
ABSTRACT
Vasoactive intestinal peptide (VIP) is a neuroendocrine peptide hormone that has potent anti-inflammatory activities. VIP signaling through its receptor VPAC1 on T cells leads to reduced proliferation and a reduction in pro-inflammatory cytokine secretion. We report here that inhibition of the VIP pathway with a peptide antagonist significantly enhances a T-cell-dependent, autologous anti-leukemia response in murine models of acute myeloid leukemia and T lymphoblastic leukemia. Subcutaneous administration of the VIP antagonist, VIPhyb, resulted in reduced tumor burden and significantly enhanced survival (30–50% survival) over vehicle-treated controls (0–20% survival). The T cells in mice treated with VIPhyb expressed lower levels of the co-inhibitory PD-1 and secreted higher levels of IFNγ. Furthermore, T cells from VIPhyb-treated survivors were protective against C1498 following adoptive transfer. These data highlight the potential for the VIP pathway as a novel target for immunomodulation in settings of hematological malignancies.
Introduction
Strategies to enhance the autologous anticancer response have been under intense study following the success of clinical trials involving patients with solid malignancies.Citation1 A variety of approaches have been explored to overcome the suppressive tumor microenvironment and circumvent tumor cell immune evasion. More specifically, many studies have aimed at enhancing antitumor T cell responses via a checkpoint blockade strategy that modulates signaling from co-inhibitory and co-stimulatory molecules. For example, treatment with a monoclonal antibody that blocks signaling through the co-inhibitory molecule CTLA-4 significantly improved survival in patients with metastatic melanoma.Citation2
Hematological malignancies have traditionally been treated with chemotherapeutics alone or in conjunction with autologous or allogeneic haematopoietic stem cell transplantation (HSCT).Citation3 The antitumor activities of allogeneic HSCT come in the form of graft-versus-leukemia effects in which donor T cells directly kill transformed recipient cells due to differences in minor and/or major histocompatibility molecules.Citation3 Despite the widespread use and success of allogeneic transplant, the associated morbidity and mortality of the procedure has prompted investigations into therapies such as checkpoint blockade that provide an enhancement of autologous anti-leukemia immune responses.
Previous work has highlighted a role for vasoactive intestinal peptide in the modulation of T cell responses in the setting of antiviral immunity.Citation4,5 Vasoactive intestinal peptide (VIP) is a 28 amino acid neuropeptide that is predominantly secreted from nerve terminals and the gastrointestinal tract from which it derives its name.Citation6,7 VIP is also secreted from a wide variety of cells in the immune system including cells of both the myeloid and lymphoid lineages.Citation6,8 The VIP receptors VPAC1 and VPAC2 are widely expressed throughout the body with differentially regulated expression on the surface of immune cells.Citation6,9 VPAC1 is constitutively expressed on lymphocytes, whereas VPAC2 is considered to be inducible following an inflammatory stimulus.Citation9-11 Signaling through these receptors involves activation of adenylyl cyclase via Gαs resulting in increased levels of intracellular cyclic AMP.Citation12 These events lead to activation of protein kinase A followed by activation of the p38 and CREB pathway.Citation12,13 VIP signaling in the immune system has potent anti-inflammatory effects on cells of both the innate and adaptive arms. Effects on T cells include a skewing of responses toward Th2 effector functions as well as the promotion of Treg development through effects on dendritic cells.Citation14-18 Additionally, the presence of VIP inhibits T cell proliferation in vitro.Citation19 One of the largest physiologic effects of VIP in myeloid and lymphoid cells is a marked reduction in a variety of pro-inflammatory cytokines, including type 1 and 2 interferons, TNF-α, IL-1, and IL-2.Citation6,8,19 Additionally, the presence of VIP leads to increases in the Th2 cytokines IL-4, IL-5, IL-13, and the anti-inflammatory IL-10.Citation6,8
Using the C1498 model of acute myeloid leukemia and the LBRM model of acute T lymphoblastic leukemia, we investigated the immunological effects of pharmacological VIP signaling blockade and their subsequent effects on tumor burden and survival. Treatment of mice with seven small subcutaneous doses of the small peptide antagonist VIPhyb resulted in significantly enhanced T cell immunity with increased numbers of CD8+ T cells, reduced PD-1 expression, and enhanced secretion of the pro-inflammatory cytokines TNF-α and IFNγ. Mice treated with VIPhyb had a significantly reduced tumor burden, which translated into significantly enhanced survival over vehicle-treated controls. Further supporting a role for T cells in the survival benefits of VIPhyb treatment was the transfer of tumor protection to Rag 1 knockout recipients receiving T cells from VIPhyb-treated donors. The data presented here highlight the potential for VIP antagonism in the treatment of hematological malignancies. In addition, VIP antagonism may hold promise as a treatment of a variety of other cancers and viral infections in which suboptimal T cell responses contribute to disease progression, morbidity, and mortality.
Methods
Mice
Six- to eight-week-old B10.BR mice were used for the LBRM model and were bred at the Emory University Animal Care Facility (Atlanta, GA). For the C1498 model, six- to eight-week-old male albino B6 mice (B6(Cg)-Tyrc-2J/J) were purchased from Jackson Laboratories. Six- to eight-week-old male Rag 1 knockout (B6.129S7-Rag1tm1Mom/J) were purchased from Jackson laboratories. VIP-PHI knockout mice were bred at Emory University.Citation20 All procedures were approved by the Emory University Institutional Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals.
Cell lines
The dsRed and luciferase-transfected acute myeloid leukemia cell line C1498 was a generous gift from Dr Bruce Blazar.Citation21 The T lymphoblastic leukemia cell line LBRM was obtained from American Type Culture Collection (ATCC) and transfected with luciferase as described previously.Citation22 C1498 and LBRM cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, and 50 μM 2-mercaptoethanol. All cell cultures were kept in a 37°C 5% CO2 humidified incubator. Cell viability was assessed before injection via trypan blue exclusion and was always greater than 95%. Cells were injected in PBS intravenously via the lateral tail vein.
VIPhyb treatment
VIPhyb peptide was purchased from New England Peptide. The desiccated peptide was reconstituted in sterile molecular grade water and diluted in sterile PBS. A 10 μg peptide dose in a 200 μL injection volume was delivered to each mouse subcutaneously. Treatment began the day before or the day of tumor injection and continued for 7 d. In some experiments, treatment was delayed until day 8 post-tumor cell injection. An equal volume of PBS was injected in control mice.
Bioluminescent imaging
Tumor burden was assessed using bioluminescent imaging. Animals were first anesthetized using a cocktail of ketamine (80 mg/kg) and xylazine (8 mg/kg). Prior to imaging, the abdomens of B10.BR mice were shaved with an electric razor. D-luciferin was injected via the intraperitoneal route at a dose of 150 mg/kg (Perkin Elmer-122799). Images were acquired on an IVIS spectrum imager (Perkin Elmer, Waltham, MA). Quantification of tumor burden was performed using Living Image software (Perkin Elmer) by creating a region of interest over each mouse. Measurements are reported as photons/s/cm2.
Adoptive transfer
Splenic T cells from VIPhyb-treated tumor survivors or naive B6 mice were purified using the EasySep T Cell Isolation Kit (StemCell Technologies-19851). 5 × 106 cells were adoptively transferred via tail vein injection into naive Rag1 knockout mice. Rag1 knockout mice receiving cells from naive donors served as controls. One week post-adoptive transfer, 1 × 106 C1498 were injected intravenously into both groups of mice. Mice were then challenged with a second dose of tumor cells and monitored for survival, tumor burden, and T cell phenotype.
Flow cytometry
Peripheral blood (cheek bleeds) and/or spleen samples were collected from mice at the time points indicated and then prepared for flow cytometric analysis. Blood samples were collected in EDTA-coated tubes. Spleens were flushed with complete media using a syringe, and clumps were removed by passing the cells through a 70-μm strainer. Red blood cells were lysed in ammonium chloride buffer. Samples were then incubated in the presence of an anti-CD16/32 Fc receptor-blocking antibody following by staining with FITC CD3, PerCP CD8, Alexa Fluor 700 CD4, PE-Cy7 CD69, APC-Cy7 CD25, PE CD62L, V450 CD44, and APC PD-1 (PharMingen-553141, 561801, 561092, 557956, 552879, 561038, 553151, 560451, 562761). Intracellular cytokine staining was performed on splenocytes using Leukocyte Activation Cocktail, BD Cytofix/Cytoperm kit, and antibodies toward CD3, CD4+, CD8+, IFNγ and TNF-α (PharMingen-554714, 554411, 560658). Samples were run on a FACS Aria flow cytometer (BD Biosciences). List mode files were analyzed using FlowJo software ver 9.8.5 (Tree Star, Inc.). Analysis of T cell markers was performed as described previously.Citation5
VIP receptor expression
VPAC1 and VPAC2 protein levels were determined by Western blot. Protein lysates were prepared from C1498 cells, and 15 μg were loaded onto a 4–20% gradient gel (BioRad-4561096). Protein samples were then transferred to a nitrocellulose membrane and probed with 1:2,000 dilutions of anti-VPAC1 and anti-VPAC2 polyclonal antibodies (Santa Cruz Biotechnologies-sc-30019, sc-30020). Expression of VPAC1 and VPAC2 mRNA was determined by semi-quantitative RT-PCR. Briefly, RNA was extracted using the RNeasy Mini Kit, and 1 μg of RNA was converted to cDNA using the Quantitect Reverse Transcription Kit (Qiagen-74104, 205310). The following primers were used in the PCR reactions: VPAC1 forward GATATGGCCCTCTTCAACAACG reverse GAAGTTGGCCATGACGCAAT VPAC2 forward CCAGATGTTGGTGGCAATGC reverse GTATGTGGATGAGATGCCAATAGG 18s rRNA forward CGGCTACCACATCCAAGGAA reverse GCTGGAATTACCGCGGCT. Products were run on a 1% agarose gel and imaged using a GelDoc XR+ system (Biorad).
CREB signaling
Phosphorylation of CREB was determined by flow cytometry and Western blot. Briefly, splenic murine T cells were isolated using the EasySep T Cell Isolation Kit (StemCell Technologies-19851) and then cultured in complete RPMI containing 0.5% fetal bovine serum overnight. Cells were then incubated at 37°C in the presence of VIPhyb for 30 min followed by stimulation with VIP for 15 min. Flow cytometry was performed using BD Phosflow reagents (BD Biosciences-558052) according to the manufacturer's protocol. Antibodies used were Alexa-488 CD3, PE-Cy7 CD4, APC CD8, and PE pS133 CREB (PharMingen-557666, 552775, 553035, 558436). Samples were run on a FACS Aria flow cytometer (Becton Dickson, San Jose, CA). Western blotting was performed under the same stimulation conditions using rabbit polyclonal antibodies to pS133 CREB and CREB at a 1:1,000 dilution (Cell Signaling Technology-9191, 9197).
T cell proliferation assay
Purified splenic T cells from B6 mice were labeled with 1 μM CFSE (Thermo Fisher-C34554) and incubated in 96-well flat-bottom plates pre-coated with functional anti-CD3 antibody (eBioscience-16–0032–81). Cells were stimulated for 48 h and stained for CD3, CD4+, and CD8+. Samples were run on a FACS Aria (Becton Dickinson), and proliferation was assessed by dilution of CFSE.
Apoptosis assay
Apoptosis in C1498 was measured in vitro by culturing cells with VIPhyb or daunorubicin HCl (Sigma Aldrich-30450). The extent of cell death was measured with an Annexin V Apoptosis Detection Kit (eBioscience-88–8007–72). Sytox blue (Life Technologies-S34857) was used in lieu of propidium iodide due to the dsRed expression in C1498. Data were acquired using a FACS Aria flow cytometer (Becton Dickson).
Statistical analysis
Data were analyzed for statistical significance using GraphPad Prism version 5.0d for Mac (GraphPad Software). Each group under study contained at least five mice with each experiment being repeated at least twice. Data are presented as ± SD. Differences in survival were calculated using the Kaplan–Meier log-rank test. Comparison of two groups was performed using an unpaired Student's t-test while comparison of three or more groups was performed using one-way ANOVA followed by a Tukey post-test for multiple comparisons. BLI imaging data was analyzed using a non-parametric test. A p-value of less than 0.05 was considered statistically significant.
Results
Early and late administration of VIPhyb significantly enhanced survival in two models of acute leukemia
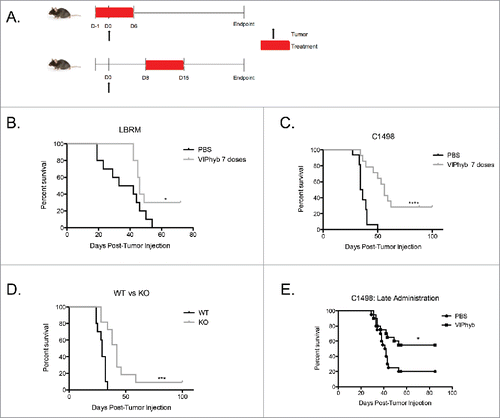
To evaluate VIPhyb as an immunotherapy for leukemia, we injected leukemia cells into syngeneic recipients and administered seven doses of VIPhyb early or late during the course of leukemia development and monitored mice for survival. shows the treatment and inoculation timelines. B10.BR mice bearing LBRM T cell lymphoblastic leukemia showed a significant enhancement of survival when treated early with VIPhyb (). Similarly, VIPhyb-treated C57Bl/6 mice bearing C1498 AML also showed significantly increased survival when VIPhyb was administered daily from the day preceding leukemic challenge to 5 d afterward (). To determine whether the effects of VIPhyb were due directly to antagonism of VIP signaling, we inoculated VIP wild type and VIP knockout mice with C1498 and monitored survival. As shown in , mice that lacked VIP survived significantly longer than mice with functional VIP. The overall survival of these mice was lower than mice treated with VIPhyb, however. Next, we sought to determine whether VIPhyb was efficacious when administered late during tumor development. To test this, we administered seven daily doses of VIPhyb or PBS in C1498-bearing mice from days 8–14 post-tumor inoculation. As shown in , there was a significant enhancement of survival following late VIPhyb administration.
Figure 1. Early and late administration of VIPhyb improves survival in AML and T cell leukemia-bearing mice. Albino B6 mice were administered 10 μg VIPhyb subcutaneously for 7seven consecutive days beginning either the day before or one week after receiving 106 C1498 or 2 × 106 LBRM cells intravenously. (A) Timeline of tumor injection and VIPhyb treatment. (B) Survival of LBRM-bearing mice receiving either PBS or VIPhyb. (n = 10 PBS, n = 10 VIPhyb) (C) Survival of C1498-bearing mice receiving either PBS or VIPhyb. (n = 16 PBS, n = 14 VIPhyb) (D) Survival of WT and VIP KO mice following C1498 inoculation. (n = 10 WT, n = 10 VIP KO) (E) Survival data from two experiments of C1498-bearing mice receiving late treatment with PBS or VIPhyb. (n = 20 PBS, n = 20 VIPhyb) *p < 0.05 and ***p < 0.001 indicate significant differences between the control and treated groups.
Early administration of VIPhyb lowered tumor burden in a lymphocyte-dependent manner
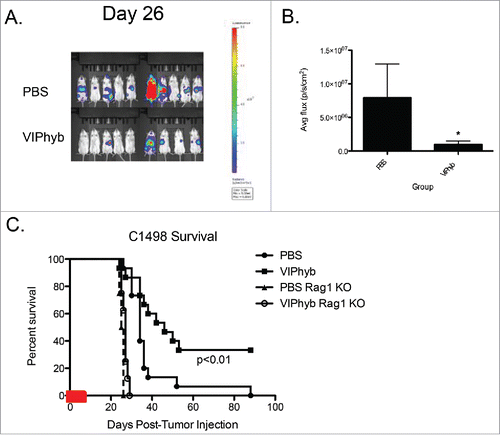
Based on the improvement in survival obtained with early VIPhyb administration, we examined the tumor burden in treated mice vs. PBS-treated controls. We used bioluminescent imaging to quantify the overall tumor burden. Mice treated with an early course of VIPhyb had significantly lower tumor burden 26 d after inoculation with leukemia compared with PBS-treated controls ( and ). To determine whether the reduced tumor burden was the result of immunological action, we repeated the experiment with Rag 1 knockout mice, which lack functional T and B cells. VIPhyb treatment was not effective at protecting Rag 1 knockout mice from C1498 tumor-associated death ().
Figure 2. VIPhyb treatment led to reduced tumor burden in mice, which required the presence of lymphocytes. C1498-bearing mice were injected i.p with luciferin, anesthetized, and imaged in an IVIS spectrum imager. Rag1 knockout mice and wild-type albino B6 mice were injected with 106 C1498 cells i.v and treated with an early course of VIPhyb or PBS. (A) Representative BLI image of late stage C1498-bearing albino B6 mice treated with an early course of either PBS or VIPhyb. The scale indicates the intensity of the signal emitted from C1498 cells. (B) Quantification of tumor burden reported as average flux emitted from each mouse's entire body. (C) Survival of C1498-bearing, VIPhyb-treated Rag1 knockout mice compared with wild-type C1498-bearing B6 albino mice treated with either PBS or VIPhyb. (n = 9 PBS, n = 10 VIPhyb, n = 4 Rag1 KO PBS, n = 8 Rag1 KO VIPhyb) The red box on the x-axis indicates the treatment period. *p < 0.05, **p < 0.01, ***p < 0.001 indicate significant differences between or among groups of mice.
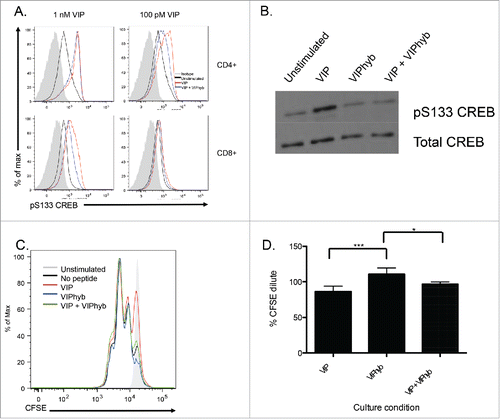
VIPhyb reduces CREB phosphorylation in CD4+ and CD8+ T cells following VIP stimulation and enhances T cell proliferation in vitro
To determine whether administration of VIPhyb prevents signaling from VIP receptors in response to exogenous VIP, T cells from B6 mice were purified and cultured in low serum medium overnight followed by VIP stimulation in the presence or absence of VIPhyb. Levels of CREB phosphorylation were then measured by phospho-flow cytometry and Western blot. CD4+and CD8+ T cells responded differently to high and low concentrations of VIP with CD8+ T cells failing to signal from concentrations as low as 100 pM (). Pre-incubation with VIPhyb was able to block CREB phosphorylation in both CD4+ and CD8+ T cells, however, only CD8+ T cells had reduced levels of CREB phosphorylation at a higher concentration of VIP indicating increased sensitivity of CD8+ T cells to VIPhyb (). shows reduced CREB phosphorylation in the total T cell compartment following incubation with VIPhyb before VIP stimulation. Next, we determined the functional effect of VIPhyb-mediated VIP signaling blockade on T cells in vitro. We cultured CFSE-labeled B6 T cells in 96-well plates pre-coated with anti-CD3 antibody for 48 h and assessed proliferation by flow cytometry. Addition of VIP to cultures resulted in a modest reduction of proliferation, whereas addition of VIPhyb enhanced proliferation in response to CD3 stimulation (). Although not significant, addition of VIPhyb to cultures containing exogenous VIP partially reversed VIP-mediated suppression of T cell proliferation ().
Figure 3. VIPhyb reduced CREB phosphorylation in T cells following VIP stimulation and enhanced proliferation in vitro. Splenic T cells were purified by negative selection and cultured in complete media containing 0.5% fetal bovine serum overnight to reduce basal CREB phosphorylation levels. Cells were then incubated in the presence of 10 μM VIPhyb before stimulation with 1 nM or 100 pM VIP. Levels of CREB phosphorylation were then assessed by Phosflow and Western blot. The effect of VIPhyb on the in vitro proliferation of T cells was assessed by culturing CFSE-labeled splenic T cells in a 96-well plate pre-coated with anti-CD3 antibody. CFSE dilution was measured 48 h later by flow cytometric analysis of live CD3+ cells. (A) Representative histograms indicating levels of CREB phosphorylation in VIP-stimulated CD4+ and CD8+ T cells. The solid gray histogram represents the isotype, the black histogram represents unstimulated samples, the red histogram represents samples stimulated with VIP, and the blue histogram represents samples pre-incubated with VIPhyb before VIP stimulation. (B) Western blot showing CREB phosphorylation in splenic T cells treated with 1 nM VIP or 10 μM VIPhyb + 1 nM VIP. (C) Histogram indicating the level of CFSE dilution in control wells and wells containing 1 nM VIP with or without 10 μM VIPhyb. The solid gray histogram represents T cells cultured in wells without anti-CD3, the black histogram represents control wells not containing peptide, the red histogram represents wells containing VIP, the blue histogram represents wells containing VIPhyb alone, and the green histogram represents wells containing VIP and VIPhyb. (D) Quantification of CFSE dilution normalized to control wells for three independent experiments. *p < 0.05, **p < 0.01 indicate significant differences among culture conditions.
C1498 cells express the VPAC2 receptor and PD-L1 but do not respond to VIPhyb
Due to the possibility of VIPhyb-mediated direct tumor growth inhibition, we next tested whether the peptide affected C1498 growth and survival. Using a semi-quantitative PCR approach, we determined that C1498 cells express mRNA for the VPAC2 receptor (). Quantitative RT-PCR confirmed these findings as the VPAC2 sequence could be successfully amplified, while the VPAC1 sequence could not (data not shown). To determine whether the gene expression results directly translated to protein expression, we performed Western blots using polyclonal antibodies to VPAC1 and VPAC2. As shown in , no bands were detected when membranes were probed for VPAC1, but probing for VPAC2 yielded a single band. These results demonstrate that C1498 cells express only the VPAC2 receptor.
Figure 4. C1498 cells express VPAC2 but are not directly affected by VIPhyb treatment. RNA and protein from cultured C1498 cells were extracted, and expression of VPAC1 and VPAC2 was determined by RT-PCR and Western blot. C1498 cells were cultured in the presence or absence of VIPhyb or daunorubicin as positive control. Survival of cells was measured by annexin V-sytox blue staining and flow cytometry while growth of the cells was measured by BLI. Expression of PD-L1 was determined by flow cytometry following 48-h culture in the presence of 10 ng/mL IFNγ and various concentrations of VIPhyb. (A) Image of PCR products run on a 1% agarose gel with 18s rRNA as control. (B) Image of Western blot probed for VPAC1 and VPAC2. (C) Growth curves of C1498 cells under various culture conditions as measured by BLI. (D) Flow cytometry plots of C1498 cells cultured with 1 μM VIPhyb, 1 μM daunorubicin, 1 nM VIP, or no drug as control. Cells in the lower right quadrant are in early apoptosis while cells in the upper right quadrant are dead. (E) Expression of PD-L1 on live C1498 cells cultured for 48 h in the presence of IFNγ and VIPhyb.
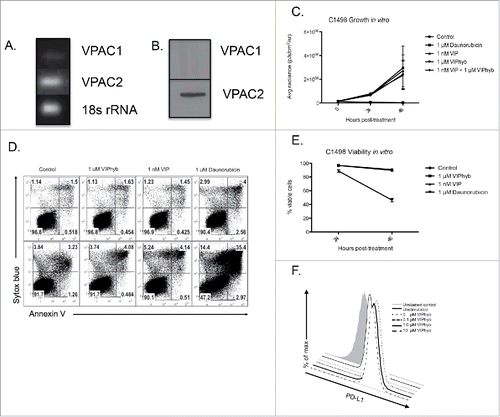
Since blockade of this receptor with VIPhyb could potentially lead to inhibition of tumor cell growth and survival, we next examined the growth and survival properties of C1498 cells in culture in the presence or absence of VIP, VIPhyb, or the chemotherapeutic daunorubicin. The growth properties of C1498 cells did not differ significantly among VIP, VIPhyb, combination, and negative control cultures as measured by BLI (). Further, cultures containing either VIP or VIPhyb did not show significant differences in survival in vitro compared with control cultures ( and ). As expected, cultures containing daunorubicin showed significant cell death beginning at 24 h ( and ).
C1498 cells are a well-established PD-L1-expressing cell line.Citation23 Upregulation of PD-L1 on the surface of tumor cells in response to pro-inflammatory cytokines is a major mechanism of tumor immune evasion. As such, we wished to determine whether culture with VIPhyb would lead to reduced expression of PD-L1 following stimulation with IFNγ. As shown in , upregulation of PD-L1 in response to IFNγ was not prevented at any of the VIPhyb concentrations tested indicating that the potential to signal through the PD-1 pathway was not inhibited in the tumor cells themselves.
Adoptive transfer of T cells from VIPhyb-treated tumor survivors protect Rag 1 knockout mice from tumor-associated death
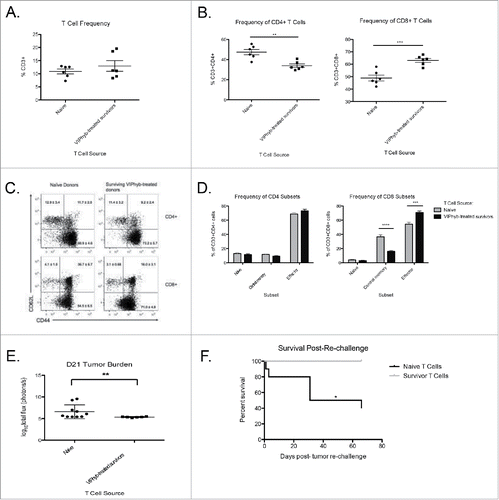
To determine whether the enhanced survival following VIPhyb treatment was T cell-mediated, we purified splenic T cells from either leukemia-naive B6 mice or VIPhyb-treated C1498 survivors and adoptively transferred them to Rag 1 knockout recipients. Seven days post-transfer, Rag 1 knockout mice were injected with C1498 and subsequently monitored for survival and T cell phenotype. As shown in , the T cell frequency in peripheral blood was not significantly different between groups. However, the frequency of T cell subsets differed significantly with the majority of T cells from VIPhyb-treated donors being CD8+ T cells (). Additionally, the T cells from VIPhyb-treated donors were predominately CD44+CD62L− effectors indicating increased levels of activation ( and ). Tumor burden was lower in mice receiving T cells from VIPhyb-treated donors 21 d post-injection (), which translated to a significant survival benefit ().
Figure 5. Adoptive transfer of T cells from VIPhyb-treated tumor survivors conferred protection to Rag1 knockout mice. Splenic T cells from VIPhyb-treated mice that survived C1498 were purified by negative selection. 5 × 106 T cells were adoptively transferred to Rag 1 knockout recipients followed by injection of 106 C1498 cells one week later. Peripheral blood samples were analyzed 14 d post-tumor inoculation for the frequencies of T cell subsets. Tumor burden was assessed 21 d post-tumor inoculation by BLI. (A) T cell frequencies in peripheral blood of tumor-bearing Rag 1 KO mice receiving naive T cells or T cells from VIPhyb-treated mice. (B) Frequencies of CD4+ and CD8+ T cells in tumor-bearing Rag 1 KO mice receiving naive T cells or T cells from VIPhyb-treated mice. (C) Flow cytometry plots showing differential expression of the markers CD44 and CD62L on peripheral blood T cells from tumor-bearing Rag 1 KO mice receiving naive T cells or T cells from VIPhyb-treated mice. (D) Quantification of T cell subsets as defined by the expression of CD44 and CD62L. (E) Comparison of tumor burden as assessed by BLI between mice receiving naive T cells and mice receiving T cells from VIPhyb-treated donors. (F) Survival of mice receiving naive T cells vs. mice receiving T cells from VIPhyb-treated donors following a second challenge of 106 tumor cells. (n = 10 naive T cells, n = 6 VIPhyb-treated T cells) *p < 0.05, **p < 0.01, ***p < 0.001 indicate significant differences between groups.
Administration of VIPhyb reduced PD-1 expression and enhanced pro-inflammatory cytokine secretion in CD4+ and CD8+ T cells
To examine the underlying mechanism whereby T cells enhance the anti-leukemia effect following VIPhyb treatment, peripheral blood was collected from PBS and VIPhyb-treated mice 15 and 30 d post-C1498 inoculation and phenotyped using flow cytometry. Of particular interest was the expression of the inhibitory molecule PD-1. The frequency of PD-1 expressing CD4+ T cells in C1498-bearing mice was significantly lower in mice that received VIPhyb treatment at day 15 post-tumor cell injection (). This effect was transient, however, as no significant difference was observed 30 d post-tumor injection (). In contrast, the frequency of PD-1-expressing CD8+ T cells from VIPhyb-treated mice trended toward significance on day 15 post-leukemia inoculation but was significantly lower on day 30 (). With an increased frequency of CD44+CD62L− CD8+ effectors in VIPhyb-treated mice () we examined the expression of PD-1 on CD4+ and CD8+ T cells with different activation statuses 15 d post-tumor inoculation. PD-1 expression on naive CD4+ T cells was not significantly lower following either one or seven doses of VIPhyb in leukemia-bearing mice (). However, expression of PD-1 on naive CD8+ T cells was significantly lower with either schedule of VIPhyb administration (). Expression of PD-1 on central memory phenotype CD4+ T cells in VIPhyb-treated mice was not significantly lower than PBS control but was significantly lower on CD8+ T cells from mice treated with seven doses of VIPhyb (). PD-1 expression on CD4+ and CD8+ T effector cells was only significantly lower than PBS control with seven doses of VIPhyb ().
Figure 6. VIPhyb treatment reduced PD-1 expression in CD4+ and CD8+ T cells and enhanced the production of pro-inflammatory cytokines. Peripheral blood and spleen samples were collected on days 15 and 30 post-tumor cell injection and analyzed by flow cytometry. (A) PD-1 expression on total peripheral blood CD4+ and CD8+ T cells at the indicated time points. (B) Frequency of CD44+CD62L− effector CD4+ and CD8+ T cells in peripheral blood 15 d post-tumor cell inoculation. (C) Expression of PD-1 on subsets of peripheral blood CD4+ and CD8+ T cells 15 d post-tumor cell injection as defined by the expression of CD44 and CD62L. Graphs compare the expression of PD-1 in mice treated with PBS, one single dose of VIPhyb, or seven consecutive daily doses of VIPhyb. (D) Expression of IFNγ and TNF-α in splenic T cells from C1498-bearing mice treated with PBS or seven doses of VIPhyb. *p < 0.05, *p < 0.01, and ***p < 0.001 indicate significant differences between groups.
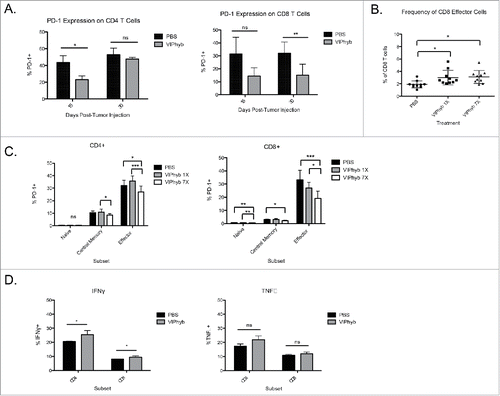
We next examined the functional capacity of T cells from the spleens of VIPhyb and PBS-treated C1498-bearing mice by intracellular cytokine staining. The frequency of IFNγ-producing cells in both the CD4+ and CD8+ compartments was significantly higher in VIPhyb-treated mice 15 d post-tumor injection (). In contrast to IFNγ production, there was no significant difference in the frequency of TNF-α-producing CD4+ or CD8+ T cells ().
Discussion
A large body of literature exists that highlights the potent anti-inflammatory actions of VIP in a wide variety of cell types. Due to these anti-inflammatory effects, VIP itself has been explored as a potential therapeutic for a variety of autoimmune disorders.Citation24 As such, it stands to reason that blockade of endogenous VIP signaling would result in stronger inflammatory responses. Previous work performed by our laboratory has explored the VIP pathway as a therapeutic target using the VIPhyb antagonist to strengthen T cell responses and promote dendritic cell maturation in settings of antiviral immune responses and allogeneic transplant.Citation4,5,25 Based on the observed effects of VIPhyb on antiviral CD8+ T cell responses in particular, we sought to determine whether VIPhyb would have a therapeutic effect by inducing autologous T cells with anti-leukemia activity. Adaptive immune responses to transformed myeloid and lymphoid cells rely largely on the CD8+ compartment, and thus we hypothesized that VIPhyb could help overcome the immunosuppressive environment created by leukemia cells. Previous work has demonstrated the therapeutic benefit of VIPhyb on the graft-vs.-leukemia effect in murine models of allo-transplantation.Citation25 Administration of VIPhyb in this setting modulated the expression of immune checkpoint molecules on donor T cells which enhanced their anti-leukemia effects.Citation25 The effects of VIP signaling blockade on host T cells was not addressed by this study, and as such we examined the effects of VIPhyb on the autologous anti-leukemia response in the absence of transplant.
Acute myeloid leukemia blasts have been shown to create an immunosuppressive environment through a wide variety of mechanisms including upregulation of IDO and secretion of factors that inhibit T cell proliferation.Citation26 The active subversion of T cell responses by AML blasts render these immunosuppressive activities a prime target for immunotherapeutics. The data presented here provide evidence that the use of a peptide antagonist to the vasoactive intestinal peptide signaling pathway enhances survival in murine models of both T cell lymphoma and acute myeloid leukemia. The survival benefits were T cell-mediated as transfer of T cells from VIPhyb-treated mice to Rag1 knockout recipients were protected from C1498 challenge. The greatest effects of VIPhyb were seen in CD8+ T cells, which failed to respond to lower concentrations of VIP and showed a greater reduction in VIP signaling in response to VIPhyb when exposed to higher VIP concentrations. This is most likely due to lower expression of the VPAC1 receptor on CD8+ T cells in comparison to CD4+ T cells.Citation9 Lower expression of the receptors would result in quicker saturation following the introduction of a peptide antagonist. This enhanced blockade accounts for the greater frequencies of CD8+ T cells in treated mice as well as the increased frequencies of effector cells. Due to the fact that CD8+ T cells are the primary effectors in antitumor immune responses this helps to explain the reduced tumor burden in treated mice.Citation27 Further supporting a T cell-mediated mechanism-of-action for VIPhyb is the lack of an effect on either growth or survival of the tumor cells in vitro despite expression of VPAC2. It has been established that many human and murine tumor cell lines overexpress VPAC1 or VPAC2, and the growth of some cell lines is supported by VIP and can be directly inhibited by VIP antagonists.Citation28-31 We did not observe similar results which supports an off-target mechanism.
The enhanced in vitro proliferation and enhanced cytokine secretion in tumor-bearing mice can be explained in part by the VIPhyb-mediated reduction in CREB signaling. Following phosphorylation of CREB by protein kinase A, CREB binds its co-activator CBP.Citation32 CBP binds to the same site on phosphorylated CREB as the RelA subunit of NF-κB, and thus phosphorylation of CREB sequesters one half of the NF-κB dimer that is required for its transcriptional activity.Citation32,33 Downstream of NF-κB are a variety of pro-inflammatory genes, including IL-2 and IFN-γ, which are necessary for T cell proliferation and functional activity.Citation34 This helps to explain the enhanced proliferation of T cells as well as the increased IFNγ secretion as VIPhyb-mediated reduction of CREB phosphorylation would enhance NF-κB activity. Stimulation through the TCR leads to secretion of VIP by T cells in vitro, and as such, stimulation with anti-CD3 would increase endogenous levels of VIP.Citation35 VIPhyb was able to significantly reduce endogenous VIP-mediated T cell suppression, but was not able to significantly prevent suppression from both endogenous and exogenous VIP. Since the source of VIP in tumor-bearing mice would be endogenous VIP it can be reasoned that VIPhyb would significantly enhance T cell proliferation in vivo. Taken together, VIPhyb treatment in tumor-bearing mice blocked CREB signaling from endogenous VIP and lead to enhanced proliferation and cytokine secretion in T cells. These effects were strongest in the CD8+ compartment contributing to reduced tumor burden and enhanced survival.
Among the observed effects of VIPhyb on T cells, the reduced expression of PD-1 on CD4+ and CD8+ subsets would have been a large contributor to the reduced tumor burden in treated mice. Several studies have demonstrated the importance of the PD-1-PD-L1 signaling axis in AML progression and anti-AML T cell responses.Citation36-38 It has been established that AML blasts upregulate PD-L1 in response to pro-inflammatory cytokines.Citation38 Additionally, the PD-1-PD-L1 signaling pathway has been shown to be a major contributor to immune evasion by C1498 in vivo.Citation23 Reduction of PD-1 signaling through the use of monoclonal antibodies has demonstrated to have profound effects on exhausted T cells in models of chronic infection and cancer and have proven highly efficacious in many clinical trials.Citation1,39,40 The functional effects of PD-1 signaling blockade include enhanced pro-inflammatory cytokine secretion, increased proliferation, and enhanced production of cytotoxic molecules.Citation39 The reduced expression of PD-1 on T cells following VIPhyb treatment along with increased T cell proliferation and secretion of IFNγ highlight the relevance of the PD-1 pathway in the survival effects and reduced tumor burden observed in treated mice. The lack of an effect of VIPhyb on the expression of PD-L1 on the tumor cells following stimulation with IFNγ points toward the interference of this pathway being specifically on the T cell axis.
We show here that blockade of VIP receptors on T cells using a peptide antagonist functions in a similar manner to currently used checkpoint blockade methods with significant enhancement of T cell function, reduction of tumor burden, and enhanced survival in two models of acute leukemia. Interestingly, late administration of VIPhyb led to an equivalent net increase in the fraction of surviving mice (compared with saline injection) as injections of VIPhyb starting 1 day before inoculation with leukemia, supporting the potency of VIP receptor blockade as an anticancer immunotherapeutic and the clinical relevance of this approach. Whether simultaneous blockade of co-inhibitory receptors and VIP receptors would result in additive or synergistic effects is a topic of future investigation. Taken together, the results presented here highlight the potential for VIP signaling blockade as an immunotherapeutic strategy in the treatment of hematological malignancies.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Kohrt HE, Tumeh PC, Benson D, Bhardwaj N, Brody J, Formenti S, Fox BA, Galon J, June CH, Kalos M et al. Immunodynamics: a cancer immunotherapy trials network review of immune monitoring in immuno-oncology clinical trials. J Immunother Cancer 2016; 4:15; PMID: 26981245; https://doi.org/10.1186/s40425-016-0118-0
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363(8):711-23; PMID: 20525992; https://doi.org/10.1056/NEJMoa1003466
- Lichtman MA Battling the hematological malignancies: the 200 years' war. Oncologist 2008; 13(2):126-38; PMID: 18305057; https://doi.org/10.1634/theoncologist.2007-0228
- Li JM, Hossain MS, Southerland L, Waller EK. Pharmacological inhibition of VIP signaling enhances antiviral immunity and improves survival in murine cytomegalovirus-infected allogeneic bone marrow transplant recipients. Blood 2013; 121(12):2347-51; PMID: 23325838; https://doi.org/10.1182/blood-2012-06-437640
- Li JM, Darlak KA, Southerland L, Hossain MS, Jaye DL, Josephson CD, Rosenthal H, Waller EK. VIPhyb, an antagonist of vasoactive intestinal peptide receptor, enhances cellular antiviral immunity in murine cytomegalovirus infected mice. PLoS One 2013; 8(5):e63381; PMID: 23723978; https://doi.org/10.1371/journal.pone.0063381
- Delgado M, Pozo D, Ganea D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev 2004; 56(2):249-90; PMID: 15169929; https://doi.org/10.1124/pr.56.2.7
- Said SI, Mutt V. Polypeptide with broad biological activity: isolation from small intestine. Science 1970; 169(3951):1217-8; PMID: 5450698; https://doi.org/10.1126/science.169.3951.1217
- Delgado M, Ganea D. Vasoactive intestinal peptide: a neuropeptide with pleiotropic immune functions. Amino Acids 2013; 45(1):rr25-39; PMID:22139413; https://doi.org/10.1007/s00726-011-1184-8
- Hermann RJ, Van der Steen T, Vomhof-Dekrey EE, Al-Badrani S, Wanjara SB, Failing JJ, Haring JS, Dorsam GP. Characterization and use of a rabbit-anti-mouse VPAC1 antibody by flow cytometry. J Immunol Methods 2012; 376(1-2):20-31; PMID: 22079255; https://doi.org/10.1016/j.jim.2011.10.009
- Metwali A, Blum AM, Li J, Elliott DE, Weinstock JV IL-4 regulates VIP receptor subtype 2 mRNA (VPAC2) expression in T cells in murine schistosomiasis. FASEB J 2000; 14(7):948-54. PMID: 10783149; https://doi.org/10.1096/fj.1530-6860
- Lara-Marquez M, O'Dorisio M, O'Dorisio T, Shah M, Karacay B. Selective gene expression and activation-dependent regulation of vasoactive intestinal peptide receptor type 1 and type 2 in human T cells. J Immunol 2001; 166(4):2522-30; PMID: 11160313; https://doi.org/10.4049/jimmunol.166.4.2522
- Chorny A, Gonzalez-Rey E, Varela N, Robledo G, Delgado M. Signaling mechanisms of vasoactive intestinal peptide in inflammatory conditions. Regul Pept 2006; 137(1-2):67-74; PMID: 16949684; https://doi.org/10.1016/j.regpep.2006.04.021
- Varela N, Chorny A, Gonzalez-Rey E, Delgado M. Tuning inflammation with anti-inflammatory neuropeptides. Expert Opin Biol Ther 2007; 7(4):461-78; PMID: 17373898; https://doi.org/10.1517/14712598.7.4.461
- Delgado M, Leceta J, Gomariz RP, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide stimulate the induction of Th2 responses by up-regulating B7.2 expression. J Immunol 1999; 163(7):3629-35. PMID: 10490956.
- Delgado M, Reduta A, Sharma V, Ganea D. VIP/PACAP oppositely affects immature and mature dendritic cell expression of CD80/CD86 and the stimulatory activity for CD4(+) T cells. J Leukoc Biol 2004; 75(6):1122-30; PMID: 15020654; https://doi.org/10.1189/jlb.1203626
- Delgado M, Chorny A, Gonzalez-Rey E, Ganea D. Vasoactive intestinal peptide generates CD4+CD25+ regulatory T cells in vivo. J Leukoc Biol 2005; 78(6):1327-38; PMID: 16204628; https://doi.org/10.1189/jlb.0605299
- Anderson P, Gonzalez-Rey E. Vasoactive intestinal peptide induces cell cycle arrest and regulatory functions in human T cells at multiple levels. Mol Cell Biol 2010; 30(10):2537-51; PMID: 20231362; https://doi.org/10.1128/MCB.01282-09
- Gonzalez-Rey E, Chorny A, Fernandez-Martin A, Ganea D, Delgado M. Vasoactive intestinal peptide generates human tolerogenic dendritic cells that induce CD4 and CD8 regulatory T cells. Blood 2006; 107(9):3632-8; PMID: 16397128; https://doi.org/10.1182/blood-2005-11-4497
- Boudard F, Bastide M, Inhibition of mouse T-cell proliferation by CGRP and VIP: effects of these neuropeptides on IL-2 production and cAMP synthesis. J Neurosci Res 1991; 29(1):29-41; PMID: 1653367; https://doi.org/10.1002/jnr.490290104
- Colwell CS, Michel S, Itri J, Rodriguez W, Tam J, Lelievre V, Hu Z, Liu X, Waschek JA. Disrupted circadian rhythms in VIP- and PHI-deficient mice. Am J Physiol Regul Integr Comp Physiol 2003; 285(5):R939-49; PMID: 12855416; https://doi.org/10.1152/ajpregu.00200.2003
- Taylor PA, Ehrhardt MJ, Lees CJ, Tolar J, Weigel BJ, Panoskaltsis-Mortari A, Serody JS, Brinkmann V, Blazar BR. Insights into the mechanism of FTY720 and compatibility with regulatory T cells for the inhibition of graft-versus-host disease (GVHD). Blood 2007; 110(9):3480-8; PMID: 17606761; https://doi.org/10.1182/blood-2007-05-087940
- Lu Y, Giver CR, Sharma A, Li JM, Darlak KA, Owens LM, Roback JD, Galipeau J, Waller EK. IFN-γ and indoleamine 2,3-dioxygenase signaling between donor dendritic cells and T cells regulates graft versus host and graft versus leukemia activity. Blood 2012; 119(4):1075-85; PMID: 22130799; https://doi.org/10.1182/blood-2010-12-322891
- Zhang L, Gajewski TF, Kline J, PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood 2009; 114(8):1545-52; PMID: 19417208; https://doi.org/10.1182/blood-2009-03-206672
- Gonzalez-Rey E, Fernandez-Martin A, Chorny A, Martin J, Pozo D, Ganea D, Delgado M. Therapeutic effect of vasoactive intestinal peptide on experimental autoimmune encephalomyelitis: down-regulation of inflammatory and autoimmune responses. Am J Pathol 2006; 168(4):1179-88; PMID: 16565493; https://doi.org/10.2353/ajpath.2006.051081
- Li JM et al. Modulation of immune checkpoints and graft-versus-leukemia in allogeneic transplants by vasoactive intestinal peptide. Cancer Res, 2016.
- Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM, Qureshi A, Dazzi F, Vyas P, Cerundolo V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013; 122(5):749-58; PMID: 23733335; https://doi.org/10.1182/blood-2013-01-480129
- Hadrup S, Donia M, Thor Straten P. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron 2013; 6(2):123-33; PMID: 23242673; https://doi.org/10.1007/s12307-012-0127-6
- Schulz S, Mann A, Novakhov B, Piggins HD, Lupp A. VPAC2 receptor expression in human normal and neoplastic tissues: evaluation of the novel MAB SP235. Endocr Connect 2015; 4(1):18-26; PMID: 25504760; https://doi.org/10.1530/EC-14-0051
- Dorsam GP, Benton K, Failing J, Batra S. Vasoactive intestinal peptide signaling axis in human leukemia. World J Biol Chem, 2011; 2(6):146-60; PMID: 21765981; https://doi.org/10.4331/wjbc.v2.i6.146
- Moody TW, Zia F, Draoui M, Brenneman DE, Fridkin M, Davidson A, Gozes I. A vasoactive intestinal peptide antagonist inhibits non-small cell lung cancer growth. Proc Natl Acad Sci U S A 1993; 90(10):4345-9; PMID: 8389448; https://doi.org/10.1073/pnas.90.10.4345
- Sharma A, Walters J, Gozes Y, Fridkin M, Brenneman D, Gozes I, Moody TW. A vasoactive intestinal peptide antagonist inhibits the growth of glioblastoma cells. J Mol Neurosci 2001; 17(3):331-9; PMID: 11859929; https://doi.org/10.1385/JMN:17:3:331
- Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J Immunol 2010; 185(11):6413-9; PMID: 21084670; https://doi.org/10.4049/jimmunol.1001829
- Parry GC, Mackman N. Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription. J Immunol 1997; 159(11):5450-6. PMID: 9548485; https://doi.org/10.1074/jbc.271.34.20828
- Ollivier V, Parry GC, Cobb RR, de Prost D, Mackman N. Elevated cyclic AMP inhibits NF-kappaB-mediated transcription in human monocytic cells and endothelial cells. J Biol Chem 1996; 271(34):20828-35; PMID: 8702838; https://doi.org/10.1074/jbc.271.34.20828
- Martinez C, Delgado M, Abad C, Gomariz RP, Ganea D, Leceta J. Regulation of VIP production and secretion by murine lymphocytes. J Neuroimmunol 1999; 93(1-2):126-38; PMID: 10378876; https://doi.org/10.1016/S0165-5728(98)00216-1
- Kong Y, Zhang J, Claxton DF, Ehmann WC, Rybka WB, Zhu L, Zeng H, Schell TD, Zheng H. PD-1(hi)TIM-3(+) T cells associate with and predict leukemia relapse in AML patients post allogeneic stem cell transplantation. Blood Cancer J 2015; 5:e330; PMID: 26230954; https://doi.org/10.1038/bcj.2015.58
- Sehgal A, Whiteside TL, Boyiadzis M, Programmed death-1 checkpoint blockade in acute myeloid leukemia. Expert Opin Biol Ther 2015; 15(8):1191-203; PMID: 26036819; https://doi.org/10.1517/14712598.2015.1051028
- Krupka C, Kufer P, Kischel R, Zugmaier G, Lichtenegger FS, Köhnke T, Vick B, Jeremias I, Metzeler KH, Altmann T et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: reversing a T-cell-induced immune escape mechanism. Leukemia 2016; 30(2):484-91; PMID: 26239198; https://doi.org/10.1038/leu.2015.214
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006; 439(7077):682-7; PMID: 16382236; https://doi.org/10.1038/nature04444
- Mittal D, Young A, Stannard K, Yong M, Teng MW, Allard B, Stagg J, Smyth MJ. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res 2014; 74(14):3652-8; PMID: 24986517; https://doi.org/10.1158/0008-5472.CAN-14-0957