
ABSTRACT
When compared with vascular cells in normal tissues, pericytes and vascular endothelial cells (VEC) in tumor blood vessels exhibit altered morphology and epigenetic programming that leads to the expression of unique antigens that allow for differential recognition by CD8+ T cells. We have previously shown that the Notch antagonist delta-like homolog 1 (DLK1) is a tumor pericyte-associated antigen expressed in setting of melanoma and a range of carcinomas. In this report, we show that therapeutic vaccination against DLK1 in murine models results in slowed tumor growth, but also to the compensatory expression of the DLK1 homolog, DLK2, by tumor-associated pericytes. Vaccines targeting both DLK1 and DLK2 resulted in superior antitumor benefits in association with improved activation and recruitment of antigen-specific Type 1 CD8+ T cells, reduced presence of myeloid-derived suppressive cells, T regulatory cell and tumor vascular normalization. The antitumor efficacy of vaccines coordinately targeting DLK1 and DLK2 was further improved by inclusion of PD-L1 blockade, thus defining a combination immunotherapy theoretically suitable for the treatment of a broad range of solid (vascularized) cancers.
Abbreviation
| DC | = | dendritic cell |
| DLK | = | delta-like homolog |
| DSL | = | Delta, Serrate, and Lag-2 |
| EGF | = | epidermal growth factor |
| FGFR1 | = | fibroblast growth factor receptor 1 |
| HIF | = | hypoxia inducible factor |
| HRE | = | hypoxia regulatory elements |
| i.d. | = | intradermal |
| i.p. | = | intraperitoneal |
| IFN | = | interferon |
| MDSC | = | myeloid-derived suppressive cells |
| MVD | = | microvessel density |
| PD-1 | = | programmed death 1 |
| PD-L1 | = | programmed death ligand 1 |
| RCC | = | renal cell carcinoma |
| RENCA | = | murine renal cell carcinoma |
| s.c. | = | subcutaneous |
| TAA | = | tumor-associated antigen |
| TAM | = | tumor-associated macrophages |
| TBVA | = | tumor-blood vessel-associated antigens |
| TIL | = | tumor infiltrating lymphocyte |
| TKI | = | tyrosine kinase inhibitor |
| TME | = | tumor microenvironment |
| Treg | = | T regulatory cell |
| VCAM1 | = | vascular cell adhesion molecule-1 |
| VEC | = | vascular endothelial cell |
| VEGF | = | vascular endothelial growth factor |
| VEGFR | = | vascular endothelial growth factor receptor |
| VN | = | vascular normalization |
Introduction
Progressive neoplasms require an expansive blood vessel network to supply nutrients and oxygen, as well as, to expel metabolic wastes and carbon dioxide, making the tumor vasculature a viable target for interventional anticancer therapy.Citation1 Indeed, therapies integrating neutralizing anti-vascular endothelial growth factor (VEGF) antibodies or tyrosine kinase inhibitors (TKIs) that interfere with proangiogenic signaling pathways have exhibited some degree of therapeutic efficacy in the pre-clinical and clinical settings.Citation2,3 However, the protective benefits resulting from anti-angiogenic therapies have typically proven to be transient in nature, with the consequent evolution of treatment-refractory disease.Citation4 Hence, there remains a great clinical need to develop novel anti-angiogenic therapies that are adaptive and support the sustained normalization of the tumor vasculature in cancer patients.
Blood vessels in solid tumors differ structurally and functionally from those found in normal tissues. In normal tissues, vascular endothelial tubes are found in close physical approximation with abluminal pericytes, where these cell types communicate via intimate cell-to-cell contact, as well as, via mutually secreted factors. This results in the coordinated proliferation and differentiation of vascular endothelial cells (VEC), and the formation of an efficient and organized mature blood vessel network. In stark contrast, in the tumor microenvironment (TME), VEC-pericyte associations are estranged, resulting in highly-permeable blood vessels and a local tissue milieu that is characterized by high interstitial fluid pressure, acidosis and hypoxia.Citation5,6 Such environmental stressors promote unique epigenetic programming among stromal cell populations that leads to abnormal genomic and proteomic profiles of tumor-associated VECs and pericytes.Citation7,8 Notably, disease-associated tumor-blood vessel-associated antigens (TBVA) may be targeted immunologically by specific CD8+ T cells that can be elicited via active, specific vaccination. Indeed, our laboratory has previously shown that TBVA-targeted vaccines are competent to promote therapeutic CD8+ T cell responses that are capable of extending overall survival in murine tumor models.Citation9
In a previous study, we demonstrated that pericytes within human renal cell carcinoma (RCC) biopsies or in vivo grown murine renal carcinomas (RENCA) express abnormally high levels of the TBVA delta-like homolog 1 (DLK1), and that therapeutic vaccination against DLK1 in RENCA-bearing mice results in tumor growth inhibition, increased frequencies of CD8+ tumor infiltrating lymphocyte (TIL) and vascular normalization (VN; Ref.Citation10). Interestingly, DLK1 (over)expression has also been correlated with the malignant transformation of adipocytes and to cancer stemness in the setting of hepatocellular carcinomas and neuroblastomas.Citation11-14 DLK1 and its homolog, delta-like homolog 2 (DLK2), belong to the Notch epidermal growth factor (EGF)-like family of receptors and ligands. DLK1 and DLK2 both contain six EGF-like repeats in the extracellular region, a single transmembrane region and a short intracellular tail.Citation15,16 Despite lacking the characteristic Delta, Serrate and Lag-2 (DSL) domain of canonical Notch ligands, DLK1 and DLK2 interact with Notch1 and serve as antagonists to DLL4- and Jagged1-mediated activation of Notch signalingCitation17-19 that are required for normal vascular maturation.Citation20
Consistent with previous reports that the Dlk1 and Dlk2 genes reciprocally regulate each other's expression,Citation16 we report that targeted therapeutic vaccination against DLK1 in RENCA-bearing mice leads to the loss of DLK1 expression in the TME, but also to a compensatory increase in the expression of DLK2 by tumor-associated vascular pericytes. We also show that combined vaccination against DLK1 and DLK2 leads to improved antitumor benefits when compared with vaccination against either single antigen in both the RENCA and B16 melanoma tumor models. The combined vaccine resulted in superior activation and recruitment of antigen-specific CD8+ TIL and to VN in the TME. In the B16 tumor model, tumor-associated antigen (TAA)-specific T cells were also activated as a consequence of combined DLK1 + DLK2-targeted vaccination, via an apparent “epitope spreading” mechanism. Furthermore, we demonstrate that the antitumor efficacy of DLK1/DLK2-targeted vaccines may be improved when further combined with programmed death ligand-1 (PD-L1) blockade, thus defining a combination immunotherapy suitable for the treatment of many solid forms of cancer.
Results
Treatment of RENCA tumor-bearing mice with DLK1 peptide-based vaccine leads to a compensatory increase in DLK2 expression in tumor-associated pericytes
We have previously reported that pericytes within murine RENCA and melanoma TME overexpress DLK1 protein when compared with their normal tissue counterparts, permitting the differential recognition of tumor-associated pericytes by DLK1-specific CD8+ T cells.Citation9,10 Indeed, treatment of RENCA-bearing Balb/c mice with DC/DLK1 peptide-based vaccines or lentivirus-based DLK1 genetic vaccines effectively promote the activation and recruitment of Type-1 anti-DLK1 CD8+ TIL in concert with removal of DLK1+ target cells in the TME and delayed tumor growth.Citation10 Since DLK1 and its homolog, DLK2, both function as Notch antagonists and may counter-regulate each other's expression,Citation17 we first investigated DLK2 expression in the tumor stroma of untreated versus DLK1-vaccinated mice. We confirmed that RENCA-bearing mice receiving DC/DLK1 peptide-based vaccines on days 10 and 17 post-tumor inoculation exhibited slowed disease progression when compared with cohorts of animals that were left untreated or that had been treated with DC control (no peptide) vaccines (). To determine whether DLK2 was differentially expressed by pericytes and VECs in tumor vs. (tumor-uninvolved) kidneys +/− treatment, tissues were enzymatically-digested, with each cell type then isolated from single-cell suspensions by fluorescence-activated cell sorting (). mRNA was extracted from the sorted cells and real-time PCR was performed to quantify the amount of DLK2 transcript in each sample. We observed that DLK2 transcript was minimally expressed by pericytes and VECs sorted from normal kidneys and untreated RENCA tumors. However, DLK2 mRNA was enriched in tumor-associated pericytes isolated from animals that had been vaccinated against DLK1 (). This observation was confirmed in immunofluorescence microscopy analysis of tumor sections, where DLK2 protein was selectively (co)expressed with NG2+, a marker that is specifically expressed by pericytes but not by RENCA cells,Citation10 within the TME of DLK1-vaccinated mice. DLK2 was not expressed within the tumors of mice in any other treatment cohort (). These data suggest that DLK2 may represent a “covert” antigen expressed by tumor-associated vascular pericytes that can be therapeutically targeted in the cancer setting, particularly under conditions in which DLK1 expression is silenced (i.e. as a consequence of targeted vaccination).
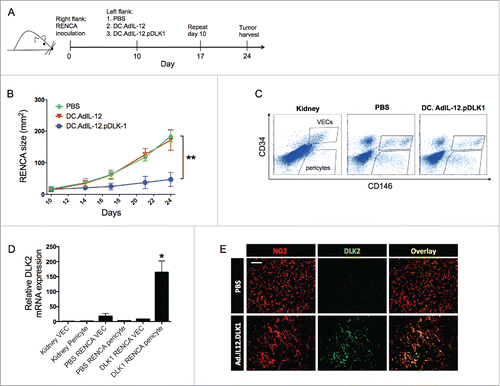
Figure 1. Vaccination of RENCA-bearing mice against DLK1 results in slowed tumor growth, the loss of DLK1+ pericytes in the TME and a compensatory increase in DLK2 expression by tumor-associated vascular pericytes. (A) Female Balb/c mice with established day 10 s.c. RENCA tumors on their right flanks were treated with s.c. injection (left flank) of PBS, 106 DCs transduced with rAd.IL12 (i.e., DC.IL12) or DC.IL12 pulsed with DLK1-derived peptide epitopes (i.e., DC.IL12.DLK1) per Materials and methods. An identical s.c. booster vaccination was provided on day 17 post-tumor inoculation. (B) Tumor growth was monitored every 3–4 d and is reported as the mean ± SEM for five animals per group. *p < 0.05, two-way ANOVA. (C) On day 21, tumor tissues and (tumor-uninvolved kidneys from matched animals) were harvested and then digested mechanically and enzymatically as described in the Materials and methods, yielding single-cell suspensions. Live cells (DAPI negative) cells were flow-sorted to select for CD45−CD146+CD34− pericytes and CD45−CD146+CD34+ VEC populations. (D) Total mRNA was isolated from sorted populations of pericytes and VECs and analyzed for DLK2 expression by quantitative real-time PCR. Relative mRNA expression was normalized to HPRT1 expression. *p < 0.05, one-way ANOVA compared with kidney pericyte DLK2 levels. In (E), RENCA tissue sections were analyzed for expression of DLK2 (green) in NG2+ pericytes (red) by immunofluorescence microscopy. Scale bar = 1mm. All data are representative of those obtained in two independent experiments.
Coordinate vaccination with lvDLK1 + lvDLK2 provides superior therapeutic benefit against established RENCA tumors
To test the hypothesis that therapeutic vaccination against both DLK1 and DLK2 would yield greater protection against tumor progression vs. single antigen-based vaccines, recombinant lentiviral vectors encoding full-length murine DLK1 (lvDLK1) or DLK2 (lvDLK2) were generated (Fig. S1). Based on a previous report that single intradermal (i.d.) injection of a lentiviral vector-based vaccine results in sustained antitumor CD8+ T cell responses in vivo,Citation21 we performed a single i.d. injection of lentiviral vectors (lvDLK1 +/− lvDLK2 or the lvNEG negative control virus) 7 d after the s.c implantation of RENCA tumor cells. Vaccines were provided on the contralateral flank to that in which the tumor was placed. Untreated animals were included as a negative control cohort. We then monitored tumor growth progression and specific T cell responses across all treatment groups. Consistent with our previous report,Citation10 we confirmed that lvDLK1-based vaccination inhibited tumor growth (). However, tumor growth was inhibited to a greater degree in animals treated with the combined lvDLK1 + lvDLK 2 vaccine. Notably, therapeutic vaccination against DLK2 alone failed to impact tumor growth. As the target DLK2 protein was not highly-expressed in RENCA tumors unless DLK1 expression was first silenced in tumor pericytes (), this result might merely reflect the lack of a salient target within the TME for recognition by DLK2 vaccine-induced CD8+ T cells in mice vaccinated using only lvDLK2. Overall, we observed that the extent of tumor growth inhibition was associated with the degree of CD8+ TIL infiltration, with the most robust CD8+Tbet+ T cell infiltrates observed in the lvDLK1 + lvDLK2 treatment cohort ().
Figure 2. Coordinate vaccination with lvDLK1 and lvDLK2 is immunogenic and therapeutic in the RENCA model. Female Balb/c mice bearing established day 7 s.c. RENCA tumors were treated with i.d. injection of PBS, 104 lvNEG, 104 lvDLK1, 104 lvDLK2 or 104 lvDLK1 + 104 lvDLK2. (A) Tumor growth was monitored every 3–4 d through day 20, at which time the animals were killed. Tumor sizes are reported as the mean ± SEM for five animals per group. *p < 0.05, two-way ANOVA. Tumors were then harvested to generate tumor sections or they were mechanically/enzymatically-digested to generate single-cell suspensions. (B) Tumor sections were analyzed by immunofluorescence microscopy for NG2+ pericytes (red) and infiltrating CD8+ T cells (green). Using Metamorph software (Materials and methods), CD8+ T cell infiltrates and NG2+ pericytes in the images were quantified and reported as mean integrated fluorescence ± SEM. *p < 0.05 and **p < 0.01, one-way ANOVA (C) Flow cytometry was also performed on the resulting cell suspensions to quantify Tbet+ CD8+ TIL. Tumor sections were analyzed for CD8+ TIL (green) infiltration and NG2+ (red) expression. In (D), splenocytes harvested from day 21 tumor-beating mice were stimulated in vitro with syngeneic DC infected with rAd.DLK1 (DC.DLK1), rAd.DLK2 (DC.DLK2) or a mixture of both DC populations (i.e., DC.DLK1 + DC.DLK2) for 5 d. Responder T cells were the isolated and restimulated with control or rAd-infected DC as indicated for 18h, and the resultant culture supernatants analyzed for IFNγ content using a cytokine-specific ELISPOT assay. *p < 0.05, one-way ANOVA. In (E) and (F), tumor sections were analyzed for NG2+ pericyte co-expression of DLK1 and DLK2, respectively, via immunofluorescence microscopy. Scale bar = 1mm. Data in A, B, C, E and F are representative of those obtained in three independent experiments. Data in D is representative of two independent experiments.
We observed that increased CD8+ T cell infiltration of tumors in lvDLK1- and lvDLK1 + lvDLK2-vaccinated animals was associated with a significant decline in NG2+ pericytes within the TME (). These results are consistent with a model in which our lentivirus-based vaccines promote the activation of specific CD8+ T cells that target DLK1+/DLK2+ (NG2+) tumor-associated pericytes in vivo. To further monitor antigen-specific CD8+ T cell responses after lentivirus-based vaccination, DCs transduced with recombinant adenovirus encoding full-length mDLK1 or mDLK2 were used to (re)stimulate splenocytes harvested from control or lv-treated animals in vitro for 5 d. CD8+ T cells were then isolated from these cultures and DLK1- and DLK2-specific responses were subsequently analyzed in interferon (IFNγ) ELISPOT assays. We found that when applied individually, the lvDLK1 and lvDLK2 vaccines promoted DLK1- or DLK2-specific responses, respectively. Predictably, the combination lvDLK1 + lvDLK2 vaccine induced both DLK1- and DLK2-specific CD8+ T cell responses in treated animals (). We were also able to confirm the induction of DLK1-specific CD8+ T cell responses in the lvDLK1 and lvDLK1 + lvDLK2 treatment cohorts using DC pulsed with defined synthetic DLK1 peptide epitopesCitation10 as a stimulus in IFNγ ELISPOT assays (Fig. S2).
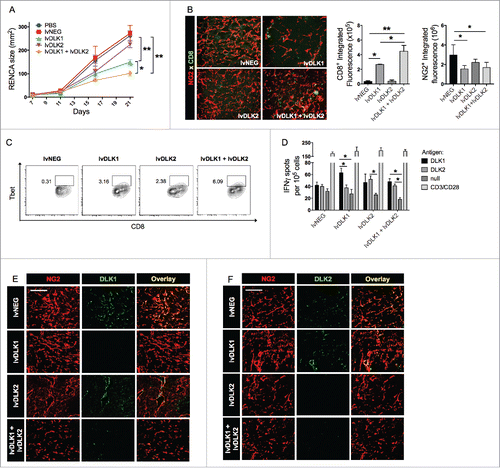
Following the observation that the lvDLK1 + lvDLK2 vaccination induced the activation of DLK1- and DLK2-specific T cells, we next investigated how the various vaccine formulations affected DLK1 and DLK2 protein expression by cells within the tumor stroma. We determined that DLK1 expression by NG2+ pericytes was diminished after vaccination with lvDLK1 (either alone or in combination with lvDLK2; ). Vaccination against lvDLK2 alone did not affect DLK1 expression in the TME, however, DLK2 became expressed by NG2+ pericytes after lvDLK1 vaccination (). Remarkably, co-vaccination using lvDLK1 and lvDLK2 resulted in NG2+ pericytes that failed to express either DLK1 or DLK2 in the therapeutic TME ().
Since TBVA are overexpressed in the TME of solid cancers independent of tumor histology,Citation9 TBVA-based vaccines should offer therapeutic benefit against a wide range of vascularized tumors, leading us to next evaluate the antitumor efficacy of DLK1- and DLK2-based vaccines against (RENCA-unrelated) B16 melanomas established s.c. in C57BL/6 mice (Fig S3a). Syngenic DCs were infected with recombinant adenovirus encoding full-length DLK1 and/or DLK2 for 48 h in vitro, then injected s.c. in the contralateral flank to that containing a progressively growing B16 melanoma. An identical booster vaccine was administered one week later. We observed that the combined DLK1 + DLK2-based vaccine yielded optimal antitumor efficacy in association with the induction of the most robust antigen-specific CD8+ T cell responses (Fig S3b). Furthermore, we noted that successful vaccination against DLK1/DLK2 in these animals was also associated with the development of CD8+ T cell responses against the (vaccine-unrelated) melanoma antigen gp100 (Fig. S3c), presumably via an “epitope spreading” mechanism.
DLK1/DLK2-based vaccines promote VN within the TME
Our ability to drive Type-1 CD8+ T cell responses against tumor-associated vascular pericytes using DLK1/DLK2-based vaccines, allows for the possibility of therapeutically proctoring a state of VN.Citation22 To investigate this further, tumor sections from treated vs. control mice were stained for the VEC marker CD31 and the pericyte marker NG2 (). Adopting the method of Weidner et al.,Citation23 we quantified microvessel densities (MVD) and we observed that tumors in mice treated with lvDLK1- or lvDLK1 + lvDLK2-based vaccine exhibited significant vascular pruning when compared with tumors in control or lvDLK2-only vaccinated mice (). In addition, when pericyte-VEC co-localization was evaluated in tumor-associated blood vessels, we observed that CD31- and NG2-associated fluorescent signals were also more closely associated with each other in the tumors of mice vaccinated with lvDLK1 or with combined lvDLK1 + lvDLK2 vs. tumors in all other treatment cohorts (). Collectively, the decreased vascular arborization and increased NG2+ pericyte coverage of CD31+ vessels observed in the tumors of lvDLK1- or lvDLK1 + lvDLK2-treated mice is consistent with therapeutic VN as described previously by Jain.Citation24 Since tumor VN is also characterized by a loss of hypoxia (and a return to normoxia) within the TME,Citation24,25 we analyzed treated vs. control tumors for expression of the hypoxia-associated biomarker HIF-2α.Citation25-27 We determined that tumors in mice vaccinated with lvDLK1 or (more so) lvDLK1 + lvDLK2 displayed significant reductions in HIF-2α transcript levels vs. control-untreated tumors (), consistent with therapeutic VN in these treatment cohorts.
Figure 3. Coordinate vaccination with lvDLK1 and lvDLK2 results in tumor VN. (A) RENCA tumor sections from were stained for NG2+ pericytes (red) and CD31+ VECs (green) to characterize blood vessel morphology. (B) Mean vessel density was determined by quantitating distinct (green) VECs or VEC clusters. (C) Colocalization of NG2+ (red) pericytes and CD31+ (green) VECs was determined using Metamorph software. (D–E) RNA extracted from Day 21 RENCA tumors was analyzed for HIF2α, VEGFR1, VEGFR2 and FGFR1 transcript levels by quantitative real-time PCR. In (E), tumor CD31+ VEC (green) were analyzed for co-expression of VCAM1 (red) by immunofluorescence microscopy. Scale bar = 1mm. *p < 0.05 and **p < 0.01, one-way ANOVA compared with the negative control (lvNEG). All data are representative of those obtained in three independent experiments.
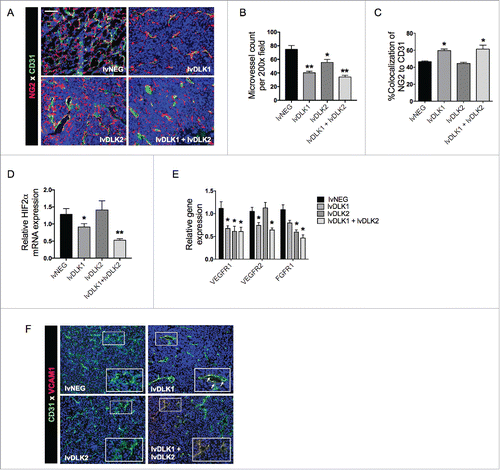
We also observed that pro-angiogenic biomarkers were repressed as a consequence of lvDLK1/2-based vaccination. In particular, expression of VEGFR1 and VEGFR2 mRNA were significantly reduced in the TME of lvDLK1-treated animals, while transcript levels of VEGFR1, VEGFR2 and fibroblast growth factor receptor 1 (FGFR1) were coordinately decreased in the TME of mice treated with the combined lvDLK1 + lvDLK2 vaccine ().
Lastly, combined vaccination against DLK1 and DLK2 led to an increase in expression of vascular cell adhesion molecule-1 (VCAM1) by tumor-associated VECs (), consistent with improved tumor infiltration by Type-1 (Tbet+) CD8+ T cells ().
lvDLK1 + lvDLK2-based vaccination decreases frequencies of immunosuppressive cell populations in the TME
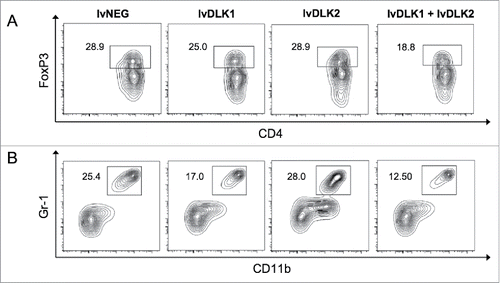
Tumor-associated hypoxia plays a critical role in the maintenance and regulatory activity of myeloid-derived suppressive cells (MDSC), as well as, regulatory T (Treg) cells in the TME.Citation28 As VN mitigates hypoxia in the TME (: Ref.Citation10,24), one might predict that vaccines promoting VN would recondition the immunologic landscape within tumors to become less suppressive. We examined this hypothesis in the TME of mice receiving vaccine-based therapy and found that levels of both MDSC () and Tregs () were reduced to a greater extent in the tumors of mice treated with combined lvDLK1 + lvDLK2 vaccination vs. all other treatment cohorts.
Figure 4. Coordinate vaccination with lvDLK1 and lvDLK2 results in a superior level of reduction in Treg and MDSC content in the therapeutic TME. Day 21 tumors from untreated or treated mice per were dissociated into single-cell suspensions and analyzed by flow cytometry for (A) CD11b+Gr1+ MDSC and (B) FoxP3+CD4+ Treg populations. Data are representative of those obtained in three independent experiments.
lvDLK1 + lvDLK2-based vaccination combined with PD-L1 blockade further enhances therapeutic efficacy
Cancer vaccines have previously been shown to be both safe and immunogenic, but rarely curative, based on a variety of immune escape mechanisms used by residual tumor cells, including elevated expression levels of immune checkpoint/co-inhibitory moleculesCitation29,30 Transcript analysis of RENCA tumors vs. normal kidney tissue (from tumor-matched animals) revealed that RENCA tissues overexpress several co-inhibitory receptors, including B7H3, Gal9, PD-L1 and PD-L2 (Fig. S4a) that would be expected to negatively impact antitumor CD8+ T cell activity in vivo.Citation29,30 Consistent with the known capacity of IFNγ to upregulate PD-L1 expression in some tumors,Citation31 we observed that culturing RENCA cells in media supplemented with rmIFNγ resulted in increased PD-L1 expression (Fig. S4b).
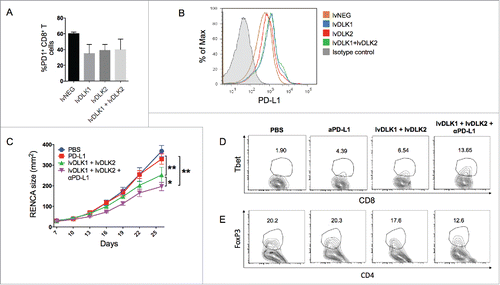
Since, we noted a dramatic influx of inflammatory CD8+ TIL in RENCA tumors sponsored by combined lvDLK1 + lvDLK2 vaccination (), we next evaluated PD1 and PD-L1 protein expression levels on TIL and tumors, respectively, in treated vs. control tumor-bearing mice. We found that PD1 protein expression on CD8+ TIL was comparable across all cohorts (). On the other hand, PD-L1 protein expression on tumor cells was upregulated in lvDLK1- and lvDLK1 + lvDLK2-vaccinated mice when compared with mice in the other treatment cohorts (). Based on these findings, we next investigated the therapeutic benefit of combining lvDLK1 + lvDLK2-based vaccination with systemic delivery of an antagonist anti-PD-L1 antibody. We observed that the degree of RENCA growth inhibition was greatest in animals that received the combined lvDLK1/lvDLK2 + anti-PD-L1 antibody treatment regimen vs. cohorts of tumor-bearing mice receiving either the lvDLK1/lvDLK2 vaccine alone or anti-PD-L1 monotherapy (). This superior level of antitumor efficacy was association with the greatest magnitude of increased recruitment/maintenance of Tbet+ CD8+ T cells within the TME () and the greatest degree of reduction in CD4+FoxP3+ TIL ().
Figure 5. PD-L1 blockade fails as a monotherapy against RENCA tumors, but improves the antitumor efficacy of combined lvDLK1 + lvDLK2 vaccination. (A) CD8+ TILs and (B) bulk RENCA single-cell suspensions (per ) were analyzed for expression of PD-1 and PD-L1, respectively, by flow cytometry. In (C), mice bearing established day 7 RENCA tumors (right flank) were vaccinated i.d. with lvDLK1 + lvDLK2 (left flank) as a therapeutic vaccine. Anti-mPD-L1 blocking antibody was injected i.p. on days 7, 10, 13 and 17 post-tumor inoculations as described in Materials and methods. Tumor growth was monitored longitudinally and is reported as the mean ± SEM for five animals per group. In (D), RENCA tumors were harvested on day 26 and dissociated single-cell suspensions analyzed for (D) Tbet+CD8+ TILs and (E) FoxP3+CD4+ Tregs by flow cytometry. *p < 0.05 and **p < 0.01.
Discussion
An ideal cancer vaccine antigen is one that is overexpressed by cells within the TME vs. normal tissues, allowing for selective T cell targeting of disease sites. We have previously reported that the non-canonical Notch ligand, DLK1, is overexpressed by vascular pericytes in murine colon and renal carcinomas, and melanomas, when compared with tumor-uninvolved kidneys in these same animals, and that vaccination against DLK1 is capable of yielding therapeutic antitumor benefits.Citation10 In the current report, we demonstrate that the expression of DLK2, a homolog of DLK1, is upregulated in tumor-associated vascular pericytes following successful DLK1-targeted vaccination, consistent with the reciprocal expression profiles of these two related Notch antagonists. Given this phenotypic adaptation by tumor pericytes, we hypothesized that a combination vaccine targeting both DLK1 and DLK2 would have greater potential to promote an extended state of VN in the TME in association with improved treatment benefits. Combined vaccination against DLK1 and DLK2 resulted in superior activation of peripheral Type-1 antigen-specific CD8+ T cell responses and more robust recruitment of T effector cells into the TME. Therapeutic TIL modified the tumor landscape by promoting the loss of DLK1+ and DLK2+ pericytes, effectively pruning immature tumor blood vessels. The normalization/maturation of the vascular network, which allows a switch from the hypoxic to a normoxic TME,Citation32 also resulted in tumors that were coordinately deficient in CD11b+Gr1+ MDSCs and FoxP3+ Tregs that are typically associated with immunosuppression.
Studies from other groups have previously shown that DLK1 and DLK2 reciprocally modulate each other's expression.Citation16,17 For instance, during murine neonatal liver development, when DLK1 expression is elevated during the first few days of life, DLK2 is absent, but once DLK1 expression starts to decline, DLK2 expression is turned on. Furthermore, forced ectopic expression of DLK1 or DLK2 in cells results in the loss of expression of the alternate homolog. The mechanism responsible for the coordinated control of DLK1 and DLK2 gene expression remains poorly-defined. When considered within the context of our DLK1-based vaccine strategy, we hypothesize that the change in the oxygenation level in the therapeutically normalized TME may play a role in flipping a transcriptional switch favoring DLK2 expression. The DLK1 promoter contains hypoxia regulatory elements (HRE), placing gene expression under the control of the HIF-1α transcription factor.Citation13 On the other hand, DLK2 transcription does not seem to be directly driven by hypoxia but is rather controlled by the Sp1 and KLF4 transcription factors,Citation33,34 whose activities may be augmented under normoxic conditions. Citation35,36
The role of DLK1 and DLK2 in blood vessel formation and maintenance in normal and disease settings remains underinvestigated. Our previous and current studies, however, suggest that DLK1 and DLK2, when expressed by tumor pericytes, contribute to pathological characteristics of the tumor blood vessel network by inhibiting Notch1 signaling in the perivascular region. Notch1 signaling in normal endothelial cells is critical in embryonic vascular development, endothelial cell fate determination and vascular homeostasis.Citation37 Notch signaling also plays a role in tumor neovascularization, as suppression of the DLL4-Notch1 interaction leads to the formation of excessive immature blood vessel branches as a result of uncontrolled endothelial proliferation.Citation38-40 Both DLK1 and DLK2 block downstream Notch1 receptor signaling by interacting with the EGF-like repeat 12 of the receptor,Citation17 known to be the binding site for canonical ligands.Citation41,42 Thus, DLK1 and DLK2 impede Notch signaling by blocking receptor activation and by competing with ligands for binding sites. Vaccine-induced removal of DLK1+ and DLK2+ pericytes from the TME leads to VN that is correlated with NICD activation and Hes1 expression in endothelial cells (Fig. S5). Consistent with a model in which activation of the Wnt/β-catenin/NOTCH signaling pathway in tumor endothelia leads to VN,Citation43 we observed that reduced mean vascular density and improved pericyte-VEC interaction in the TME are biologic correlates associated with therapeutic vaccination against DLK1/DLK2 in tumor-bearing mice.
In order for cancer vaccines to work effectively, effector T cells must extravasate through the tumor blood vessel wall and into the interstitial space to interact with antigen-presenting stromal cell or tumor cell targets. However, conditions within the TME may suppress VEC expression of adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and VCAM1, that may limit tumor infiltration by circulating (antitumor) T cells.Citation44 This situation may be remedied by combined administration of anti-angiogenic agents (such as the TKIs axitinib, dasatinib or sunitinib) along with a tumor-specific vaccine.Citation45-47 Our DLK1/DLK2-targeted vaccine has an advantage over conventional TAA-targeted vaccine due to the proximal accessibility of circulating T effector cells to tumor-associated vascular pericyte targets in contrast to tumor cells that require T cells to enter, transmigrate and functionally persist within the TME to mediate meaningful therapeutic action. Furthermore, our vaccine promotes sustainable immune-mediated VN that would be envisioned to improve the deliverability of co-delivered chemotherapies or adoptively-transferred (TCR- or CAR-engineered) T cells, theoretically without invoking the development of drug resistance or the adverse side-effects associated with anti-VEGF monoclonal antibodies and TKIs. However, it is possible that low-doses of anti-angiogenic drugs, such as sunitinib, might further reduce immunoregulatory cell populations in support of an even more robust pro-inflammatory TME when combined with DLK1/DLK2-targeted vaccines.Citation48
When taken in the context to existing paradigms for temporal cascades of T cell infiltration into the TME,Citation49,50 we would propose that the vaccine-induced DLK1- and DLK2-specific T cells might represent an initial wave of effector TILs that trim and promote the maturation of tumor-associated blood vessels, leading to VN. The normalized TME is conducive to DC maturation and the subsequent cross-priming of “second set” CD8+ T cell responses reactive against TAA acquired by DC within the TME.Citation10,51 This effectively drives a broadening in the antitumor T cell repertoire based on an “epitope spreading” paradigm,Citation51,52 such as we observed in the case of DLK1/DLK2-based therapeutic vaccines leading to the development of anti-gp100 CD8+ T cell responses in the B16 melanoma model (Fig. S2).
One of the anticipated problems with immunotherapy is the development of immune evasion mechanisms in the tumor that would lead to treatment resistance. Vaccines induce antitumor protection by expanding the size and specificity of the tumor-specific T response, with superior vaccines also promoting TIL recruitment. However, chronic inflammation in the TME (mediated by T cells or NK cells) may lead to upregulated expression of immune checkpoint molecules such PD-L1.Citation31,53,54 in the tumor, which can subvert or delete protective PD-1+ T effector cells. Consistent with this paradigm, we observed that effective vaccination against DLK1/DLK2 led to increased numbers of inflammatory (IFNγ+) TIL and to upregulated PD-L1 expression in the TME. While we did not discern any significant benefit to tumor-bearing mice upon treatment with anti-PD-L1 monotherapy, we observed that the addition of PD-L1 blockade to our DLK1/DLK2-based vaccine regimen resulted in improved Type-1 CD8+ T cell-mediated control of tumor growth vs. either monotherapy. Our data support a model in which PD1/PD-L1 blockade and anti-vascular vaccines complement each other in their ability to promote, recruit and sustain therapeutic antitumor immunity within the TME. However even this “optimized” treatment strategy did not achieve tumor regression, suggesting room for further translational improvement by combination with additional immune checkpoint antagonists (such as anti-B7H3, anti-Gal9 or -Tim3 and anti-PD-L2 or -PD1) in extended treatment protocols, before the translation of such approaches into the clinic.
Materials and methods
Animals and cell lines
Female 6–8 week old Balb/c and C57BL/6 mice were purchased from Jackson Laboratory and were maintained in a pathogen-free animal facility. All animals were handled under aseptic conditions per an Institutional Animal Care and Use Committee (IACUC)-approved protocol and in accordance with recommendations for the proper care and use of laboratory animals. The RENCA (CRL-2947) and B16-F10 (CRL-6475) cell lines were purchased from American Type Culture Collection. These cell lines were free of Mycoplasma contamination and were maintained in complete medium [CM: RPMI-1640 media (Gibco, Cat. No. 21870–076) supplemented with 10% heat-inactivated fetal bovine serum (Sigma, Cat. No. F442), Pen/Strep [100 μg/mL streptomycin and 100 U/mL penicillin (Gibco, Cat. No. 15140–22)] and 10 mmol/L L-glutamine (Gibco, Cat. No. 25030–081)] at 5% CO2 tension in a 37°C humidified incubator.
Generation of bone marrow-derived DCs and DC-based vaccines
Bone marrow precursors isolated from the tibias and femurs of mice were cultured for 5 d in CM supplemented with 1,000 U/mL rIL-4 and 1,000 U/mL rGM-CSF (both from Peprotech) to generate DCs. The resulting DCs were then purified using CD11c microbeads (Miltenyi, Cat. No. 130–108–338) according to the manufacturer's instructions. To generate DC/adenovirus-based vaccines, purified DCs were infected with rAd.mIL-12p70 at a multiplicity of infection (MOI) of 50, alone or in combination with or rAd.mDLK1 or rAd.mDLK2 (MOI = 100 each) and cultured for 2 d in rIL-4 and rGM-CSF supplemented CM, before harvest and use as vaccines. All recombinant adenoviral vectors were produced and provided by the University of Pittsburgh Cancer Institute's Vector Core Facility (a shared resource). To generate DC/peptide-based vaccines, DCs transduced with Ad.mIL-12p70 (DC.IL12; as described above) were pulsed with a pool of the DLK1158–166, DLK1161–169 and DLK1259–270 peptides (10 μM each; ref.Citation10) for 2 h. DLK1 peptides were synthesized by the University of Pittsburgh Cancer Institute's Peptide Synthesis Facility (a shared resource) and analyzed for purity by the University of Pittsburgh Cancer Institute's Protein Sequencing Facility (a shared resource).
Generation of recombinant lentiviral vector
Lentiviral vectors (lv) encoding mDLK1 (lvDLK1), mDLK2 (lvDLK2) and a reverse RGS5 sequence (used as a negative control vector designated as lvNEG) were generated as described previouslyCitation10 Briefly, full-length murine DLK1, DLK2 and reverse RGS5 (control) were cloned into pLenti6/V5 D-TOPO using the Lentiviral Directional TOPO Expression Kit (Invitrogen, Cat. No. K495000). To produce the lentiviral vectors, 293FT cells (Invitrogen, Cat. No. R700–07) were transfected with the pLenti plasmids with DLK1, DLK2 or control inserts using ViraPower Packaging Mix (Invitrogen, Cat. No.44–2050) combined with Lipofectamine 2,000 (Invitrogen, Cat. No. 11668019) according to the manufacturer's instructions. The lentiviral vectors were collected using a Fast-Trap Virus Purification and Concentration kit (Millipore, Cat. No. FTLV00003) 48 h after the transduction. Lentiviral titers were determined via blasticidin-resistance assay (Invitrogen, Cat. No. R210–01) in HT1080 cells (kindly provided by Dr. Chuanyue Wu, University of Pittsburgh, Pittsburgh, PA) according to the manufacturer's instructions.
Animal experiments
Balb/c mice received subcutaneous (s.c.) injection of 106 RENCA cells on the right flank. Six days after tumor inoculation, the animals were randomized into cohorts of five mice, with each cohort exhibiting comparable mean tumor sizes. For the DC/peptide-based vaccine experiments, RENCA-bearing mice were injected s.c. on the left flank (i.e., contralateral to tumor) with 100 μL PBS, 106 DC.IL12 or 106 DC.IL12 that had been pre-loaded with an equimolar (10 μM) mixture of DLK1 peptides on Days 7 and 14 post-tumor inoculation. For the lentiviral vaccine experiments, RENCA-bearing mice (on day 10 post-tumor inoculation) received intradermal injection of 50 μL PBS, 104 transduction units (TU) of lvNEG, 104 TU lvDLK1, 104 TU lvDLK2 or 104 TU lvDLK1 + 104 TU lvDLK2. The dose of 104 TU was selected for analysis based on previous results using lvDLK1-based therapeutic vaccines in the RENCA model (10). For the checkpoint inhibitor experiments, 200 μg PD-L1 blocking antibody (BioXCell, Cat. No. BE0101) was administered via i.p. injection into lentivector-vaccinated RENCA-bearing mice on days 7, 10, 13 and 17 after tumor injection. C57BL/6 mice received s.c. injections of 105 B16-F10 cells on the right flank. Six days post-tumor inoculation, the animals were randomized into cohorts of five. On days 7 and 14 post-tumor inoculation, tumor-bearing mice received s.c. injection of 100 μL PBS, 106 DC.IL12, 106 DC.IL12.DLK1, 106 DC.IL12.DLK2 or 106 DC.IL12.DLK1 + 106 DC.IL12.DLK2 on their left flank. Tumor size was assessed every 3–4 d using a vernier caliper. Data are reported as mean tumor area ± SEM.
Stromal cell isolation
Kidneys and tumors were isolated from control or vaccinated mice 21 d after initial tumor inoculation. After the tissues were mechanically minced and enzymatically digested with 0.5 mg/mL collagenase IA (Sigma, Cat. No. C5894), 0.5 mg/mL collagenase II (Sigma, Cat. No. C1764), 0.5 mg/mL collagenase IV (Sigma, Cat. No. C1889) and 20 U/mL DNase I (Sigma, Cat. No. D5025), the resulting single cell suspensions were labeled with anti-mouse CD45-APC (BD Biosciences, Cat. No. 559864), anti-mouse CD34-FITC (eBioscience, Cat. No.11–0341–82) and anti-mouse CD146-PE (BD Biosciences, Cat. No. 562196). The cells were flow sorted into pericytes (CD45negCD34negCD146+) and VECs (CD45negCD34+CD146+) using a FACSAria II Cell Sorter (BD Biosciences). Sorted cells were > 95% pure for the specified phenotype.
Real time PCR
mRNA from whole murine RCC tumors and murine kidney- and murine RCC-associated pericytes and VECs was isolated using the RNeasy Plus Micro kit (Qiagen, Cat. No. 74034) according to the manufacturer's instructions. cDNA was then generated from the RNA samples using the High Capacity RNA-to-cDNA kit (Applied Biosystems, Cat. No. 2017–08–31) and real-time PCR was performed using the Fast SYBR Green Master Mix (Applied Biosystems, Cat. No. 2017–07–31). PCR reactions were performed in duplicate on a StepOnePlus real-time PCR thermocycler (Applied Biosystems) using the primers listed in and cycling conditions of 95°C for 20 min, then 40 cycles of 95°C for 3 sec and 60°C for 30 seconds followed by post-amplification melting curve analysis at 95°C for 15 sec and 60°C for 1 min. Assays were normalized to HPRT1 gene (Qiagen, Cat. No. 249900) and the results were analyzed by the 2−ΔΔCt method.
Table 1. Murine qPCR primers.
Fluorescent imaging of tumors
Tumor tissue samples were fixed in 2% paraformaldehyde for 2 h and dehydrated in 30% sucrose for 24 h at 4°C. Six-micron tissue sections were analyzed using specific pAb against mNG2 (gift from Dr. William Stallcup, Sanford Burnham Prebys Medical Discovery Institute), mDLK1 (Santa Cruz Biotechnology, Cat. No. sc-25437), mDLK2 (Aviva Systems Biology, Cat. No. ARP49784_P050), mCD31 (BD Biosciences, Cat. No.550274), mVCAM1 (R&D, Cat. No. AF643), mNICD1 (Abcam, Cat. No. ab8925), mHes1 (Miltenyi, Cat. No. AB5702) and mCD8 (BD Biosciences, Cat. No. 558733) paired with secondary antibodies donkey anti-guinea pig Cy3 (Jackson ImmunoResearch, Cat. No. 706–165–148), donkey anti-goat Cy3 (Jackson ImmunoResearch, Cat. No. 705–165–147), donkey anti-rabbit Alexa Fluor 488 (Molecular Probes, Cat. No. A-21206) or donkey anti-rat Alexa Fluor 488 (Molecular Probes, Cat. No. A-21208) via immunofluorescence microscopy using a cold CCD camera (Olympus Magnafire; Olympus; Olympus, Central Valley, PA) to collect wide-field images. The Metamorph software (Molecular Devices, Downingtown, PA) was used to determine fluorescence signals over background and to measure individual structures as the integration of pixels (total number of positive pixels in structure above background) multiplied by the brightness of each pixel in gray scale. The same software was used to measure co-localization of specific labeled markers.
In vitro evaluation of Ag-specific CD8+ T cell responses
To evaluate specific CD8+ T cells responses in vaccinated tumor-bearing mice, spleens were harvested from animals 14 d after lentiviral injection or 7 d after the second DC/adenovirus vaccination. Splenocytes were re-stimulated in vitro with syngeneic DCs transduced with mDLK1 and mDLK2 recombinant adenovirus for 5 d (1:10 DC:splenocyte ratio). Afterwards, CD8+ T cells were isolated from the splenocytes using a CD8+ T cell enrichment kit (StemCell Technologies, Inc., Cat. No. 19753A) and were then assessed for antigen-specific secretion of IFNγ (Mabtech, Cat. No. 3321–3–1000 and 3321–6–250) or granzyme B (R&D, Cat. No. SEL1865) via ELISPOT, or by intracellular staining (for IFNγ BD Biosciences, Cat. No. 554714) and flow cytometry according to manufacturer's protocol. To test for anti-DLK1 and anti-DLK2-specific responses, syngeneic DCs transduced with rAd encoding mDLK1 or mDLK2, respectively, were used as target cells. To test for Ag-specific responses in melanoma models, C57BL/6 DCs were pulsed with 1 μM human gp10025–33 (Anaspec, Cat. No. AS-62589) or 1 μM mouse TRP2180–188 (AnaSpec, Cat. No. AS-61058), abd used as stimulator cells. As positive controls, purified T cells or splenocytes were stimulated with 2 μg/mL conA (Sigma, Cat. No. C0412) or 0.5 μg anti-mCD3e antibody (eBioscience, Cat. No. 16–0031–82) along with 0.5 μg anti-mCD28 antibody (eBioscience, Cat. No. 16–0281–82).
Flow cytometry
Spleens and tumors were harvested from mice 14 d after lentiviral injection or 7 d after the second DC/adenovirus vaccination, with single-cell suspensions then prepared as described previously.Citation9,10 Fluorescently-labeled antibodies against the following target molecules were used for cell surface staining: CD3e-BUV395 (BD Biosciences, Cat. No. 563565), CD4-PEDazzle (Biolegend, Cat. No. 100566), CD8a-FITC (BD Biosciences, Cat. No. 553031), CD8-PECy7 (BD Biosciences, Cat. No.552877), CD11b-APC (eBioscience, Cat. No.17–0112–82), Gr-1-PE (BD Biosciences, Cat. No. 553128), PD-1-FITC (eBioscience, Cat. No. 11–9985–81) and PD-L1-PE (Biolegend, Cat. No. 124308). Cell staining was performed in FACS buffer (PBS with 5% FBS and 0.1% NaN3). To stain for intracellular cytokine, the BD Cytofix/Cytoperm kit (BD Biosciences, Cat. No. 554714) was used with anti-IFNγ-PE (BD Biosciences, Cat. No. 554412). To stain for intracellular transcription factors, FoxP3/Transcription factor staining buffer set (eBioscience, 2017–11) was used with the anti-FoxP3-APC (eBioscience, Cat. No.17–5773–82) and anti-Tbet-APC (Biolegend, Cat. No. 644814) antibodies.
Statistical analyses
Comparisons between groups were performed using a two-tailed Student's t-test or analysis of variance (ANOVA; one-way or two-way) with Tukey's post-hoc analysis. All data were analyzed using GraphPad Prism software, version 6.07 (GraphPad Software, Inc., La Jolla, CA). Differences between groups with a p value <0.05 were considered significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1290035_supplementary_data.zip
Download Zip (3.9 MB)Funding
This project used the UPCI Peptide Synthesis and Vector Core Facilities supported in part by NIH P30 CA047904. The performance of this work was also supported by NIH R01 CA169118 (W.J.S.).
References
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971;285(21):1182-6; PMID:4938153; http://dx.doi.org/10.1056/NEJM197111182852108
- Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 2011;91(3):1071-121; PMID: 21742796; http://dx.doi.org/10.1152/physrev.00038.2010
- Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov 2007;6(4):273-86; PMID: 17396134; http://dx.doi.org/10.1038/nrd2115
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008;8(8):592-603; PMID:18650835; http://dx.doi.org/10.1038/nrc2442
- Bergers G and S Song The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol 2005;7(4):452-64; PMID:16212810; http://dx.doi.org/10.1215/S1152851705000232
- Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol 2002;160(3):985-1000; PMID:11891196; http://dx.doi.org/10.1016/S0002-9440(10)64920-6
- Ghilardi C, Chiorino G, Dossi R, Nagy Z, Giavazzi R, Bani M. Identification of novel vascular markers through gene expression profiling of tumor-derived endothelium. BMC Genomics 2008;9:201; PMID:18447899; http://dx.doi.org/10.1186/1471-2164-9-201
- Hofmeister V, Schrama D, Becker JC. Anti-cancer therapies targeting the tumor stroma. Cancer Immunol Immunother 2008;57(1):1-17; PMID:17661033; http://dx.doi.org/10.1007/s00262-007-0365-5
- Zhao X, Bose A, Komita H, Taylor JL, Chi N, Lowe DB, Okada H, Cao Y, Mukhopadhyay D, Cohen PA et al. Vaccines targeting tumor blood vessel antigens promote CD8(+) T cell-dependent tumor eradication or dormancy in HLA-A2 transgenic mice. J Immunol 2012;188(4):1782-8; PMID:22246626; http://dx.doi.org/10.4049/jimmunol.1101644
- Chi Sabins N, Taylor JL, Fabian KP, Appleman LJ, Maranchie JK, Stolz DB, Storkus WJ. DLK1: a novel target for immunotherapeutic remodeling of the tumor blood vasculature. Mol Ther 2013;21(10):1958-68; PMID:23896726; http://dx.doi.org/10.1038/mt.2013.133
- Espina AG, Méndez-Vidal C, Moreno-Mateos MA, Sáez C, Romero-Franco A, Japón MA, Pintor-Toro JA. Induction of Dlk1 by PTTG1 inhibits adipocyte differentiation and correlates with malignant transformation. Mol Biol Cell 2009;20(14):3353-62; PMID: 19477929; http://dx.doi.org/10.1091/mbc.E08-09-0965
- Begum A, Kim Y, Lin Q, Yun Z. DLK1, delta-like 1 homolog (Drosophila), regulates tumor cell differentiation in vivo. Cancer letters 2012;318(1):26-33; PMID: 22142700; http://dx.doi.org/10.1016/j.canlet.2011.11.032
- Kim Y, Lin Q, Zelterman D, Yun Z. Hypoxia-regulated delta-like 1 homologue enhances cancer cell stemness and tumorigenicity. Cancer research 2009;69(24):9271-80; PMID:19934310; http://dx.doi.org/10.1158/0008-5472.CAN-09-1605
- Xu X, Liu RF, Zhang X, Huang LY, Chen F, Fei QL, Han ZG. DLK1 as a potential target against cancer stem/progenitor cells of hepatocellular carcinoma. Mol Cancer Ther 2012;11(3):629-38; PMID:22238367; http://dx.doi.org/10.1158/1535-7163.MCT-11-0531
- Smas CM, Sul HS. Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell 1993;73(4):725-734; PMID:8500166; http://dx.doi.org/10.1016/0092-8674(93)90252-L
- Nueda ML, Baladrón V, García-Ramírez JJ, Sánchez-Solana B, Ruvira MD, Rivero S, Ballesteros MA, Monsalve EM, Díaz-Guerra MJ, Ruiz-Hidalgo MJ et al. The novel gene EGFL9/Dlk2, highly homologous to Dlk1, functions as a modulator of adipogenesis. J Mol Biol 2007;367(5):1270-80; PMID:17320102; http://dx.doi.org/10.1016/j.jmb.2006.10.020
- Sanchez-Solana B, Nueda ML, Ruvira MD, Ruiz-Hidalgo MJ, Monsalve EM, Rivero S, García-Ramírez JJ, Díaz-Guerra MJ, Baladrón V, Laborda J. The EGF-like proteins DLK1 and DLK2 function as inhibitory non-canonical ligands of NOTCH1 receptor that modulate each other's activities. Biochim Biophys Acta 2011;1813(6);1153-64; PMID:21419176; http://dx.doi.org/10.1016/j.bbamcr.2011.03.004
- Baladron V, Ruiz-Hidalgo MJ, Nueda ML, Díaz-Guerra MJ, García-Ramírez JJ, Bonvini E, Gubina E, Laborda J. dlk acts as a negative regulator of Notch1 activation through interactions with specific EGF-like repeats. Exp Cell Res 2005;303(2):343-59; PMID:15652348; http://dx.doi.org/10.1016/j.yexcr.2004.10.001
- Traustadóttir GÁ, Jensen CH, Thomassen M, Beck HC, Mortensen SB, Laborda J, Baladrón V, Sheikh SP, Andersen DC. Evidence of non-canonical NOTCH signaling: Delta-like 1 homolog (DLK1) directly interacts with the NOTCH1 receptor in mammals. Cellular Signalling 2016;28(4):246-254; PMID:26791579; http://dx.doi.org/10.1016/j.cellsig.2016.01.003
- Sainson RCA, Harris AL. Regulation of angiogenesis by homotypic and heterotypic notch signalling in endothelial cells and pericytes: from basic research to potential therapies. Angiogenesis 2008;11(1):41-51; http://dx.doi.org/10.1007/s10456-008-9098-0
- He Y, Zhang J, Donahue C, Falo LD Jr. Skin-derived dendritic cells induce potent CD8(+) T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity 2006;24(5):643-56; PMID:16713981; http://dx.doi.org/10.1016/j.immuni.2006.03.014
- Wentink MQ, Huijbers EJ, De Gruijl TD, Verheul HM, Olsson AK, Griffioen AW. Vaccination approach to anti-angiogenic treatment of cancer. Biochim Biophys Acta 2015;1855(2):155-171; PMID:25641676; http://dx.doi.org/10.1016/j.bbcan.2015.01.005
- Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis — correlation in invasive breast carcinoma. N Engl J Med 1991;324(1):1-8; PMID:1701519; http://dx.doi.org/10.1056/NEJM199101033240101
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005;307(5706):58-62; PMID:15637262; http://dx.doi.org/10.1126/science.1104819
- Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol 2005;25(13):5675-86; PMID:15964822; http://dx.doi.org/10.1128/MCB.25.13.5675-5686.2005
- Dengler VL, Galbraith M. Espinosa Jí, Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol 2014;49(1):1-15; PMID: 24099156; http://dx.doi.org/10.3109/10409238.2013.838205
- Bertout JA, Majmundar AJ, Gordan JD, Lam JC, Ditsworth D, Keith B, Brown EJ, Nathanson KL, Simon MC. HIF2α inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc Natl Acad Sci USA 2009;106(34):14391-6; PMID:19706526; http://dx.doi.org/10.1073/pnas.0907357106
- Kumar V, Gabrilovich DI. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 2014;143(4):512-9; PMID:25196648; http://dx.doi.org/10.1111/imm.12380
- Van der Burg SH, Arens R, Ossendorp F, Van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer 2016;16(4):219-233; PMID: 26965076; http://dx.doi.org/10.1038/nrc.2016.16
- Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest 2015;125(9):3401-12; PMID:26214521; http://dx.doi.org/10.1172/JCI80009
- Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, Yoshioka Y, Baba T, Konishi I, Mandai M. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer 2015;112(9):1501-9; PMID:25867264; http://dx.doi.org/10.1038/bjc.2015.101
- Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, Xu L, Hicklin DJ, Fukumura D, di Tomaso E et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation. Cancer Cell 2004;6(6):553-563; PMID: 15607960; http://dx.doi.org/10.1016/j.ccr.2004.10.011
- Rivero S, Díaz-Guerra MJ, Monsalve EM, Laborda J, García-Ramírez JJ. DLK2 is a transcriptional target of KLF4 in the early stages of adipogenesis. J Mol Biol 2012;417(1-2):36-50; PMID:22306741; http://dx.doi.org/10.1016/j.jmb.2012.01.035
- Rivero S, Ruiz-García A, Díaz-Guerra MJ, Laborda J, García-Ramírez JJ. Characterization of a proximal Sp1 response element in the mouse Dlk2 gene promoter. BMC Mol Biol 2011;12:52; PMID:22185379; http://dx.doi.org/10.1186/1471-2199-12-52
- Culver C, Melvin A, Mudie S, Rocha S. HIF-1alpha depletion results in SP1-mediated cell cycle disruption and alters the cellular response to chemotherapeutic drugs. Cell Cycle 2011;10(8):1249-60; PMID:21412054; http://dx.doi.org/10.4161/cc.10.8.15326
- Jean JC, George E, Kaestner KH, Brown LA, Spira A, Joyce-Brady M. Transcription factor Klf4, induced in the lung by oxygen at birth, regulates perinatal fibroblast and myofibroblast differentiation. PLoS One 2013;8(1):e54806; PMID:23372771 ; http://dx.doi.org/10.1371/journal.pone.0054806
- Hofmann JJ, Iruela-Arispe ML. Notch signaling in blood vessels: who is talking to whom about what? Circ Res 2007;100(11):1556-68; PMID: 17556669; http://dx.doi.org/10.1161/01.RES.0000266408.42939.e4
- Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007;445(7129):776-80; PMID:17259973; http://dx.doi.org/10.1038/nature05571
- Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006;444(7122):1032-7; PMID:17183313; http://dx.doi.org/10.1038/nature05355
- Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, Kowalski J, Watts RJ, Callahan C, Kasman I et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006;444(7122):1083-7; PMID:17183323; http://dx.doi.org/10.1038/nature05313
- Rebay I, Fleming RJ, Fehon RG, Cherbas L, Cherbas P, Artavanis-Tsakonas S. Specific EGF repeats of Notch mediate interactions with Delta and Serrate: implications for Notch as a multifunctional receptor. Cell 1991;67(4):687-99; PMID:1657403; http://dx.doi.org/10.1016/0092-8674(91)90064-6
- Cordle J, Redfieldz C, Stacey M, Van der Merwe PA, Willis AC, Champion BR, Hambleton S, Handford PA. Localization of the delta-like-1-binding site in human Notch-1 and its modulation by calcium affinity. J Biol Chem 2008;283(17):11785-93; PMID:18296446; http://dx.doi.org/10.1074/jbc.M708424200
- Reis M, Czupalla CJ, Ziegler N, Devraj K, Zinke J, Seidel S, Heck R, Thom S, Macas J, Bockamp E et al. Endothelial Wnt/β-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J Exp Med 2012;209(9):1611-27; PMID:22908324; http://dx.doi.org/10.1084/jem.20111580
- Griffioen AW. Anti-angiogenesis: making the tumor vulnerable to the immune system. Cancer Immunol Immunother 2008;57(10): 1553-8; PMID:18438662; http://dx.doi.org/10.1007/s00262-008-0524-3
- Bose A, Lowe DB, Rao A, Storkus WJ. Combined vaccine+axitinib therapy yields superior antitumor efficacy in a murine melanoma model. Melanoma Res 2012;22(3):236-43; PMID:22504156; http://dx.doi.org/10.1097/CMR.0b013e3283538293
- Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, Ko JS, Cohen PA, Finke JH, Storkus WJ. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer 2011;129(9):2158-70; PMID:21170961; http://dx.doi.org/10.1002/ijc.25863
- Lowe DB, Bose A, Taylor JL, Tawbi H, Lin Y, Kirkwood JM, Storkus WJ. Dasatinib promotes the expansion of a therapeutically superior T-cell repertoire in response to dendritic cell vaccination against melanoma. Oncoimmunology 2014;3(1):e27589; PMID:24734217; http://dx.doi.org/10.4161/onci.27589
- Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, Wood L, Elson P, Garcia J, Dreicer R et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clinical cancer research : an official journal of the American Association for Cancer Research 2008;14(20):6674-82; PMID:18927310; http://dx.doi.org/10.1158/1078-0432.CCR-07-5212
- Mrass P, Takano H, Ng LG, Daxini S, Lasaro MO, Iparraguirre A, Cavanagh LL, von Andrian UH, Ertl HC, Haydon PG et al. Random migration precedes stable target cell interactions of tumor-infiltrating T cells. J Exp Med 2006;203(12):2749-2761; PMID:17116735; http://dx.doi.org/10.1084/jem.20060710
- Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, Amigorena S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J Exp Med 2007;204(2):345-356; PMID:17261634; http://dx.doi.org/10.1084/jem.20061890
- Facciponte JG, Ugel S, De Sanctis F, Li C, Wang L, Nair G, Sehgal S, Raj A, Matthaiou E, Coukos G et al. Tumor endothelial marker 1-specific DNA vaccination targets tumor vasculature. J Clin Invest 2014;124(4):1497-511; PMID:24642465; http://dx.doi.org/10.1172/JCI67382
- Seavey MM, Maciag PC, Al-Rawi N, Sewell D, Paterson Y. An anti-vascular endothelial growth factor receptor 2/fetal liver kinase-1 Listeria monocytogenes anti-angiogenesis cancer vaccine for the treatment of primary and metastatic Her-2/neu+ breast tumors in a mouse model. J Immunol 2009;182(9):5537-46; PMID:19380802; http://dx.doi.org/10.4049/jimmunol.0803742
- Blattman JN, Wherry EJ, Ha SJ, Van der Most RG, Ahmed R. Impact of epitope escape on PD-1 expression and CD8 T-cell exhaustion during chronic infection. J Virol 2009;83(9):4386-94; PMID: 19211743; http://dx.doi.org/10.1128/JVI.02524-08
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013;5(200):200ra116; PMID:23986400; http://dx.doi.org/10.1126/scitranslmed.3006504
- Jayaraman P, Sada-Ovalle I, Beladi S, Anderson AC, Dardalhon V, Hotta C, Kuchroo VK, Behar SM. Tim3 binding to galectin-9 stimulates antimicrobial immunity. J Exp Med 2010;207(11):2343-54; PMID:20937702; http://dx.doi.org/10.1084/jem.20100687