
ABSTRACT
The colorectal tumor microenvironment contains a diverse population of myeloid cells that are recruited and converted to immunosuppressive cells, thus facilitating tumor escape from immunoediting. We have identified a genetically and functionally distinct subset of dynamic bone marrow myeloid cells that are characterized by histidine decarboxylase (HDC) expression. Lineage tracing in Hdc-CreERT2;R26-LSL-tdTomato mice revealed that in homeostasis, there is a strong bias by HDC+ myeloid cells toward the CD11b+Ly6Ghi granulocytic lineage, which was accelerated during azoxymethane/dextran sodium sulfate (AOM/DSS)-induced colonic carcinogenesis. More importantly, HDC+ myeloid cells strongly promoted colonic tumorigenesis, and colon tumor progression was profoundly suppressed by diphtheria toxin A (DTA)-mediated depletion of HDC+ granulocytic myeloid cells. In addition, tumor infiltration by Foxp3+ regulatory T cells (Tregs) was markedly impaired following HDC+ myeloid cell depletion. We identified an HDC+ myeloid-derived Cxcl13/Cxcr5 axis that mediated Foxp3 expression and Treg proliferation. Ablation of HDC+ myeloid cells or disruption of the Cxcl13/Cxcr5 axis by gene knockdown impaired the production and recruitment of Tregs. Cxcl13 induction of Foxp3 expression in Tregs during tumorigenesis was associated with Stat3 phosphorylation. Overall, HDC+ granulocytic myeloid cells affect CD8+ T cells directly and indirectly through the modulation of Tregs and thus appear to play key roles in suppressing tumoricidal immunity.
Abbreviations
| 5-FU | = | 5-fluorouracil |
| AOM | = | azoxymethane |
| BAC | = | Bacterial artificial chromosome |
| BrdU | = | 5-bromo-2′-deoxyuridine |
| CRC | = | colorectal cancer |
| DSS | = | dextran sodium sulfate |
| DTA | = | diphtheria toxin A |
| ELISPOT | = | enzyme-linked ImmunoSpot |
| FACS | = | fluorescence-activated cell sorting |
| FBS | = | fetal bovine serum |
| Foxp3 | = | foxhead box P3 |
| HDC | = | histidine decarboxylase |
| IMC | = | immature myeloid cells |
| i.p. | = | intraperitoneal |
| i.v. | = | intravenous |
| MDSC | = | myeloid-derived suppressor cell |
| OCT | = | optimal cutting temperature |
| PFA | = | paraformaldehyde |
| qRT-PCR | = | quantitative RT-PCR |
| RBC | = | red blood cell |
| rCxcl13 | = | recombinant Cxcl13 |
| Stat3 | = | signal transducer and activator of transcription 3 |
| Tgfb1 | = | transforming growth factor 1 |
| Treg | = | regulatory T cell |
| WT | = | wild type |
Introduction
Chronic inflammation has long been recognized as an important risk factor in the development of colorectal cancer (CRC).Citation1 Inflammation can directly stimulate tumor growth and spread, but the suppression of tumor immunity is also needed to establish a favorable tumor niche.Citation2 In the setting of cancer treatment, many therapies can now be used to shrink or inhibit the growth of tumors, but the long-term prognosis of most patients is closely linked to the reestablishment of antitumor immunity.Citation3 With the remarkable success in recent years of immune checkpoint drugs, there has been a growing interest in the mechanisms of immune response to these drugs, and thus the cell types responsible in general for immunosuppression.
Many stromal cells in the tumor microenvironment can contribute to the suppression of tumor immunity, but two of the leading candidates are myeloid cells and regulatory T cells (Tregs). Myeloid cells, arising in the bone marrow from haematopoietic stem cells and myeloid progenitors,Citation4,5 frequently expand in the setting of tumorigenesis. They represent a diverse group of cells and are commonly subdivided into two major categories: monocytic (mononuclear) myeloid cells and granulocytic (polymorphonuclear) myeloid cells.Citation3 Monocytic cells are typically precursors of macrophages and dendritic cells, whereas the granulocytic cells comprise neutrophils, eosinophils, basophils, and mast cells.Citation6 However, in the setting of cancer, immature myeloid cells (IMCs, CD11b+Gr1+) tend to emerge with properties favoring tumor growth. Among these, the myeloid-derived suppressor cells (MDSCs), including M-MDSCs and PMN-MDSCS, overlap with monocytic and granulocytic myeloid cells, but represent two immunophenotypically distinct cell populations.Citation3 The granulocytic MDSCs (PMN-MDSCs) can be identified based on their higher expression of Ly6G and lower expression of Ly6C, in contrast to M-MDSCs which express Ly6C but lack Ly6G expression.Citation7 Although both myeloid lineages have been implicated in cancer,Citation8-10 studies by our group and others have shown the predominant expansion and recruitment of granulocytic (CD11b+Ly6Ghi) myeloid cells, which often express Cxcr2 and Cxcr4, in murine colon cancer models.Citation11,12 In a recent study, we linked these CD11b+Ly6Ghi (PMN-MDSCs and neutrophils) to the gene Histidine decarboxylase (Hdc), which is expressed higher in granulocytic compare to monocytic myeloid cells, raising the possibility that HDC might be a general marker of the granulocytic myeloid lineage.Citation13
In the bone marrow and spleen, myeloid cells are spatially separated from other lineages and tend to congregate together to form myeloid clusters,Citation14 while T cells mature in the thymus and then reside in the spleen and peripheral lymphoid tissue. Nevertheless, both lineages are activated and recruited in the setting of chronic inflammation and cancer, with the level of immunity and clinical outcomes depending on the interactions between them.Citation15 Indeed, interactions between myeloid cells and CD4+ T cells resulting in enhanced tumor-promoting immunity have been reported in neoplasia of the intestine as well as other organs.Citation9,16 Communication between T cells and myeloid cells has been reported in both directions. For instance, IL-17-producing γδ T cells can expand and recruit neutrophils to tumors, while myeloid cells can secret cytokines and chemokines that modulate the function of T cells.Citation17,18 Among the various subclasses of T cells, CD4+ Tregs are suppressor T cells that modulate adaptive immune responses, maintaining tolerance to self-antigens and inhibiting cytotoxic CD8+ T cell responses.Citation19,20 Treg function can be influenced by tumor myeloid cells through chemokine secretion,Citation17,21,22 and, in colon cancer, through the increased expression of Foxhead box P3 (Foxp3), leading to the expansion of Tregs and a poorer prognosis.Citation23
A number of studies have begun to explore strategies for inhibiting tumor-associated myeloid cells and Tregs because of their likely role in modulating cytotoxic CD8+ T cell function.Citation24 Indeed, many widely used cancer therapies, including both chemotherapy and radiation therapy, have been shown to actually increase circulating myeloid cells, as might be expected in the setting of tissue injury.Citation3 In theory, many tumor ablative therapies should enhance antitumor immune responses, given the effects on exposing tumor antigens, but a simultaneous expansion of myeloid suppressor cells might negate such immune stimulation. Thus, therapeutic resistance may occur in part due to the expansion of myeloid cells secondary to injury and repair. Current immunotherapies such as immune checkpoint blockers and monoclonal antibodies have shown great promise, but many tumors have shown resistance, which could be due in part to myeloid suppressor cells.Citation3 There is a growing interest to clarify the precise contribution of the various myeloid lineages to tumor progression.Citation25 In the current study, we compared the role and function of the HDC+ myeloid lineage to the remainder of the HDC− myeloid cells during carcinogenesis, using a combination of genetic lineage tracing and DTA-mediated cell depletion. Our data show that HDC+ myeloid cells are specifically amplified during colonic carcinogenesis, strongly suppress CD8+ T cells and act in part through supporting Tregs. Our data further reveal HDC+ cells to be the most immune suppressive myeloid subpopulation and suggest that targeted inhibition or depletion of these cells might be beneficial in cancer therapy.
Results
HDC labels a distinct population of myeloid cells
Myeloid cells are a heterogeneous collection of leukocytes, fundamental to innate immunity and mobilized from the bone marrow in response to injury or carcinogenesis.Citation5 We and others have identified a subpopulation of myeloid cells that express CD11b, Gr1, and HDC, which are associated with colon cancer progression.Citation13,26 To better characterize this tumor-promoting myeloid cell subpopulation, we used gene microarrays to compare the transcriptomes of HDC+ and HDC− CD11b+Gr1+ myeloid cells by sorting bone marrow cells from Hdc-GFP transgenic mice.Citation13 In contrast to HDC−CD11b+Gr1+ myeloid cells that were more monocytic, HDC+ myeloid cells expressed significantly higher levels of Cxcr2 and Cxcr4 and lower levels of Ccr2 and Cx3cr1 (). In addition, HDC+ myeloid cells showed gene expression patterns suggestive of more active proliferation, with the upregulation of cell cycle positive regulators (Ccnd2, Ccnd3, and Cdc14a), and downregulation of cell cycle inhibitors such as Cdc14b and Cdkn1a (Fig. S1A). P21, an essential component of the P53-mediated senescence pathway,Citation27 was downregulated in HDC+ myeloid cells (Fig. S1A), while Pd-l1, a crucial immune checkpoint ligand, was upregulated in HDC+ myeloid cells (). Cell cycle analysis using Ki-67 and Hoechst staining confirmed that HDC+ myeloid cells were largely in non-G0 cell cycle phases and thus actively cycling ().
Figure 1. HDC marks a distinct granulocytic myeloid population. (A and B) Gene microarray comparison between HDC+ and HDC− CD11b+Gr1+ bone marrow myeloid cells (n = 3 per group), showing differential expression of chemokine receptors and Pdl-1. (C) Proportion of G0 and Non-G0 cells from HDC+ or HDC− bone marrow, spleen, and small intestine myeloid cells (n = 5 per group) as measured by flow cytometry using Ki-67 and Hoechst 33342 staining. Showing representative flow plots (left) and quantification (right). (D and E) Lineage tracing in Hdc-CreERT2;Rosa-tdTomato mice (n = 3) under homeostatic condition. FACS at different time points for tdTomato+ myeloid cells from spleen (D) and intestine (E) 6 d after tamoxifen withdrawal. (F and G) Lineage tracing in Hdc-CreERT2;Rosa-tdTomato mice (n = 5) in carcinogenic conditions in AOM/DSS-treated mice. FACS at different time points for tdTomato+ cells from spleen (F) and intestine (G) 6 d after tamoxifen withdrawal. *p < 0.05; **p < 0.01; ***p < 0.001, data are mean ± SEM, representing three independent experiments analyzed with two-tailed Student's t-test (C) and one-way analysis of variation (ANOVA) with Dunnett's pos-hoc test (D–G).
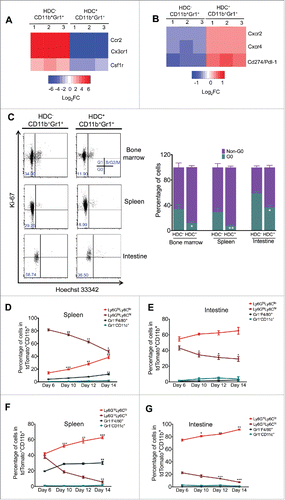
To trace the cellular fate of the HDC+ myeloid lineage, we generated Hdc-CreERT2 mice and crossed them to Rosa26-LSL-tdTomato reporter mice (Fig. S1B). By inducing Hdc-CreERT2;Rosa26-LSL-tdTomato mice with a tamoxifen-containing diet, the tdTomato+ myeloid populations, isolated from spleen and intestine, expressed significantly higher levels of Hdc mRNA compare to tdTomato− myeloid cells, confirming the specificity of the Hdc-CreERT2 transgene (Fig. S1C). We next conducted lineage tracing by inducing Hdc-CreERT2;Rosa26-LSL-tdTomato mice with a tamoxifen diet for 10 d, followed by a tamoxifen-free washout for 6 d (Fig. S1D). The majority of traced myeloid cells (tdTomato+CD11b+) cells in the spleen were first observed at day 6 after tamoxifen withdrawal, and the bulk of these cells were Ly6GloLy6Chi monocytes, with a smaller proportion of Ly6GhiLy6Clo granulocytes. There were fewer Gr1−F4/80+ macrophages and Gr1−CD11c+ dendritic cells (). Over the next 8 d, there was an increase in traced granulocytes along with a decrease in traced monocytes. Interestingly, in the small intestine, most of the tdTomato+ cells were granulocytes, and the labeling was steadily increased while traced monocytes declined (). Thus, this baseline lineage tracing study suggests that HDC-CreERT2 marks both granulocytic and monocytic lineages, but specifically targets granulocytic progenitors that have proliferative potential, leading to an expansion of granulocytes over time.
Next, we traced the HDC+ myeloid lineage during initiation of murine AOM/DSS inflammatory colorectal carcinogenesis. HDC-CreERT2;tdTomato mice were treated with AOM (10 mg/kg) followed 10 d later by DSS (2%) for another 10 d, then induced by a tamoxifen diet for 10 d. After a 6-d washout, the spleen and intestinal myeloid cells were analyzed (Fig. S1E). At the first time point (6 d post-washout), traced CD11b+Ly6GhiLy6Clo granulocytic cells resembling PMN-MDSCs and neutrophils were found markedly expanding in the spleen, with continued increases in number over the next 8 d (). Furthermore, these Ly6GhiLy6Clo cells were also increased in the small intestine, comprising the majority of traced myeloid cells in this AOM/DSS model.Citation12,28 This suggests that AOM/DSS treatment leads to a systemic and preferential expansion of the HDC+ granulocytic myeloid lineage (Fig. S1G).
To further characterize HDC+ myeloid cells, we examined the expressions of known cell surface markers and genes previously associated with distinct populations of myeloid cells. Our data showed that HDC+ (tdTomato+) myeloid cells had higher expression of CXCR4 and CXCR2 and lower expression of CCR2 compared with HDC− (tdTomato−) myeloid cells in both the steady-state and tumor (Figs. S1D, S1F, and S1H). Interestingly, the tumor HDC+ myeloid cells also expressed higher levels of CD80, a B7 family member that defines immunosuppressive myeloid cells (Fig. S1H).Citation29 In addition, other immunosuppression-associated genes, including Cox-2, Arg-1, and Mmp9, were highly abundant in the HDC+ tumor myeloid population (Fig. S1I). In addition, HDC+CD11b+Gr1+ myeloid cells exhibited a more active cell cycle and higher Pdl-1 gene expression (Figs. S1A and B). Given these findings, we next sought to compare directly the immunosuppressive ability of tdTomato+ and tdTomato− myeloid cells from AOM/DSS-treated Hdc-CreERT2;Rosa26-LSL-tdTomato tumor-bearing mice (after 10 weeks, three cycles of DSS). We flow sorted splenic tdTomato+ and tdTomato− CD11b+Gr1+ myeloid cells, and incubated them with naive splenic CD8+ T cells. The tumor tdTomato+ myeloid cells demonstrated greater CD8+ T cell suppressive ability compared with the remainder of the tdTomato− myeloid cells, as shown by Enzyme-Linked ImmunoSpot (ELISPOT) analysis of IFNγ and Granzyme B (Fig. S1J). Moreover, tdTomato+ myeloid cells were dramatically expanded following 5-fluorouracil (5-FU) treatment (Fig. S1K). Taken together, these data indicate that HDC+ granulocytic myeloid cells may represent a distinct myeloid population with strong immunosuppressive abilities that is expanded during for colorectal carcinogenesis and in response to cytotoxic therapy.
HDC+ myeloid cells promote the development of intestinal tumors in APCmin/+ mice
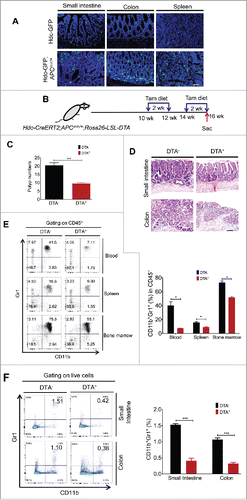
In addition to establishing that HDC+ myeloid cells are expanded in the AOM/DSS colon carcinogenesis model, we also wanted to explore the role of HDC+ cells in a genetic model of intestinal neoplasia. Interestingly, truncating APC mutations have also been shown to lead to inflammatory infiltrates (including myeloid cells) in the small intestine, which likely contribute to the growth of intestinal polyps in APCmin/+ mice.Citation30 Strikingly, performing immunofluorescence analysis on frozen sections of 18 weeks old APCmin/+;Hdc-GFP mice, we found an abundant accumulation of HDC-GFP+ cells in small intestinal polyps and colon mucosa, as well as an expansion of HDC-GFP+ cells in the spleen (). This suggests that HDC-GFP+ cells play a role during cancer initiation. Since myeloid cells have also been implicated in the progression of APCmin/+ tumor,Citation31 we next sought to investigate the role of HDC+ myeloid cells herein by crossing APCmin/+ mice with Hdc-CreERT2;Rosa26-LSL-DTA mice expressing inducible DTA. Two cycles of tamoxifen diet were administered at 10–12 weeks and 14–16 weeks of age, respectively, and mice were killed immediately following the last tamoxifen diet treatment (). Depletion of HDC+ myeloid cells in Hdc-CreERT2; APCmin/+;Rosa26-LSL-DTA mice inhibited the development of crypt expansion and neoplastic lesions, resulting in a significantly decreased number of intestinal polyps and much more regular intestinal histology (). Of note, the total number of CD11b+Gr1+ myeloid cells in CD45+ leukocytes was significantly decreased in the blood, spleen, and bone marrow (). The number of CD11b+Gr1+ myeloid cells in the small intestine and colon was also markedly reduced (). Thus, our data suggest that myeloid cells, specifically the HDC+ myeloid lineage, strongly promote adenomatous polyposis initiated by APC mutation.
Figure 2. HDC+ myeloid cells promote intestinal adenomatous polyposis in APCmin/+ mice. (A) Representative fluorescence images showing accumulation of HDC+ (GFP+) cells in small intestine, colon, and spleen of APCmin/+;Hdc-GFP mice (n = 3). Scale bar = 50 µm (B) Experimental protocol for induction of HDC+ myeloid cell depletion in Hdc-CreERT2;APCmin/+;Rosa26-LSL-DTA mice by tamoxifen diet administration. (C) Number of intestinal polyps in 16-weeks-old Hdc-CreERT2;APCmin/+;Rosa26-LSL-DTA mice treated with tamoxifen diet, comparing genotyped DTA− (n = 4) and DTA+ (n = 5) mice. (D) Representative H&E images of small intestine and colon histological sections from mice analyzed in (C). Scale bar = 50 µm. (E) Percentages of CD11b+Gr1+ myeloid cells in CD45+ cells from blood, spleen, and bone marrow, respectively, from mice analyzed in (C). Showing flow plots (left) and quantification (right) (F) Percentages of myeloid cells in digested intestinal tissue of mice analyzed in (C), with representative FACS plots (left), and quantification (right). *p < 0.05; ***p < 0.001, data are mean ± SEM, representing two to three independent experiments. Data were analyzed with two-tailed Student's t-test (E and F) and Mann–Whitney test (C).
HDC+ myeloid cells promote colorectal carcinogenesis
We next examined whether HDC+ myeloid cells are essential for promoting tumor-prone immunity in the AOM/DSS colorectal carcinogenesis model. The Hdc-CreERT2;Rosa26-LSL-tdTomato mice were crossed to Rosa26-LSL-DTA and treated with AOM (10 mg/kg) followed 10 d later by three cycles of 2% DSS (10 d each) (). The mice were given tamoxifen in the chow for 20 d after each cycle of DSS to activate DTA expression and thus deplete HDC+ myeloid cells. Three cycles of tamoxifen were needed, in view of the relatively short life span of myeloid cells and their rapid replacement from haematopoietic stem and progenitor cells (). Indeed, colon tumor size was markedly reduced by HDC+ myeloid ablation, and overall tumor numbers were decreased around 3-fold (). The decreased tumor growth was strongly associated with a reduction in tumor-infiltrating myeloid cells, as DTA expression efficiently depleted most of the tdTomato+ cells from colonic tumor tissue (). Accordingly, the numbers of CD11b+Gr1+ myeloid cells in bone marrow (), spleen (), and blood () were all decreased in the DTA+ animals, as well as splenomegaly (), which is a common sign of myeloid cell expansion in tumor-bearing animals. Moreover, to confirm the functional significance of HDC+ myeloid cell depletion, we performed adoptive transfer of splenic HDC+CD11b+Gr1+ myeloid cells from AOM/DSS-treated donors into induced DTA mice and found that this significantly rescued tumor numbers ().
Figure 3. HDC+ myeloid cells promote colitis-associated colorectal carcinogenesis. (A) Experimental protocol for HDC+ myeloid depletion and rescue in Hdc-CreERT2;Rosa26-tdTomato;Rosa26-DTA mice subject to AOM/DSS colorectal carcinogenesis. (B) Macroscopic tumor images and tumor quantification from groups of AOM/DSS-treated mice as per protocol shown in (A), including DTA genotype negative mice (DTA−, n = 8), DTA genotype positive mice (DTA+, n = 10), and DTA+ mice treated with HDC+ myeloid cell adoptive transfer (HDC+ spleen myeloid cells from AOM/DSS-treated mice, n = 5). (C) Representative fluorescence images showing depletion of tdTomato+ cells in colon tumor frozen sections from mice analyzed in (B). Scale bar = 50 µm. (D) FACS plots and bar graphs showing the percentage of CD11b+Gr1+ myeloid cells in CD45+ bone marrow leukocytes of DTA− or DTA+ AOM/DSS tumor mice analyzed in (B). (E) Macroscopic images of spleens and quantitation of splenic myeloid cells in DTA− or DTA+ AOM/DSS-treated tumor mice analyzed in (B). (F) Percentage of CD11b+Gr1+ myeloid cells in circulating leukocytes in DTA+ and DTA− mice analyzed in (B). (G) Representative FACS plots (left) and quantitation (right) of tumor-associated CD8+ T cells from mice analyzed in (B). *p < 0.05; **p < 0.01; ***p < 0.001, data are mean ± SEM, representing two independent experiments. Data were analyzed with Mann–Whitney test (B), two-tailed Student's t-test (D, E, and F), and one-way analysis of variation (ANOVA) with Dunnett's post-hoc test (H).
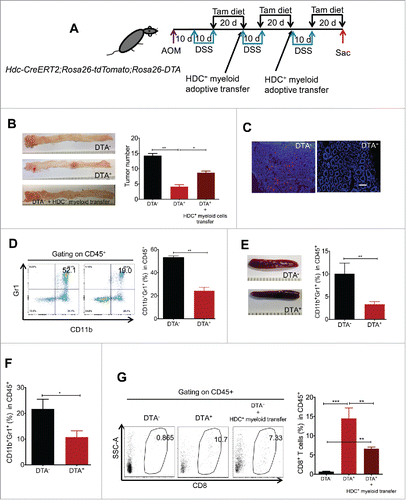
CD8+ cytotoxic T cells were quite rare in colonic tumors of AOM/DSS tumor-bearing wild-type (WT) mice. Ablation of HDC+ myeloid cells markedly reduced overall PD-L1 expression in CD11b+Gr1+ cells (Figs. S2A and B), and thus in theory should allow greater expansion and tumor infiltration by CD8+ T cells. Indeed, DTA-mediated ablation of HDC+ myeloid cells resulted in a marked (> 20-fold) increase in CD8+ T cells in colonic tumors (). Furthermore, adoptive transfer of HDC+CD11b+Gr1+ myeloid cells (from the spleen of AOM/DSS-treated mice) into DTA-treated mice partially suppressed CD8+ T cell infiltration (). Moreover, in vitro functional analysis of sorted CD8+ T cells from the colons of either DTA− or DTA+ mice revealed that DTA-mediated depletion of HDC+ myeloid cells led to greater T cell activation, with over 3-fold increases in IFNγ and 2-fold increases of Granzyme B (Fig. S2C), while adoptive transfer of HDC+CD11b+Gr1+ myeloid cells from tumor-bearing mice suppressed CD8+ T cell activation (Fig. S2C). Interestingly, although adding back HDC+ myeloid cells decreased activated CD8+ T cells and increased colon tumor number (), it never fully rescued these parameters compared with the non-depletion group. Given that the adoptive transfer of HDC+ myeloid cells was performed at a somewhat late point during cancer development, we wondered whether the lower efficacy of this intervention in stimulating tumor growth might indicate a need for interactions between HDC+ myeloid cells and other immune regulatory cells.
HDC+ myeloid cells derived Cxcl13 recruits Foxp3+ Treg cells in colorectal carcinogenesis
In addition to myeloid suppressor cells, another group of immunosuppressive cells is Tregs, which can suppress CD8+ T cells and also show cross-talk with myeloid cells.Citation32 Tregs can support myeloid suppressor cells by upregulating the expression of immunosuppressive molecules such as B7-H1.Citation33 Myeloid cells, in turn, are thought to modulate Foxp3+ Tregs, but the mechanisms have not been well defined. To begin to study interactions between HDC+ myeloid cells and other immune cells, we compared serum chemokine profiles between DTA− and DTA+ AOM/DSS-induced tumor-bearing mice. A number of chemokines associated with myeloid cell recruitment, such as CXCL1, CCL2, and CCL20, were significantly decreased in the DTA+ group (). The Treg trafficking-associated chemokine CCL22,Citation34 which was highly presented in the sera of tumor-bearing mice, but depletion of HDC+ myeloid cells resulted in no significant change in CCL22 serum levels, suggesting that HDC+ myeloid cells are probably not the major source of CCL22 production. However, another chemokine, CXCL13, previously associated with Treg regulation through the receptor CXCR5, was more than 5-fold downregulated in the sera of tumor-bearing DTA+ mice (). Quantitative RT-PCR (qRT-PCR) on sorted tumor CD45+ cells before and after DTA induction showed that the loss of HDC+ myeloid cells significantly decreased Cxcl13 mRNA expression (), while the change of Cxcl13 was not associated with transforming growth factor 1 (Tgfb1) or IL-10 perturbation in haematopoietic cells (Fig. S3A). These findings were consistent with the gene expression profile data, which showed that HDC+ and HDC− myeloid cells expressed similar levels of Tgfb1 and IL-10 mRNA (see GSE79728). This decrease in Cxcl13 expression was accompanied by a marked decrease in tumor-infiltrating Foxp3+ Tregs, as revealed by Foxp3 immunofluorescence staining of tumor frozen sections (). The Cxcr5 receptor on tumor Treg cells, accordingly, was also downregulated in Tregs from DTA+ mice (Fig. S3B). To confirm the role of the Cxcl13/Cxcr5 axis in the recruitment of Foxp3+ Tregs, we injected AOM/DSS-treated Foxp3-GFP mice with a Cxcl13 neutralizing antibody. Cxcl13 immunoneutralization led to a significant reduction in the percentage of Foxp3+ cells in tumor CD4+ lymphocytes (), and Cxcr5 mRNA expression on Foxp3+ cells isolated from tumor tissue was downregulated compared with tumor-bearing mice treated with the isotype control antibody (Fig. S3C). Taken together, these data indicate that the Cxcl13/Cxcr5 axis is essential for Foxp3+ Treg recruitment during AOM/DSS colitis-associated carcinogenesis.
Figure 4. HDC+ myeloid cells recruit Foxp3+ Tregs in colitis-associated colon carcinogenesis model. (A) Comparison of circulating chemokine levels in DTA− (n = 5) and DTA+ (n = 6)AOM/DSS tumor-bearing mice sera. (B) Expression of Cxcl13 mRNA from sorted CD45+ cells in colon tumor tissue, comparing DTA− and DTA+ (n = 3 per group). (C) Representative fluorescence staining showing Foxp3+ cells in colon tumor tissue in DTA− and DTA+ AOM/DSS-treated mice (n = 3 per group). Scale bar = 50 µm. (D) Percentages of Foxp3-GFP+ cells in colon CD4+ cells in AOM/DSS-treated mice with (n = 3) or without (n = 4) rCxcl13 neutralization antibody. (E) Migration of Foxp3+ cells in response to Cxcl13. Showing quantitation of migrated Foxp3+ cell numbers by flow cytometry after co-culture of 5,000 splenic Foxp3-GFP+ cells (from spleens of AOM/DSS-treated Foxp3-GFP mice) in transwell plates with either HDC+ or HDC− myeloid cells. N = 3 to 5 per group. *p < 0.05; **p < 0.01; ***p < 0.001, data are mean ± SEM, representing two to three independent experiments. Data were analyzed with two-tailed Student's t-test (A, B, and D) and Mann–Whitney test (E).
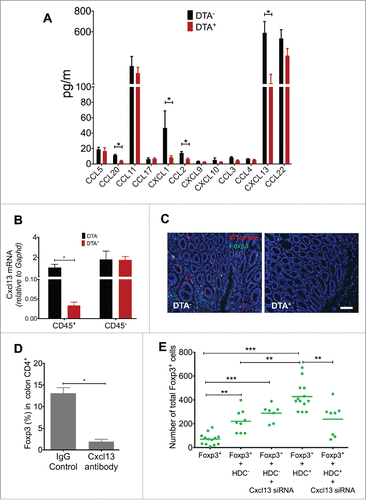
To investigate further the role of HDC+ myeloid cells in mobilizing Tregs, we performed transwell culture experiments with Foxp3-GFP+ Tregs on the top well and CD11b+Gr1+ myeloid cells (HDC+ vs. HDC−) on the bottom wells. Migration of GFP+ Tregs was quantified by flow cytometry after co-culture. In some groups, HDC+ myeloid cells were transfected with a specific Cxcl13 siRNA to knock down Cxcl13 expression. We found that Cxcl13-expressing HDC+ myeloid cells, rather than Cxcl13 deficient HDC+ myeloid cells or HDC− myeloid cells, recruited the greatest number of GFP+ Tregs (). Although HDC− myeloid cells also marginally increased GFP+ Foxp3 Tregs mobilization, Cxcl13 knockdown did not change Treg mobilization in co-culture, indicating other mechanisms might be involved (). Moreover, in the DTA+ mouse colon, where Treg infiltration was largely dampened by the ablation of HDC+ myeloid cells, the infusion of recombinant Cxcl13 (rCxcl13) protein rescued the recruitment of GFP+ Foxp3 Tregs to colonic tissue (Fig. S3D). Collectively, these data establish the importance of the Cxcl13/Cxc5r axis in mediating the recruitment of Tregs by HDC+ myeloid cells in the setting of colon carcinogenesis.
Cxcl13/Cxcr5 axis increases Treg Foxp3 expression and is dependent on Stat3 phosphorylation
Having established Cxcl13 from HDC+ myeloid cells as an important Treg chemoattractant factor in the AOM/DSS colon tumor model, we next investigated how HDC+ myeloid-derived Cxcl13 influenced Treg function. Foxp3 gene expression is essential for Treg differentiation and function in tumor models,Citation35 and there is a close association between Stat3 activity and chemokine receptor expression in Tregs.Citation36 We thus hypothesized that Cxcl13 might induce Foxp3 expression and Tregs expansion in part through Stat3 phosphorylation. To test this hypothesis, we first analyzed Stat3 phosphorylation and Foxp3 gene expression in Foxp3-GFP+ cells from the colon tumor tissue of AOM/DSS-treated Foxp3-GFP;Hdc-CreERT2;Rosa26-tdTomato;Rosa26-DTA mice induced with tamoxifen. Again, three cycles of 2% DSS were given, alternating with tamoxifen diet (). Controls for this experiment were Rosa26-DTA genotype negative (DTA−) littermates, which we assumed reflected baseline expression of phospho-Stat3 and Foxp3. The phosphorylation of Stat3 was decreased by 4-fold upon ablation of HDC+ myeloid cells, and this could be rescued by either adoptive transfer of HDC+ myeloid cells (from spleen of AOM/DSS-treated donors) or infusion of rCxcl13 (). Moreover, Foxp3 mRNA expression was decreased by >5-fold in Foxp3-GFP+ cells after depletion of HDC+ myeloid cells, and this reduction could also be partially restored by replenishing HDC+ myeloid cells or by rCxcl13 infusion (Fig. S4A). Interestingly, similar to the effects of HDC+ myeloid cell transfer (), rCxcl13 infusion in DTA+ mice increased tumor number while decreasing tumor infiltration by CD8+ cytotoxic T cells (Figs. S4B and C).
Figure 5. HDC+ myeloid-derived Cxcl13 augment Foxp3 expression through Stat3 phosphorylation. (A) Mouse experimental protocol. (B and C) Levels of phospho-Stat3 in Foxp3-GFP+ cells isolated by flow sorting from colon tumors. (B) Shows representative flow histogram graphs of phospho-Stat3, while (C) shows quantitation of phospho-Stat3+Foxp3+ cells (n = 4 DTA−, 5 DTA+, 4 DTA+ + HDC+ myeloid transfer, 5 DTA+ + rCxcl13). (D) Foxp3+ cell proliferation measured by BrdU incorporation assay. Foxp3-GFP+ cells cultured in vitro with either HDC+ or HDC− myeloid cells (n = 5 to 8), with a knockdown in some groups of Cxcr5 or Cxcl13 by siRNA in Foxp3-GFP+ cells or HDC+ myeloid cells, respectively. rCxcl13 was added to Foxp3-GFP+ cells cultured without myeloid cells. Cells were isolated from spleens of AOM/DSS- and tamoxifen- treated Foxp3-GFP;Hdc-CreERT2;Rosa26-tdTomato mice. (E and F) Immunofluorescence images (E) and quantification (F) of CD4+ splenic T cells co-cultured with either HDC+ or HDC− splenic myeloid cells. CD4+ cells were sorted from AOM/DSS-treated Foxp3-GFP mice and myeloid cells were isolated from AOM/DSS- and tamoxifen- treated Hdc-CreERT2;Rosa26-tdTomato mice. Cxcr5 or Cxcl13 were knocked down by siRNA before co-culture with CD4+ cells or HDC+ cells, respectively. Cells were stained with PE-conjugated antibody against phospho-Stat3 and (E) show representative staining of Foxp3-GFP and Phospho-Stat3-PE. The Foxp3-GFP+ and Phospho-Stat3+ cells were counted as shown in (F). (G) BrdU proliferation assay of cultured Foxp3-YFP+ splenic cells from AOM/DSS-treated Foxp3-YFP-Cre-Stat3+/+ or Foxp3-YFP-Cre-Stat3−/− mice, with or without Cxcl13 for 24 h (n = 5 to 9 per group). **p < 0.01; ***p < 0.001, data are mean ± SEM, representing two to three independent experiments. Data were analyzed with one-way analysis of variation (ANOVA) with Dunnett's post-hoc test (C, D, and G) and Mann–Whitney test (F).
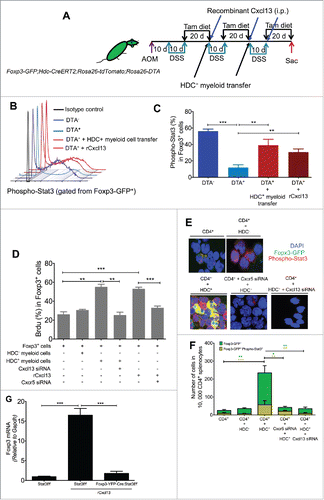
To show that Cxcl13 was acting directly on Foxp3+ Tregs, we sorted splenic CD4+ cells from AOM/DSS-treated Foxp3-GFP mice and co-cultured them with myeloid cells or rCxcl13. After a 1-h incubation with BrdU, flow cytometry analysis of BrdU-labeled Foxp3-GFP+ cells revealed significantly increased proliferation of Foxp3-GFP+ cells when cultured with either HDC+ myeloid cells or rCxcl13. These effects were abolished by RNAi knockdown of Cxcl13 in HDC+ myeloid cells or Cxcr5 in Foxp3-GFP+ cells (). Interestingly, in addition to inducing Treg proliferation, HDC+ myeloid cells or rCxcl13 also increased Foxp3-GFP expression and phospho-Stat3 fluorescence intensity (as revealed by a fluorescence-conjugated antibody against phospho-Stat3) in vitro (). The increase in nuclear accumulation of Foxp3 and phospho-Stat3 observed with HDC+ myeloid cell co-culture was also abrogated by knockdown of either myeloid-derived Cxcl13 or Cxcr5 in CD4+ T cells (). Finally, to further demonstrate that Cxcl13/Cxcr5 signaling acts through Stat3, we harvested CD4+Foxp3-GFP+ spleen cells from Foxp3-YFP-Cre;Stat3f/f mice treated with AOM/DSS. Foxp3 expression and cell proliferation were measured in CD4+ cells cultured in the presence of rCxcl13. Recombinant Cxcl13 stimulated Foxp3+ cell proliferation and increased Foxp3 mRNA expression, but only in the presence of Stat3 ( and Fig. S4D).
Discussion
The role of myeloid cells in mediating the link between inflammation and cancer is now well established, but a better understanding of the mechanisms involved has been constrained by the marked heterogeneity of myeloid cells.Citation37,38 Further, while the majority of studies have focused on the role of monocytic myeloid cells in tumorigenesis,Citation39,40 the Ly6Ghi granulocytic myeloid cells are extremely abundant in colonic tumors and have not been as well characterized. In the current study, we have defined an HDC-expressing sub-population of myeloid cells that predominate in colon carcinogenesis and expand during chemotherapy-induced stress. HDC+ myeloid cells highly expressing Pd-l1 have a strong ability to suppress CD8+ T cells, and appear to be an immunophenotypically and functionally distinct myeloid population. Thus, the ablation of HDC+ myeloid cells in vivo leads to increased CD8+ T cell tumor infiltration and decreased colonic tumor growth, while adoptive transfer of HDC+ myeloid cells inhibits CD8+ T cell infiltration and stimulates tumor growth. Interestingly, HDC+ myeloid cells mediated tumor growth both in colitis-associated CRC and in the APCmin/+ model of intestinal polyposis, where the ablation of HDC+ myeloid cells decreased the formation of intestinal adenomatous polyps and the grade of intestinal epithelial dysplasia. The effect of HDC+ myeloid cells was likely related in part to the induction of Tregs. The ablation of HDC+ myeloid cells also reduced Treg infiltration, which was restored by HDC+ myeloid cell transfer or the infusion of rCxcl13. Finally, we showed that Cxcl13, produced by HDC+ myeloid cells, acts through Cxcr5 to stimulate Foxp3 expression and Treg proliferation through a phospho-Stat3 pathway.
In contrast to more commonly studied myeloid cell lineages, HDC+ myeloid cells express higher levels of Cxcr2 and Cxcr4, and lower levels of Ccr2 or Cx3cr1,Citation41,42 consistent with a predominantly granulocytic lineage.Citation12,31 In addition, tumor-associated HDC+ myeloid cells expressed higher levels of CD80, Cox-2, Arg-1, and Mmp9, which have previously been associated with the development of immuosuppressive myeloid cells. Furthermore, the abundance of HDC+ myeloid cells in the AOM/DSS CRC model supports previous observations that Cxcr2-expressing CD11b+Ly6GhiLy6Clo myeloid cells comprise the majority of the tumor-associated myeloid population.Citation12 Indeed, lineage tracing of the HDC+ myeloid lineage in the settings of homeostasis and AOM/DSS carcinogenesis demonstrated the proliferative nature of HDC+ myeloid cells and the expansion of the granulocytic lineage during colorectal tumorigenesis. At a gene expression level, HDC+ myeloid cells exhibit a strong proliferative potential, with high expression of positive cell cycle genes and decreased expression of cell cycle repressors. This greater proliferative potential appears to allow HDC+ myeloid cells to expand and contribute to a permissive tumor microenvironment.
While HDC+ myeloid cells can likely contribute to tumor growth in several ways, including through direct growth stimulation as previously shown,Citation11,13,26 the current study shows a clear contribution to the suppression of antitumor immunity.Citation4 HDC+ myeloid cells express higher levels of the immune checkpoint ligand Pdl-l gene, and depletion of HDC+ myeloid cells by conditional induction of DTA decreased almost half of the myeloid-derived PD-L1 expression and increased more than 8-fold the colonic infiltration of IFNγ- and Granzyme B-secreting CD8+ T cells, which could be partially reversed by adoptive transfer of HDC+ myeloid cells from the AOM/DSS-treated donors. At least some of this immunosuppressive effect was likely direct, given our studies showing that HDC+ myeloid cells were superior in suppressing CD8+ T cell activity in vitro compared with HDC− myeloid cells, consistent with previous studies.Citation11 However, we also observed a reduction in Tregs and HDC− myeloid cells with HDC+ myeloid ablation, suggesting the presence of a more involved immune regulatory network, primarily implicating Tregs as a key downstream effector cell.
We found that HDC+ myeloid cells exhibited increased production of several chemokines, compared with HDC− myeloid cells, and one of these chemokines, Cxcl13, was of particular interest due to its association with Treg cell migration.Citation43 The Cxcl13 receptor Cxcr5 is highly expressed by tumor-infiltrating Tregs and consistent with a Cxcl13/Cxcr5 axis mediating Treg recruitment. Expression of Cxcr5 was decreased by HDC+ myeloid ablation or Cxcl13 neutralization. Cxcl13 was shown to be a key mediator by in vivo Cxcl13 neutralization studies, where immunoneutralization reduced the recruitment of Foxp3+ T cells to tumor tissue, and transwell culture studies, where siRNA knockdown of Cxcl13 in HDC+ myeloid cells reduced Treg migration.
Stat3, widely expressed by haematopoietic cells, is elevated in the tumor tissue of advanced cancers and has shown promise as a target for cancer therapy.Citation44,45,46 Elevated Stat3 phosphorylation in Treg cells is usually associated with increased trafficking and acquisition of greater immune suppression properties, and an interaction between Stat3 and Foxp3 in tumor Tregs has been recently proposed.Citation47 Given our observations that Cxcl13 modulated Foxp3 expression, we wondered if it might be related to the status of Stat3 phosphorylation.Citation47,48 Strikingly, ablation of HDC+ myeloid cells reduced phospho-Stat3 in vivo, which was rescued by Cxcl13 infusion, and in vitro studies showed that Cxcl13 was able to directly increase phospho-Stat3 nuclear accumulation in Tregs, which was abolished by Cxcr5 knockdown. Finally, conditional knockout of Stat3 in Foxp3+ T cells abrogated the ability of Cxcl13 to stimulate Treg proliferation and Foxp3 gene expression. Taken together, HDC+ myeloid cells induce the expansion and recruitment of Treg cells, a key immunosuppressive cell type in the tumor microenvironment, through Cxcl13 secretion.
In summary, we have identified and characterized HDC+CD11b+Gr1+ myeloid cells as a distinct immunosuppressive and pro-tumorigenic myeloid subpopulation. In the setting of cancer, HDC-expressing myeloid cells acquire greater immune suppressive capacity and promote the accumulation of Cxcr5-expressing Tregs at the tumor site through the secretion of Cxcl13. HDC+ myeloid-derived Cxcl13 acts on Tregs to enhance Foxp3 expression and cellular proliferation in part through Stat3 phosphorylation. Targeted ablation of HDC+ myeloid cells leads to an increase in CD8+ T cells and a reduction in Tregs and colonic tumor growth. It has been recognized that cancer development from early dysplastic lesions to more advanced invasion stages is accompanied by tissue infiltration of variety of immune cells. Myeloid heterogeneity thus appears to be a critical issue closely associated with cancer development and treatment.Citation49 In this regard, targeting individual immune population might be a promising approach for prevention or treatment of cancer. Our data suggest that the HDC+ myeloid sub-population is likely to be of continued interest in the therapy of early stage cancer. Moreover, recent studies have cast light on the critical role of immunosuppressive myeloid cells in checkpoint blockade and chemotherapy resistance.Citation50,51 Given the superior immunosuppressive capacity and abundance of HDC+ myeloid cells in the tumor microenvironment, and their robust response to 5-FU treatment, we propose that targeting HDC+ myeloid cells, interruption of HDC+ myeloid-Treg interaction, or antagonizing the Cxcl13-Cxcr5 axis, might be useful strategies to explore in the future.
Material and methods
Mouse models
All animal experiment protocols have been approved by Columbia University Institutional Animal Care and Use Committee. Hdc-CreERT2 line was generated by using Bacterial artificial chromosome (BAC) recombineering protocol from clone RP23–474H6. The Hdc-CreERT2 transgenic founders were backcrossed to C57BL/6 mice for at least six generations to generate C57BL/6 background. Hdc-GFP,Citation13 Foxp3-GFP,Citation52 Foxp3-YFP-Cre,Citation53 were described previously. The conditional Stat3 knockout mice (B6.129S1-Stat3tm1Xyfu/J), Rosa26-LSL-tdTomato (B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J), and Rosa26-LSL-DTA (Gt(ROSA)26Sortm1(DTA)Lky/J) were obtained from The Jackson Laboratory (Bar Harbour, ME). All mice were backcrossed and maintained at C57BL/6 background and housed in the special pathogen-free facilities.
In vivo treatments
Tamoxifen-containing chow (TD. 130872) was purchased from Harlan Laboratories (Madison, WI). AOM (Sigma) was injected intraperitoneally (i.p.) (10 mg/kg). 2% DSS (w/v, molecular weight 36,000–50,000; MP Biomedicals, Solon, OH) was dissolved in drinking water provided ad libitum for 10 d per cycle. To neutralize Cxcl13, AOM/DSS-treated mice were injected i.p. with 200 µg anti-Cxcl13 antibody (R&D Systems) or Rat IgG control twice a week after each cycle of DSS water. Recombinant mouse Cxcl13 (Biolegend, San Diego, CA) was injected i.p. at 50 µg/mouse. 5-FU (Sigma) was diluted in PBS and injected intravenously (i.v.) at 250 mg/kg.
Flow cytometry analysis and cell sorting
Bone marrow cells were collected by flushing from the mouse long bones with Ca2+- and Mg2+-free HBSS (Gibco) added 2% heat-inactivated fetal bovine serum (FBS, Invitrogen). Mouse spleen tissue was squeezed by the rubber end of a plunger from syringe against a cell strainer (BD Biosciences). Blood cells were collected from the submandibular vein in EDTA-containing Tubes (BD Diagnostics). Red blood cell lysis was performed by using red blood cell (RBC) lysis solution (Biolegend, San Diego, CA). Intestinal tissue and colon tumor were enzymatically digested for 30 min with constant shaking at 37˚C in RPMI1640 medium (Gibco) supplemented with 10% FBS (Invitrogen) and enzyme mixture including collagenase IV (Worthington Biochemical Corp, Lakewood, NJ), Dispase II (Gibco), and DNAse I (Roche). The resulting digested cells were filtered and subjected to 40/80 Percoll gradient (GE Healthcare, Pittsburgh, PA) to enrich for leukocytes. All cells were passed through a 70-µm cell strainer to acquire monolayer cell suspension before antibody staining. Cells were stained by combinations of mouse antibodies including CD45 (30-F11), CD11b (M1/70), Gr1 (RB6–8C5), Ly6G (1A8), Ly6C (HK1.4), F4/80 (BM8), CD11c (N418), CXCR4 (L276F12), CD80 (16–10A1), CXCR2 (SA044G4), CCR2 (SA203G11), CD8 (53–6.7), CD4 (GK1.5), PD-L1 (10F.9G2), and phospho-Stat3 (Y705, eBioscience). For intracellular staining, sorted GFP+ and GFP− myeloid cells (isolated from Hdc-GFP mice) cells were fixed and permeabilized (BD cytofix/cytoperm solutions) before staining with Ki-67 (16A8) and Hoechst 33342 (BD PharMingen) to reveal the cell cycle. The same fixation and permeabilization protocols were applied to stain phospho-Stat3 in sorted Foxp3-GFP+ cells. All flow cytometry antibodies, except phospho-Stat3, were purchased from Biolegend. All flow cytometry analyses were performed on LSRII or LSRFortessa (BD Biosciences). BD Influx cell sorter was used for sorting cells for further experiments.
Cell culture
Flow-sorted CD11b+Gr1+ myeloid cells or CD4+ T cells were cultured in RPMI 1640 medium supplemented with 10% FBS, antibiotics, and specific growth factors. For transwell experiments, sorted myeloid cells (HDC+ or HDC− from AOM/DSS-treated, tamoxifen diet-induced Hdc-CreERT2-Rosa26-tdTomato mice) were placed in the lower chamber of HTS Transwell (Corning), while Foxp3-GFP+ spleen Tregs were added in the upper chamber. The mobilization of Foxp3-GFP+ Tregs was measured by FACS counting the numbers of GFP+ Tregs in lower chamber after 12 h of culture. In some experiments, Cxcl13 or Cxcr5 were knocked down by using RNAi (TriFECTa® DsiRNA, Integrated DNA Technologies, Coralville, Iowa) according to manufacturer's manual. To measure the cell proliferation in some co-culture experiments, 10 µM BrdU (BD PharMingen) was added 1-h before harvest the cells, the BrdU incorporation ratio was determined by using BD PharmingenTM BrdU Flow Kits according to manufacturer's instructions.
Chemokine profiling and ELISPOT analysis
Sera from AOM/DSS tumor-bearing mice were collected and isolated. The chemokines were then detected by using LEGENDplex™ Mouse Proinflammatory Panel (Biolegend). The cytotoxicity of CD8+ T cells were determined by using ELISPOT assay. In brief, sorted colon CD8+ T cells were cultured at 1:1 ratio with irradiated naïve spleen leukocytes and stimulated with 1 mg/mL of anti-CD3/CD28 antibodies (Biolegend) for 48 h. The dots of IFNγ and Granzyme B were detected by using Mouse IFNγ/Granzyme B Dual-Color FluoroSpot Kit (R&D systems). In some experiments, CD8+ T cells were co-cultured with either HDC+ or HDC− myeloid cells for 12 h before ELISPOT assay.
Immunofluorescence microscopy and histopathology
Frozen slides from 4% paraformaldehyde (PFA) fixed, optimal cutting temperature (OCT) compound embedded mouse spleen, small intestine, and colon tissue were permeabilized in 0.05% TritonX-100/PBS and blocked with 5% normal goat serum. Primary antibodies were applied (anti-GFP, 1:200, Abcam; anti-RFP, 1:200, Rockland; anti-Foxp3, 1:100, Biolegend). Secondary antibodies (AlexaFluor, Invitrogen) were used to reveal the staining. DAPI counterstained slides were mounted with Prolong anti-fade mounting medium (Invitrogen). In some experiments, cultured cells were sorted and cytospinned onto gelatin-coated slides (Fisher Scientific), fixed and stained with anti-GFP and anti-phospo-Stat3 primary antibodies followed by secondary antibodies staining. All fluorescence staining slides were scanned with A1 laser scanning confocal equipped with an Elipse Ti microscope stand (Nikon Instruments). To reveal the tissue histopathology, hematoxylin and eosin staining was performed on formalin-fixed paraffin-embedded tissue.
Myeloid cells adoptive transfer
Myeloid cells (CD11b+Gr1+), including HDC+ or HDC−, were flow sorted from spleen of Hdc-GFP or Hdc-CreERT2-Rosa26-tdTomato (tamoxifen diet-induced) mice treated with AOM/DSS (10 weeks after treatment). Cells were injected i.v. into AOM/DSS-treated hosts at indicated time points.
Gene microarray analysis
Bone marrow CD11b+Gr1+ myeloid cells were isolated from Hdc-GFP mice femurs and tibias by flow sorting. Total mRNA was extracted by using Rneasy Micro kit (Qiagen) according to the manufacturer's instructions. 3′ IVT Expression Kit (Affymetrix) was used to label RNA. Microarray studies were performed by using the GeneChip mouse genome 430 2.0 array (Affymetrix). The quality of arrays was examined by using a battery of quality test. Sample normalization and statistical significance estimation were calculated by using the GCRMA algorithm and Linear Models for MicroArrays (Limma), respectively.Citation54,55 Data analysis was performed on Bioconductor platform in the R statistical computing environment. A significance cutoff of a Benjamini–Hochberg false discovery rate ≤ 0.05 was used.Citation56-58 Microarray data have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE79728 for HDC-GFP+CD11b+Gr1+ versus HDC-GFP−CD11b+Gr1+ myeloid cells.
Quantitative RT-PCR
Flow-sorted cells were lysed and mRNA was isolated by using Rneasy Micro kit (Qiagen) according to manufacturer's instructions. Reverse transcription was performed by using SuperScript® III First-Strand Synthesis System (Life Technologies, Grand Island, NY). Gene expression was determined by using PrimeTime qRT-PCR assays (Integrated DNA Technologies, Coralville, Iowa), including mouse Hdc (Mm.PT.58.41686379.g), Cox2 (Ptgs2, Mm.PT.58.9154407), Arg1 (Mm.PT.58.8651372), Mmp9 (Mm.PT.58.10100097), Cxcl13 (m.PT.58.31389616), Cxcr5 (Mm.PT.58.45859964), Foxp3 (Mm.PT.58.23713819.g), and Gapdh (Mm.PT.39a.1). All PCR experiments were performed on Applied Biosystems Prism 9700 PCR machine (Applied Biosystems).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was supported by NIH NIDDK to T.C.W. (5R01DK048077). Huan Deng was supported by a postdoctoral fellowship grant from National Science Foundation of China (81300347). Moritz Middelhoff was supported by a postdoctoral fellowship grant from the Mildred-Scheel-Stiftung, Deutsche Krebshilfe, Germany (70111870). We thank Pavel Tishchenko, Spandan Shah, Caisheng Lu, Wei Wang, and Siu-Hong Ho from CCTI Flow Cytometry Core (supported in part by the Office of the Director, National Institutes of Health under awards S10OD020056 and S10RR027050). We thank Ms. Sarah Stokes, Ms. Ashlesha Muley, and Dr Samuel Asfaha for helping on generating the Hdc-CreERT2 construct. We thank Dr Chyuan-Sheng Lin at Columbia University Transgenic Mouse Shared resource for his transgenic advice and service. We also gratefully acknowledge Dr Joji Fujisaki at Columbia University for sharing the Foxp3-GFP and Foxp3-YFP-Cre mice.
KONI_A_1290034_supplementary_data.zip
Download Zip (7.1 MB)References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID: 21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013; 19:1423-37; PMID: 24202395; http://dx.doi.org/10.1038/nm.3394
- Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer 2016; 16:447-62; PMID: 27339708; http://dx.doi.org/10.1038/nrc.2016.54
- Schouppe E, De Baetselier P, Van Ginderachter JA, Sarukhan A. Instruction of myeloid cells by the tumor microenvironment: Open questions on the dynamics and plasticity of different tumor-associated myeloid cell populations. Oncoimmunology 2012; 1:1135-45; PMID: 23170260; http://dx.doi.org/10.4161/onci.21566
- Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest 2015; 125:3365-76; PMID: 26325033; http://dx.doi.org/10.1172/JCI80006
- Iwasaki H, Akashi K. Myeloid lineage commitment from the hematopoietic stem cell. Immunity 2007; 26:726-40; PMID: 17582345; http://dx.doi.org/10.1016/j.immuni.2007.06.004
- Youn JI, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, Villagra A, Antonia S, McCaffrey JC, Fishman M et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol 2013; 14:211-20; PMID: 23354483; http://dx.doi.org/10.1038/ni.2526
- Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol 2016; 37:208-20; PMID: 26858199; http://dx.doi.org/10.1016/j.it.2016.01.004
- Ortiz ML, Kumar V, Martner A, Mony S, Donthireddy L, Condamine T, Seykora J, Knight SC, Malietzis G, Lee GH et al. Immature myeloid cells directly contribute to skin tumor development by recruiting IL-17-producing CD4+ T cells. J Exp Med 2015; 212:351-67; PMID: 25667306; http://dx.doi.org/10.1084/jem.20140835
- Feng PH, Lee KY, Chang YL, Chan YF, Kuo LW, Lin TY, Chung FT, Kuo CS, Yu CT, Lin SM et al. CD14(+)S100A9(+) monocytic myeloid-derived suppressor cells and their clinical relevance in non-small cell lung cancer. Am J Respir Crit Care Med 2012; 186:1025-36; PMID: 22955317; http://dx.doi.org/10.1164/rccm.201204-0636OC
- Dubeykovskaya Z, Si Y, Chen X, Worthley DL, Renz BW, Urbanska AM, Hayakawa Y, Xu T, Westphalen CB, Dubeykovskiy A et al. Neural innervation stimulates splenic TFF2 to arrest myeloid cell expansion and cancer. Nat Commun 2016; 7:10517; PMID: 26841680; http://dx.doi.org/10.1038/ncomms10517
- Katoh H, Wang D, Daikoku T, Sun H, Dey SK, Dubois RN. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013; 24:631-44; PMID: 24229710; http://dx.doi.org/10.1016/j.ccr.2013.10.009
- Yang XD, Ai W, Asfaha S, Bhagat G, Friedman RA, Jin G, Park H, Shykind B, Diacovo TG, Falus A et al. Histamine deficiency promotes inflammation-associated carcinogenesis through reduced myeloid maturation and accumulation of CD11b+Ly6G+ immature myeloid cells. Nat Med 2011; 17:87-95; PMID: 21170045; http://dx.doi.org/10.1038/nm.2278
- Kwak HJ, Liu P, Bajrami B, Xu Y, Park SY, Nombela-Arrieta C, Mondal S, Sun Y, Zhu H, Chai L et al. Myeloid cell-derived reactive oxygen species externally regulate the proliferation of myeloid progenitors in emergency granulopoiesis. Immunity 2015; 42:159-71; PMID: 25579427; http://dx.doi.org/10.1016/j.immuni.2014.12.017
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID: 19197294; http://dx.doi.org/10.1038/nri2506
- Khamri W, Walker MM, Clark P, Atherton JC, Thursz MR, Bamford KB, Lechler RI, Lombardi G. Helicobacter pylori stimulates dendritic cells to induce interleukin-17 expression from CD4+ T lymphocytes. Infect Immun 2010; 78:845-53; PMID: 19917709; http://dx.doi.org/10.1128/IAI.00524-09
- Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, Cerwenka A. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol 2012; 189:5602-11; PMID: 23152559; http://dx.doi.org/10.4049/jimmunol.1201018
- Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJ, Ciampricotti M, Hawinkels LJ, Jonkers J et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015; 522:345-8; PMID: 25822788; http://dx.doi.org/10.1038/nature14282
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 2013; 210:1695-710; PMID: 23897981; http://dx.doi.org/10.1084/jem.20130579
- Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF et al. Regulatory T Cells in tumor-associated tertiary lymphoid structures suppress anti-tumor t cell responses. Immunity 2015; 43:579-90; PMID: 26341400; http://dx.doi.org/10.1016/j.immuni.2015.08.006
- Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res 2008; 68:5439-49; PMID: 18593947; http://dx.doi.org/10.1158/0008-5472.CAN-07-6621
- Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, Wang H, Chen J, Wang H. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS One 2011; 6:e19495; PMID: 21559338; http://dx.doi.org/10.1371/journal.pone.0019495
- Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, Maeda Y, Hamaguchi M, Ohkura N, Sato E et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med 2016; 22:679-84; PMID: 27111280; http://dx.doi.org/10.1038/nm.4086
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011; 480:480-9; PMID: 22193102; http://dx.doi.org/10.1038/nature10673
- Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer 2015; 15:409-25; PMID: 26105538; http://dx.doi.org/10.1038/nrc3958
- Ahn B, Kohanbash G, Ohkuri T, Kosaka A, Chen X, Ikeura M, Wang TC, Okada H. Histamine deficiency promotes accumulation of immunosuppressive immature myeloid cells and growth of murine gliomas. Oncoimmunology 2015; 4:e1047581; PMID: 26451324; http://dx.doi.org/10.1080/2162402X.2015.1047581
- Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8:729-40; PMID: 17667954; http://dx.doi.org/10.1038/nrm2233
- Wei TT, Lin YT, Tseng RY, Shun CT, Lin YC, Wu MS, Fang JM, Chen CC. prevention of colitis and colitis-associated colorectal cancer by a novel polypharmacological histone deacetylase inhibitor. Clin Cancer Res 2016; 22:4158-69; PMID: 27528734; http://dx.doi.org/10.1158/1078-0432.CCR-15-2379
- Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol 2008; 8:467-77; PMID: 18500231; http://dx.doi.org/10.1038/nri2326
- Li Y, Kundu P, Seow SW, de Matos CT, Aronsson L, Chin KC, Kärre K, Pettersson S, Greicius G. Gut microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis 2012; 33:1231-8; PMID: 22461519; http://dx.doi.org/10.1093/carcin/bgs137
- Jamieson T, Clarke M, Steele CW, Samuel MS, Neumann J, Jung A, Huels D, Olson MF, Das S, Nibbs RJ et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest 2012; 122:3127-44; PMID: 22922255; http://dx.doi.org/10.1172/JCI61067
- Fujimura T, Kambayashi Y, Aiba S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology 2012; 1:1433-4; PMID: 23243619; http://dx.doi.org/10.4161/onci.21176
- Fujimura T, Ring S, Umansky V, Mahnke K, Enk AH. Regulatory T cells stimulate B7-H1 expression in myeloid-derived suppressor cells in ret melanomas. J Invest Dermatol 2012; 132:1239-46; PMID: 22189788; http://dx.doi.org/10.1038/jid.2011.416
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10:942-9; PMID: 15322536; http://dx.doi.org/10.1038/nm1093
- Yuan XL, Chen L, Li MX, Dong P, Xue J, Wang J, Zhang TT, Wang XA, Zhang FM, Ge HL et al. Elevated expression of Foxp3 in tumor-infiltrating Treg cells suppresses T-cell proliferation and contributes to gastric cancer progression in a COX-2-dependent manner. Clin Immunol 2010; 134:277-88; PMID: 19900843; http://dx.doi.org/10.1016/j.clim.2009.10.005
- Kluger MA, Luig M, Wegscheid C, Goerke B, Paust HJ, Brix SR, Yan I, Mittrücker HW, Hagl B, Renner ED et al. Stat3 programs Th17-specific regulatory T cells to control GN. J Am Soc Nephrol 2014; 25:1291-302;PMID:24511136;http://dx.doi.org/10.1681/ASN.2013080904
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014; 41:14-20; PMID: 25035950; http://dx.doi.org/10.1016/j.immuni.2014.06.008
- Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016; 7:12150; PMID: 27381735; http://dx.doi.org/10.1038/ncomms12150
- Ren G, Zhao X, Wang Y, Zhang X, Chen X, Xu C, Yuan ZR, Roberts AI, Zhang L, Zheng B et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFalpha. Cell Stem Cell 2012; 11:812-24; PMID: 23168163; http://dx.doi.org/10.1016/j.stem.2012.08.013
- Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 2011; 11:762-74; PMID: 21984070; http://dx.doi.org/10.1038/nri3070
- Hart KM, Usherwood EJ, Berwin BL. CX3CR1 delineates temporally and functionally distinct subsets of myeloid-derived suppressor cells in a mouse model of ovarian cancer. Immunol Cell Biol 2014; 92:499-508; PMID: 24613975; http://dx.doi.org/10.1038/icb.2014.13
- Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest 2007; 117:185-94; PMID: 17200718; http://dx.doi.org/10.1172/JCI28549
- Lee BP, Chen W, Shi H, Der SD, Forster R, Zhang L. CXCR5/CXCL13 interaction is important for double-negative regulatory T cell homing to cardiac allografts. J Immunol 2006; 176:5276-83; PMID: 16621993; http://dx.doi.org/10.4049/jimmunol.176.9.5276
- Jones LM, Broz ML, Ranger JJ, Ozcelik J, Ahn R, Zuo D, Ursini-Siegel J, Hallett MT, Krummel M, Muller WJ. STAT3 establishes an immunosuppressive microenvironment during the early stages of breast carcinogenesis to promote tumor growth and metastasis. Cancer Res 2016; 76:1416-28; PMID: 26719528; http://dx.doi.org/10.1158/0008-5472.CAN-15-2770
- Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 2007; 7:41-51; PMID: 17186030; http://dx.doi.org/10.1038/nri1995
- Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mulé J, Kerr WG et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 2005; 11:1314-21; PMID: 16288283; http://dx.doi.org/10.1038/nm1325
- Hossain DM, Panda AK, Manna A, Mohanty S, Bhattacharjee P, Bhattacharyya S, Saha T, Chakraborty S, Kar RK, Das T et al. FoxP3 acts as a cotranscription factor with STAT3 in tumor-induced regulatory T cells. Immunity 2013; 39:1057-69; PMID: 24315995; http://dx.doi.org/10.1016/j.immuni.2013.11.005
- Pallandre JR, Brillard E, Crehange G, Radlovic A, Remy-Martin JP, Saas P, Rohrlich PS, Pivot X, Ling X, Tiberghien P et al. Role of STAT3 in CD4+CD25+FOXP3+ regulatory lymphocyte generation: implications in graft-versus-host disease and antitumor immunity. J Immunol 2007; 179:7593-604; PMID: 18025205; http://dx.doi.org/10.4049/jimmunol.179.11.7593
- Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013; 501:346-54; PMID: 24048067; http://dx.doi.org/10.1038/nature12626
- Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, Valle LD, Trillo-Tinoco J, Maj T, Zou W et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res 2015; 3:1236-47; PMID: 26025381; http://dx.doi.org/10.1158/2326-6066.CIR-15-0036
- De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu S, Ghosh A, Pink M, Tchaicha J et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 2016; 539:443-7; PMID: 27828943; http://dx.doi.org/10.1038/nature20554
- Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, Gao W, Saito TI, Lo Celso C, Tsuyuzaki H et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 2011; 474:216-9; PMID: 21654805; http://dx.doi.org/10.1038/nature10160
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR Jr et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 2008; 28:546-58; PMID: 18387831; http://dx.doi.org/10.1016/j.immuni.2008.02.017
- Wu Z, Irizarry RA. Stochastic models inspired by hybridization theory for short oligonucleotide arrays. J Comput Biol 2005; 12:882-93; PMID: 16108723; http://dx.doi.org/10.1089/cmb.2005.12.882
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43:e47; PMID: 25605792; http://dx.doi.org/10.1093/nar/gkv007
- Benjamini Y, Hochberg Y. Controlling the false discovery rate; A practical and powerful approach to multiple testing. J Roy Stat Soc Ser B 1995; 57:289-300; http://dx.doi.org/10.2307/2346101
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5:R80; PMID: 15461798; http://dx.doi.org/10.1186/gb-2004-5-10-r80
- Huber, W., Carey, V.J., Gentleman, R., Anders, S., Carlson, M., Carvalho, B.S., Bravo, H.C., Davis, S., Gatto, L., Girke, T., et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 2015; 12, 115–121.