes, and all procedures were approved by the Research Ethics Board of the Guangzhou Institutes of Biomedicine and Health (GIBH).
Cells and culture conditions
HEK-293T cells were maintained in Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY, USA). A549 (human lung adenocarcinoma), H23 (human lung adenocarcinoma) and H460 (human large cell lung cancer) cell lines were obtained from ATCC (Manassas, VA, USA) and maintained in RPMI-1640. Luciferase-GFP expressing cell lines (A549GL, H23GL and H460GL) were generated through transfection of the parental cell line with a lentiviral supernatant containing luciferase-2A-GFP and were sorted for GFP expression on a FACS Aria™ cell sorter (BD Biosciences, San Jose, CA, USA). H23-PSCA-GL or H23-MUC1-GL cells were generated by transfecting H23 cells with lentiviral supernatant. DMEM and RPMI-1640 media were supplemented with 10% heat-inactivated FBS (Gibco/Life Technologies), 10 mM HEPES, 2 mM glutamine (Gibco/Life Technologies) and 1% penicillin/streptomycin. All cells were cultured at 37 °C in an atmosphere of 5% carbon dioxide.
Flow cytometry
All samples were analyzed using an LSR Fortessa or C6 flow cytometer (BD Biosciences), and data were analyzed using FlowJo software (FlowJo, LLC, Ashland, OR, USA). The following antibodies were used: PSCA (clone 7F5) from Santa Cruz Biotechnology (Dallas, TX, USA) and anti-human CD227 (MUC-1, clone 16ACitation52) from BioLegend (San Diego, CA, USA). The clone 16A binds to glycopeptide RPAPGS(GalNAc)TAPPAHG of MUC1 (MUC1-Tn) with high affinity.Citation52 The scFv of HMFG2 (used to engineer the CAR) can recognize a range of tumor-associated MUC1 glycoforms, such as Tn, STn, T and ST (binds to MUC1-Tn and MUC1-STn with higher affinity). HMFG2 has the broadest capacity for strong binding to tumor-associated MUC1 glycoforms.Citation34 So, the CAR-MUC1 T cells we generated can recognize the 16A positively stained cells.
In vitro tumor killing assays and cytokine release assays
A549GL, H23GL, H23-PSCA-GL or H23-MUC1-GL target cells were incubated with GFP T, CAR-PSCA T or CAR-MUC1 T cells at the indicated ratios in triplicate wells of U-bottomed 96-well plates. Supernatants were collected from wells with E:T ratios of 1:1 and subjected to an analysis of IL-2 and IFNγ concentrations using ELISA kits (eBioscience, San Diego, CA, USA). Cells were treated with 100 µL/well of the luciferase substrate D-luciferin (potassium salt, 150 µg/mL; Cayman Chemical, Ann Arbor, MI, USA), and target cell viability was monitored using a microplate reader at a 450-nm excitation wavelength. Background luminescence was negligible (< 1% of the signal from wells containing only target cells); therefore, the viability percentage (%) was equal to the experimental signal/maximal signal×100, and the killing percentage was equal to 100 − viability percentage.
PDX models for CAR T cell treatment
We used 6–8-week-old NSI mice according to protocols approved by the Institutional Animal Care and Use Committee of GIBH. All mice were maintained in specific pathogen-free (SPF)-grade cages and provided with autoclaved food and water. Surgical tumor samples were obtained from the Sun Yat-Sen University Cancer Center (Guangzhou, China) with informed consent of the patients; tumors were cut into 2 mm × 2 mm pieces and directly transplanted subcutaneously without matrigel or other additives into 3–6 immunodeficient NSI mice within a 30-min period. Tumors that reached an approximate size <1,000 mm3 were removed and passaged to other NSI mice. On days 7 and 10, according to the doses used in other reports,Citation53,54 5 × 106 total T cells were injected through the tail vein into each NSCLC-burdened NSI mouse. Tumors were measured every 4 d with a caliper to determine the subcutaneous growth rate. The tumor volume was calculated using the following equation: (length × width × width)/2.
Reverse Transcription (RT-PCR)
mRNA was extracted from cells using RNeasy mini kit (Qiagen, Stockach, Germany) and reverse transcribed into cDNA using the PrimeScript™ RT reagent Kit (Takara, Shiga, Japan). The following primers were used:
β-ACTIN – forward: 5′-AGAGCTACGAGCTGCCTGAC-3′
β-ACTIN – reverse: 5′-AGCACTGTGTTGGCGTACAG-3′
scFv of CAR-PSCA – forward: 5′-CTCTGTGGGGGATAGGGTCA-3′
scFv of CAR-PSCA – reverse: 5′-TCACAAGATTTGGGCTCGCT-3′
scFv of CAR-MUC1 – forward: 5′-TCGGTGGAGGAACCAAACTG-3′
scFv of CAR-MUC1 – reverse: 5′-CCTCCCTTTCACAGACTCCG-3′
Immunohistochemistry
Tumor tissue sections were fixed with 10% paraformaldehyde, embedded in paraffin, sectioned at a thickness of 4 μm, and stained using a standard hematoxylin and eosin technique. Paraffin sections were also immunostained with antibodies specific for E-cadherin (ZA0565), vimentin (ZA-0511), PSCA (ZA-0158), MUC1 (ZM-0391) and HLA (Abcam, Cambridge, UK) overnight at 4 °C, followed by secondary staining with secondary goat anti-mouse or goat anti-rabbit Ig (PV-9000) (ZSGB-BIO, Beijing, China). Images of all sections were obtained with a microscope (DMI6000B; Leica Microsystems, Wetzlar, Germany).
Statistics
Data are presented as means ± standard errors of the means. Student's t-test was used to determine the statistical significance of differences between samples, and a p value < 0.05 was considered to indicate a significant difference. All statistical analyses were performed using Prism software, version 5.0 (GraphPad, Inc., San Diego, CA, USA).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
XW, YL and JL contributed to the conception and design, collection and/or assembly of data, data analysis and interpretation and manuscript writing. LQ, YX, RZ and QL contributed to the provision of study material or patients, collection and/or assembly of data. BL, SL, SW and QW provided animal care and administrative supports. YY and DP contributed to the conception and design and provided financial support. YY, MP, FY, YL, XZ, YW and PL contributed to the conception and design. XW and PL contributed to the conception and design, data analysis and interpretation, manuscript writing, and final approval of manuscript and provided financial support. All authors read and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (NSFC: 81272329, 81522002, 81570156 and 81327801), Strategic Priority Research Program of the Chinese Academy of Sciences (XDA01020310), the Natural Science Fund for Distinguished Young Scholars of Guangdong Province (2014A030306028), the Guangdong Provincial Applied Science and Technology Research & Development Program (2016B020237006), the Guangdong Provincial Outstanding Young Scholars Award (2014TQ01R068), the Guangdong Provincial Basic Research Program (2015B020227003), the Guangdong Provincial Research and Commercialization Program (2014B090901044), the Guangdong Province and Chinese Academy of Sciences Joint Program for Research and Commercialization Program (2013B091000010), the Guangzhou Basic Research Program (201510010186), the MOST funding of the State Key Laboratory of Respiratory Disease, and the National Basic Research Program of China (973 Program) (2011CB504004 and 2010CB945500), the Frontier and key technology innovation special grant from the Department of Science and Technology of Guangdong province, (2014B020225005), the Guangzhou Science Technology and Innovation Commission Project (201504010016).
References
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65:87-108; PMID:25651787; http://dx.doi.org/10.3322/caac.21262
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69-90; PMID:21296855; http://dx.doi.org/10.3322/caac.20107
- Qin A, Coffey DG, Warren EH, Ramnath N. Mechanisms of immune evasion and current status of checkpoint inhibitors in non-small cell lung cancer. Cancer Med 2016; 5:2567-78; PMID:27416962; http://dx.doi.org/10.1002/cam4.819
- Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res 2016; 5:288-300; PMID:27413711; http://dx.doi.org/10.21037/tlcr.2016.06.07
- Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 2015; 21:560-2; PMID:25939061; http://dx.doi.org/10.1038/nm.3854
- Wang S, Tsui ST, Liu C, Song Y, Liu D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J Hematol Oncol 2016; 9:59; http://dx.doi.org/10.1186/s13045-016-0290-1
- Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol 2016; 9:34; PMID:27071706; http://dx.doi.org/10.1186/s13045-016-0268-z
- Wu J, Savooji J, Liu D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J Hematol Oncol 2016; 9:19; PMID:26951079; http://dx.doi.org/10.1186/s13045-016-0251-8
- Alexander GS, Palmer JD, Tuluc M, Lin J, Dicker AP, Bar-Ad V, Harshyne LA, Louie J, Shaw CM, Hooper DC et al. Immune biomarkers of treatment failure for a patient on a phase I clinical trial of pembrolizumab plus radiotherapy. J Hematol Oncol 2016; 9:96; PMID:27663515; http://dx.doi.org/10.1186/s13045-016-0328-4
- Dang TO, Ogunniyi A, Barbee MS, Drilon A. Pembrolizumab for the treatment of PD-L1 positive advanced or metastatic non-small cell lung cancer. Expert Rev Anticancer Ther 2016; 16:13-20; PMID:26588948; http://dx.doi.org/10.1586/14737140.2016.1123626
- Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Eng J Med 2015; 373:123-35; PMID:26028407; http://dx.doi.org/10.1056/NEJMoa1504627
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Eng J Med 2015; 372:2018-28; PMID:25891174; http://dx.doi.org/10.1056/NEJMoa1501824
- Ma W, Gilligan BM, Yuan J, Li T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol 2016; 9:47; PMID:27234522; http://dx.doi.org/10.1186/s13045-016-0277-y
- Geyer MB, Brentjens RJ. Review: Current clinical applications of chimeric antigen receptor (CAR) modified T cells. Cytotherapy 2016; 18:1393-409; PMID:27592405; http://dx.doi.org/10.1016/j.jcyt.2016.07.003
- Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood 2016; 127:3312-20; PMID:27207800; http://dx.doi.org/10.1182/blood-2016-02-629063
- Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol 2016; 13:370-83; PMID:27000958
- Li K, Pan X, Bi Y, Xu W, Chen C, Gao H, Shi B, Jiang H, Yang S, Jiang L et al. Adoptive immunotherapy using T lymphocytes redirected to glypican-3 for the treatment of lung squamous cell carcinoma. Oncotarget 2016; 7:2496-507; PMID:26684028; http://dx.doi.org/10.18632/oncotarget.6595
- Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, Han W. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci 2016; 59:468-79; PMID:26968708; http://dx.doi.org/10.1007/s11427-016-5023-8
- Situ D, Wang J, Ma Y, Zhu Z, Hu Y, Long H, Rong T. Expression and prognostic relevance of MUC1 in stage IB non-small cell lung cancer. Med Oncol 2011; 28 Suppl 1:S596-604; PMID:21116877; http://dx.doi.org/10.1007/s12032-010-9752-4
- Kawaguchi T, Sho M, Tojo T, Yamato I, Nomi T, Hotta K, Hamada K, Suzaki Y, Sugiura S, Kushibe K et al. Clinical significance of prostate stem cell antigen expression in non-small cell lung cancer. Jpn J Clin Oncol 2010; 40:319-26; PMID:20085909; http://dx.doi.org/10.1093/jjco/hyp181
- Reiter RE, Gu Z, Watabe T, Thomas G, Szigeti K, Davis E, Wahl M, Nisitani S, Yamashiro J, Le Beau MM et al. Prostate stem cell antigen: a cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci USA 1998; 95:1735-40; PMID:9465086; http://dx.doi.org/10.1073/pnas.95.4.1735
- Zou Q, Yang L, Yang Z, Huang J, Fu X. PSCA and Oct-4 expression in the benign and malignant lesions of gallbladder: implication for carcinogenesis, progression, and prognosis of gallbladder adenocarcinoma. BioMed Res Int 2013; 2013:648420
- Zhao X, Wang F, Hou M. Expression of stem cell markers nanog and PSCA in gastric cancer and its significance. Oncol Lett 2016; 11:442-8; PMID:26870231; http://dx.doi.org/10.3892/ol.2015.3884
- The MICAD Research Team. [125I]Anti-prostate stem cell antigen antibody. Molecular Imaging and Contrast Agent Database (MICAD) 2004; PMID:20641839
- Ross S, Spencer SD, Holcomb I, Tan C, Hongo J, Devaux B, Rangell L, Keller GA, Schow P, Steeves RM et al. Prostate stem cell antigen as therapy target: tissue expression and in vivo efficacy of an immunoconjugate. Cancer Res 2002; 62:2546-53; PMID:11980648
- Matsueda S, Kobayashi K, Nonaka Y, Noguchi M, Itoh K, Harada M. Identification of new prostate stem cell antigen-derived peptides immunogenic in HLA-A2(+) patients with hormone-refractory prostate cancer. Cancer Immunol Immun 2004; 53:479-89; PMID:14634796; http://dx.doi.org/10.1007/s00262-003-0464-x
- Matsueda S, Yao A, Ishihara Y, Ogata R, Noguchi M, Itoh K, Harada M. A prostate stem cell antigen-derived peptide immunogenic in HLA-A24- prostate cancer patients. Prostate 2004; 60:205-13; PMID:15176050; http://dx.doi.org/10.1002/pros.20038
- Gu Z, Yamashiro J, Kono E, Reiter RE. Anti-prostate stem cell antigen monoclonal antibody 1G8 induces cell death in vitro and inhibits tumor growth in vivo via a Fc-independent mechanism. Cancer Res 2005; 65:9495-500; PMID:16230414; http://dx.doi.org/10.1158/0008-5472.CAN-05-2086
- Abate-Daga D, Lagisetty KH, Tran E, Zheng Z, Gattinoni L, Yu Z, Burns WR, Miermont AM, Teper Y, Rudloff U et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum Gene Ther 2014; 25:1003-12; PMID:24694017; http://dx.doi.org/10.1089/hum.2013.209
- Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Mælandsmo GM et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov 2014; 4:998-1013; PMID:25185190; http://dx.doi.org/10.1158/2159-8290.CD-14-0001
- Dong R, Qiang W, Guo H, Xu X, Kim JJ, Mazar A, Kong B, Wei JJ. Histologic and molecular analysis of patient derived xenografts of high-grade serous ovarian carcinoma. J Hematol Oncol 2016; 9:92; PMID:27655386; http://dx.doi.org/10.1186/s13045-016-0318-6
- Ye W, Jiang Z, Li GX, Xiao Y, Lin S, Lai Y, Wang S, Li B, Jia B, Li Y et al. Quantitative evaluation of the immunodeficiency of a mouse strain by tumor engraftments. J Hematol Oncol 2015; 8:59; PMID:26022250; http://dx.doi.org/10.1186/s13045-015-0156-y
- Olafsen T, Gu Z, Sherman MA, Leyton JV, Witkosky ME, Shively JE, Raubitschek AA, Morrison SL, Wu AM, Reiter RE. Targeting, imaging, and therapy using a humanized antiprostate stem cell antigen (PSCA) antibody. J Immunother 2007; 30:396-405; PMID:17457214; http://dx.doi.org/10.1097/CJI.0b013e318031b53b
- Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, Arif S, Mather SJ, Taylor-Papadimitriou J, Burchell JM et al. Retargeting of human t cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol 2008; 180:4901-9; PMID:18354214; http://dx.doi.org/10.4049/jimmunol.180.7.4901
- Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med 2016; 22:26-36; PMID:26735408; http://dx.doi.org/10.1038/nm.4015
- Djureinovic D, Hallström BM, Horie M, Mattsson JS, La Fleur L, Fagerberg L, Brunnström H, Lindskog C, Madjar K, Rahnenführer J et al. Profiling cancer testis antigens in non-small-cell lung cancer. JCI Insight 2016; 1:e86837; PMID:27699219; http://dx.doi.org/10.1172/jci.insight.86837
- Beatty GL, O'Hara M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: Defining the challenges and next steps. Pharmacol Ther 2016; 166:30-9; PMID:27373504; http://dx.doi.org/10.1016/j.pharmthera.2016.06.010
- Di S, Li Z. Treatment of solid tumors with chimeric antigen receptor-engineered T cells: current status and future prospects. Sci China Life Sci 2016; 59:360-9; PMID:26968709; http://dx.doi.org/10.1007/s11427-016-5025-6
- Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, Liu H, Wang LL, Rowley DR, Pfizenmaier K et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther 2013; 21:1611-20; PMID:23732988; http://dx.doi.org/10.1038/mt.2013.110
- Zhou X, Li J, Wang Z, Chen Z, Qiu J, Zhang Y, Wang W, Ma Y, Huang N, Cui K et al. Cellular immunotherapy for carcinoma using genetically modified EGFR-specific T lymphocytes. Neoplasia 2013; 15:544-53; PMID:23633926; http://dx.doi.org/10.1593/neo.13168
- Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov 2016; 6:133-46; PMID:26503962; http://dx.doi.org/10.1158/2159-8290.CD-15-0583
- Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 2013; 31:71-5; PMID:23242161; http://dx.doi.org/10.1038/nbt.2459
- Hillerdal V, Ramachandran M, Leja J, Essand M. Systemic treatment with CAR-engineered T cells against PSCA delays subcutaneous tumor growth and prolongs survival of mice. BMC Cancer 2014; 14:30; PMID:24438073; http://dx.doi.org/10.1186/1471-2407-14-30
- Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, Burbridge SE, Box C, Eccles SA, Maher J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol 2012; 32:1059-70; PMID:22526592; http://dx.doi.org/10.1007/s10875-012-9689-9
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015; 21:581-90; PMID:25939063; http://dx.doi.org/10.1038/nm.3838
- Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012; 119:696-706; PMID:22117050; http://dx.doi.org/10.1182/blood-2011-03-344275
- Hombach AA, Heiders J, Foppe M, Chmielewski M, Abken H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4(+) T cells. Oncoimmunology 2012; 1:458-66; PMID:22754764; http://dx.doi.org/10.4161/onci.19855
- Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, Dotti G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010; 24:1160-70; PMID:20428207; http://dx.doi.org/10.1038/leu.2010.75
- Perna SK, De Angelis B, Pagliara D, Hasan ST, Zhang L, Mahendravada A, Heslop HE, Brenner MK, Rooney CM, Dotti G et al. Interleukin 15 provides relief to CTLs from regulatory T cell-mediated inhibition: implications for adoptive T cell-based therapies for lymphoma. Clin Cancer Res 2013; 19:106-17; PMID:23149818; http://dx.doi.org/10.1158/1078-0432.CCR-12-2143
- Perna SK, Pagliara D, Mahendravada A, Liu H, Brenner MK, Savoldo B, Dotti G. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin Cancer Res 2014; 20:131-9; PMID:24097874; http://dx.doi.org/10.1158/1078-0432.CCR-13-1016
- Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood 2010; 115:3508-19; PMID:20190192; http://dx.doi.org/10.1182/blood-2009-09-241398
- Song W, Delyria ES, Chen J, Huang W, Lee JS, Mittendorf EA, Ibrahim N, Radvanyi LG, Li Y, Lu H et al. MUC1 glycopeptide epitopes predicted by computational glycomics. Int J Oncol 2012; 41:1977-84; PMID:23023583; http://dx.doi.org/10.3892/ijo.2012.1645
- O'Hear C, Heiber JF, Schubert I, Fey G, Geiger TL. Anti-CD33 chimeric antigen receptor targeting of acute myeloid leukemia. Haematologica 2015; 100:336-44; PMID:25480499; http://dx.doi.org/10.3324/haematol.2014.112748
- Qin H, Cho M, Haso W, Zhang L, Tasian SK, Oo HZ, Negri GL, Lin Y, Zou J, Mallon BS et al. Eradication of B-ALL using chimeric antigen receptor-expressing T cells targeting the TSLPR oncoprotein. Blood 2015; 126:629-39; PMID:26041741; http://dx.doi.org/10.1182/blood-2014-11-612903