
ABSTRACT
Platelets promote metastasis, among others by coating cancer cells traveling through the blood, which results in protection from NK cell immune-surveillance. The underlying mechanisms, however, remain to be fully elucidated. Here we report that platelet-coating reduces surface expression of NKG2D ligands, in particular MICA and MICB, on tumor cells, which was mirrored by enhanced release of their soluble ectodomains. Similar results were obtained upon exposure of tumor cells to platelet-releasate and can be attributed to the sheddases ADAM10 and ADAM17 that are detectable on the platelet surface and in releasate following activation and at higher levels on platelets of patients with metastasized lung cancer compared with healthy controls. Platelet-mediated NKG2DL-shedding in turn resulted in impaired “induced self” recognition by NK cells as revealed by diminished NKG2D-dependent lysis of tumor cells. Our results indicate that platelet-mediated NKG2DL-shedding may be involved in immune-evasion of (metastasizing) tumor cells from NK cell reactivity.
Introduction
NK cell activation is guided by the principles of “missing self” and “induced self” recognition, implying that NK cells kill target cells with low/absent expression of MHC class I (missing self) and/or expression of ligands for activating NK receptors (induced self).Citation1-3 The C-type lectin-like NKG2D receptor is a prototypical activating immunoreceptor that is crucial for the ability of NK cells to recognize their target cells and exert effector functions.Citation4 Its ligands (NKG2DL) are induced upon cellular stress including malignant transformation and comprise, in humans, the 2 MIC molecules A and B (MICA, MICB) and ULBP1–6. Notably, NKG2DL surface expression levels on a given tumor cell directly correlate with the extent of NK cell antitumor reactivity.Citation5,6 We and others described that surface-expressed NKG2DL can be cleaved, resulting in reduced surface expression and enhanced levels of the soluble ectodomains.Citation7-9 While various mechanisms have been implied in NKG2DL shedding and release, “a disintegrin and metalloproteinase domain-containing protein” (ADAM) 10 and ADAM17 appear to play a prominent role for this mechanism of tumor immune-evasion, as was reported particularly for MICA, MICB and ULBP2.Citation10-13 Notably, expression of ADAM10/17 has also been described for multiple other cell types which may influence NK cell immune-surveillance of tumor cells, and this includes platelets.Citation14
It is meanwhile well established that platelets play an important role in tumor dissemination. Among others, platelets mediate endothelial adhesion and transmigration, provide growth factors/chemokines and facilitate epithelial to mesenchymal transition of tumor cells.Citation15,16 Moreover, there is long-standing evidence that platelets also influence the interaction of tumor cells with the immune system in general and evasion from NK cell immune-surveillance in particular:Citation17 Among others, it was shown that metastasis formation is inhibited in the absence of platelets,Citation18,19 and additional depletion of NK cells reverts this anti-metastatic effect of thrombocytopenia,Citation20 indicating that platelets protect (metastasizing) tumor cells from NK cell reactivity. Upon entering the blood stream, metastasizing tumor cells are rapidly surrounded – i.e. coated – by platelets,Citation15 which initially was assumed to provide a mechanical shielding from NK cell attack.Citation20 Meanwhile, evidence for specific molecular mechanisms by which platelets influence tumor-NK cell interaction is accumulating.Citation15 This comprises, among others, weakening of “missing self” and “induced self” NK cell recognition by conferment of healthy, non-malignant MHC class I and downregulation of NKG2D by platelet-derived TGF-β, respectively.Citation21,22
In this study we provide evidence for a further mechanism by which platelets may impair “induced self” recognition of tumor cells: exposure to platelets and platelet-releasate, which express and contain the sheddases ADAM10 and ADAM17, respectively, reduced surface expression of NKG2DL, enhanced their release in soluble form and ultimately impaired NKG2D-mediated NK cell immune-surveillance. Together with our observation that platelets of lung cancer patients express significantly higher ADAM10/17 levels than healthy controls, these data provide clear evidence that platelet-mediated NKG2DL-shedding facilitates immune-evasion of (metastasizing) tumor cells from NK cell reactivity.
Results and discussion
Platelet-coating mediates NKG2DL shedding from the tumor cell surface, resulting in impaired NK cytotoxicity
As a first step we investigated whether the interaction with platelets influences the expression of the NKG2DL MICA, MICB and ULBP1–3, for which antibodies with validated specificity were available, on tumor cells. To this end, we used 2 colon (HCT-116, COLO-678) and 2 breast (MDA-MB-231, T-47D) cancer cell lines and cultured them in the presence or absence of freshly isolated platelets of healthy donors, which resulted in platelet-coating of the tumor cells as described previously by us and others (data not shown).Citation15,21-23 Subsequent FACS analysis revealed that platelet-coating caused a substantial reduction of NKG2DL surface expression, in particular of MICA and MICB (). Effects appeared to be dependent on levels of constitutive NKG2DL expression on a given tumor cell line and are in agreement with observations that NKG2DL expression patterns and release vary widely among different tumor cell types and cell lines.Citation8,24 Expression of EGFR or CD133 as control antigens remained unaltered, which excluded that platelet-coating technically interfered with flow cytometric analysis. Reduction of NKG2DL surface expression was paralleled by an increase of their soluble ectodomains in culture supernatants, which pointed toward platelet-induced shedding as underlying mechanism (). Alterations of soluble NKG2DL levels appeared to be more pronounced than modulation of surface levels. Besides methodological differences between FACS and ELISA that certainly play a role, tumor cell-inherent mechanisms, e.g. NKG2DL upregulation upon altered proliferation could (partially) have compensated for increased shedding.Citation25,26 Notably, besides low levels of ULBP1 and ULBP3, no relevant release of soluble NKG2DL was observed by platelets alone. Also for ULBP1 and ULBP3, the levels detectable with platelets alone were clearly lower than with tumor cells alone, and more than additive levels were detected upon tumor-platelet interaction (Supplementary Figure 1A). Surprisingly, for ULBP2, a decrease of soluble protein detectable by ELISA following platelet-coating was observed. A potential explanation is provided by our finding that the detectable levels of recombinant ULBP2 (but not ULBP1 or ULBP3) in our ELISA were reduced in the presence of platelet-derived factors (Supplementary Figure 1B), suggesting that platelets may contain proteases that degrade ULBP2 but not other ULBP molecules. The nature of these putative proteases was not elucidated in our study, and it remains unclear whether their activity merely technically prevents detection of the true amount of released protein or directly affects the levels of surface protein. The latter would potentially constitute a yet unknown mechanism among the various factors like microRNAs, polyubiquitination and vesicle-mediated excretion that reportedly influence expression of NKG2DL on tumor cells and is presently under study.Citation9
Figure 1. Platelet-induced shedding of NKG2DL impairs NK antitumor reactivity. The indicated tumor cells were cultured for 48 h alone or in the presence of platelets (ratio tumor cells/platelets of 1:150) to enable coating. After washing to remove surplus platelets and soluble factors, (A) surface expression of MICA, MICB, ULBP1–3 and EGFR/CD133 as control were analyzed by flow cytometry. Open peaks, tumor cells alone; shaded peaks, platelet-coated tumor cells; dotted lines, isotype controls. (B) Levels of sNKG2DL in supernatants were analyzed by ELISA. Statistically significantly different results are indicated by #. (C) Tumor cells were incubated with pNKC in the presence or absence of anti-NKG2D F(ab´)2 fragments or isotype control (both 5 µg/ml). Target cell lysis was determined by 4 h chromium release assays. Results obtained at the indicated E:T ratios (left) and the calculated reduction of lysis (δ) by NKG2D blockade (at the lowest E:T ratio, right) are displayed. Representative data of one experiment from a total of at least 4 with similar results are shown.
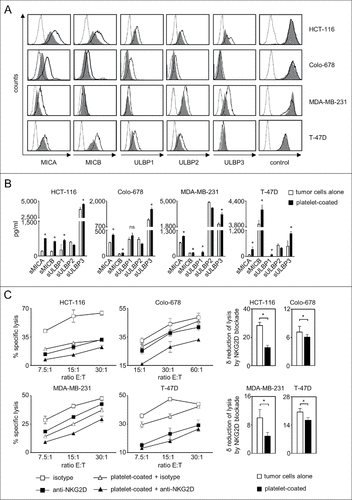
Next we determined whether altered NKG2DL expression translated in impaired NK cell reactivity. Lysis of tumor cells that had been cultured in the presence of platelets by polyclonal NK cells (pNKC) was significantly (except T-47D at the E:T ratio of 30:1 in the depicted exemplary experiment) reduced with all used tumor cell lines. Addition of blocking NKG2D F(ab)´2 fragments always caused a significant reduction of cytotoxicity, and lysis rates of tumor cells pretreated with platelets were always lower than that with untreated tumor cells upon NKG2D blockade. This indicates that other platelet-derived factors beyond those affecting the NKG2D-NKG2DL system like MHC class I or GITRL, which are transferred from platelets to tumor cells, contribute to impairment of NK cell reactivity.Citation17 However, our finding that the extent (δ), by which NKG2D blockade reduced lysis of platelet-exposed tumor cells was significantly lower compared with that observed with untreated tumor cells underlines the specific functional relevance of reduced NKG2DL expression ().
Platelets express and release ADAM10 and ADAM17 which reportedly act as sheddases of MICA and MICB
Next we set out to determine whether and how ADAM10 and ADAM17 as major mediators of NKG2DL cleavage are expressed/released by platelets. Analyses in a model of megakaryopoiesis revealed a profound upregulation of ADAM10 mRNA, while ADAM17 mRNA was present but rather downregulated in the course of megakaryocytic differentiation (). In line, FACS analysis using previously characterized antibodies with proven specificityCitation10 revealed high ADAM10 expression on the platelet surface with ADAM17 being expressed at lower levels. Surprisingly and in contrast to many other immunomodulatory molecules that are activation-dependently expressed by platelets,Citation23,27 no clear association of ADAM10/17 levels with activation state was observed upon stimulation of platelets with the classical agonists ADP, collagen and thrombin (). This was confirmed by results obtained in Western blot analyses. Analysis of equal amounts of protein (20 µg each) revealed substantial levels of both sheddases already in resting platelets. After 5 minutes of activation, the amount of ADAM10/17 detectable in lysate of platelet pellets containing equal amounts of protein was found to be reduced, while in turn a smaller band corresponding to a soluble form was detectable in the respective platelet-releasate (). These results not only confirm the presence of ADAM10 and ADAM17 in platelets, but also indicate that the sheddases are released upon activation, which is in line and extends available data regarding the expression and release of these 2 proteases by platelets.Citation28-31 FACS analysis of tumor cells and platelets alone and following their interaction revealed higher ADAM10/17 levels on tumor cells after platelet-coating (Supplementary Figure 2A). Notably, analyses of ADAM10 and ADAM17 mRNA levels in tumor cells did not reveal an induction of the sheddases in the tumor cells themselves upon exposure to releasate or platelet-coating (Supplementary Figure 2B).
Figure 2. ADAM10 and ADAM17 expression in thrombopoietic cells. (A) CD34+ haematopoietic progenitor cells were cultured for 12 d in the presence of thrombopoietin to induce megakaryocyte differentiation. On days 0, 6, 9, and 12 mRNA expression of ADAM10 and ADAM17 was analyzed by qPCR. Representative data of one from a total of 3 biologic experiments performed in technical replicates are shown. (B) Platelets from healthy donors were either left untreated (resting) or activated with ADP, collagen or thrombin until aggregation was visible (for a maximum of 30 s to prevent clumping) as described previously.Citation23 Then FACS analysis of ADAM10 or ADAM17 expression was performed. (C) Platelets were either left untreated (resting) or activated by exposure to the platelet superagonist TRAP-6 (5 µM) for 5 minutes. Then ADAM10 and ADAM17 levels in platelet pellets and releasate was analyzed by Western blot. In (B-C), representative data of one experiment from a total of at least 4 with similar results are shown. (D) Resting platelets from healthy donors and patients with NSCLC were analyzed by FACS as described in (B). Statistically significant differences (p<0.05, Kolmogorov-Smirnov test) are indicated by #.
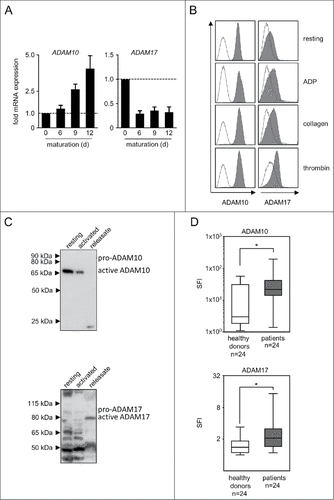
Platelets in general and also metalloproteases like ADAM10/17 in particular influence cancer biology by multiple mechanisms far beyond NKG2DL shedding.Citation15,32 Considering that platelets from patients with malignant disease were reported to display an altered phenotype compared with platelets from healthy donors,Citation33 we comparatively studied whether ADAM10 and ADAM17 are differentially expressed in platelets of patients with metastasized non-small cell lung cancer (NSCLC) and healthy donors. NSCLC cells, alike a broad range of other tumor entities, reportedly express NKG2DL,Citation24 and we found substantial NKG2DL surface expression in a panel of 8 lung cancer cell lines (Supplementary Figure 3). FACS analysis of platelets from NSCLC patients displayed significantly (both p<0.05, Kolmogorov-Smirnov test) higher levels of both ADAM10 and ADAM17 compared with healthy controls (). Notably, these data do not provide direct evidence for the pathophysiological relevance of platelet-derived sheddases in general or for NKG2DL shedding and evasion from NK immune-surveillance in particular. Nevertheless, these observations may serve to put our findings into a greater perspective. Overall and specifically regarding the role of ADAM10/17 in NKG2DL shedding it remains to be investigated whether tumors simply take advantage of the hosts' platelets, exploiting their ability to induce local immunosuppression and chaperoning tumor cells during blood borne spread to future metastatic sites, or whether the malignant platelet phenotype is a result of reprogrammed megakaryopoiesis.Citation33 However, both concepts may reflect mechanisms of tumor-host-interaction, and a quantitative influence of cancer on megakaryopoiesis is evidenced by paraneoplastic thrombocytosis that is observed in many cancer patients.Citation34 This influence may also explain differences in cytokine and chemokine content, which reportedly differ in healthy platelets and platelets from tumor patients.Citation33 Notably, a substantial inter-patient variability was observed in our analysis of platelet ADAM expression, which could indicate that the “malignant phenotype” of platelets in individual patients may mirror specific disease characteristics. However, if and how tumors truly edit host megakaryopoiesis in a qualitative way and whether this also holds true for ADAM molecules remains to be determined in future studies.
Platelet-releasate mediates shedding of NKG2DL thereby impairing NK cytotoxicity
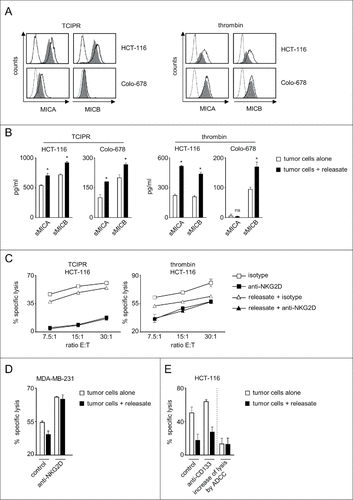
As the many soluble factors released by platelets upon activation largely influence cancer pathophysiology and we had detected sheddases that reportedly affect NKG2DL molecule expression in platelet-releasate,Citation10,11 we set out to determine whether also releasate alone influenced tumor cell NKG2DL expression and the associated NK cell reactivity. To this end we exposed tumor cells to platelet-releasate that was either obtained by incubation with tumor cells (tumor cell-induced platelet-releasate, TCIPR) or upon activation with thrombin. These analyses were focused on MIC molecules, as our antecedent analyses had revealed pronounced effects particularly with this NKG2DL subgroup for the cell lines used in our study (). FACS analysis confirmed that also releasate alone caused a profound downregulation of MICA and MICB expression on the tumor cell surface, which was mirrored by enhanced release of the soluble ectodomains (, ).
Figure 3. Platelet-releasate mediates shedding of NKG2DL and impairs NK cytotoxicity. The indicated tumor cells were cultured for 48 h alone or in the presence of thrombin-induced releasate or TCIPR generated by activation of platelets with the same tumor cells that were subsequently treated with the respective releasate. Then supernatants were collected and tumor cells were washed to remove surplus platelets and soluble factors. Subsequently (A) surface expression of MICA and MICB on tumor cells was analyzed by flow cytometry. Open peaks, tumor cells alone; shaded peaks, tumor cells cultured with releasate; dotted lines, isotype controls; (B) levels of sMICA and sMICB in supernatants were analyzed by ELISA. Statistically significantly different results are indicated by #. (C) The indicated tumor cells were incubated with pNKC in the presence or absence of anti-NKG2D F(ab´)2 fragments or isotype control (both 5 µg/ml). Target cell lysis was determined by 4 h chromium release assays. (D) NK cell lysis of tumor cells (E:T ratio 3.75:1) was analyzed in the presence of absence of an agonistic NKG2D antibody or isotype control (both at 10 µg/ml) as described in C. (E) NK lysis of tumor cells in the presence or absence of an Fc-optimized CD133 antibody or isotype control (10 µg/ml each) was determined as described in C. Results obtained at an E:T ratio 7.5:1 and the calculated increase of lysis as induced by ADCC are shown. Representative data of one experiment from a total of at least 4 with similar results are shown.
Inhibition of ADAM10 and ADAM17 during culture of tumor cells alone or upon exposure to releasate clearly reduced NKG2DL release, and clearly more pronounced effects were observed with a broad spectrum matrix metalloproteinase inhibitor (MPI) as compared with inhibition of ADAM10 or ADAM17 alone. The finding that the amount of soluble MIC molecules detectable in the presence and absence of releasate declined to a comparable level provides further evidence for the contribution of metalloprotease activity to NKG2DL release upon tumor-platelet interaction (Supplementary Figure 2C).
To elucidate the functional relevance of altered NKG2DL expression we again used a NKG2D blocking approach in functional experiments. Cytotoxicity assays revealed significantly (with TCIPR and thrombin releasate) lower pNKC lysis rates when tumor cells had been pretreated with releasate. Notably, using releasate in contrast to platelet-coated tumor cells excluded an influence of physical shielding as well as membrane-bound platelet factors. The latter may comprise the aforementioned MHC class I and GITRL, but also others like CD40 ligand and TWEAK, which have been identified as factors by which platelets influence host-immune interaction.Citation27,35 In this setting, blocking NKG2D again significantly (with both releasates at all E:T ratios) reduced lysis rates of both, tumor cells pretreated with releasate and untreated tumor cells, but in contrast to the analyses shown in comparable levels were now reached with naïve and releasate-exposed tumor cells (). One may interprete these results in a way that the partial reduction of NKG2DL surface levels caused by exposure to releasate is mirrored by a corresponding reduction of lysis rates. Blocking the contribution of NKG2D to NK reactivity completely results in even lower lysis rates. These are then similar with untreated and releasate-treated tumor cells, as differences in NKG2DL expression caused by platelet-induced reduction of NKG2DL surface levels no longer account.
Notably, exposure of tumor cells to platelet-releasate can affect NK reactivity by multiple mechanisms. We thus aimed to obtain further support for the specific functional relevance of altered expression of NKG2DL on tumor cells in our experimental system. This could ideally be achieved by using a completely NKG2DL-negative cancer cell line as target, with which no effect of platelet-releasate on (NKG2D-mediated) NK reactivity should occur. However, we were not able to identify a NKG2DL-negative cell line among a total of > 30 different cell lines (, Supplementary Figure 3 and data not shown). Therefore we used an approach in which NK cell lysis of tumor cells pretreated or not with releasate was analyzed in the presence of an agonistic NKG2D antibody. While releasate caused the expected statistically significant reduction of lysis compared with untreated tumor cells in the presence of an isotype control antibody, NKG2DL-independent maximal triggering of NKG2D by the agonistic antibody profoundly increased lysis rates and abrogated the significant difference caused by releasate (). Further evidence that the effect of platelet-releasate is in fact, at least in major part, due to reduction of NKG2DL levels was provided by the observation that pretreatment with releasate, while reducing constitutive NK reactivity, did not significantly affect the increase of lysis caused by induction of antibody dependent cellular cytotoxicity (ADCC) upon treatment of cancer cells with a Fc-optimized CD133 antibody that triggers CD16 as an independent NK cell receptor ().Citation36 Together, the observations that full stimulation of NKG2D abrogates the effect of releasate and that induction of NK cell reactivity by an independent NK cell receptor was not affected by releasate in our experimental system, provide clear evidence that the effects of releasate were mediated essentially via altered NKG2DL expression. They also exclude potential effects that might be caused by transfer of immunoregulatory molecules e.g., via platelet-derived exosomes.
Notably, tumor cells underwent several washing steps following exposure to releasate (and also after platelet-coating ()) before use in the functional assays shown in . Thereby, potential effects of TGF-β which is contained in platelet-releasate and can downregulate NKG2D expression on NK cells,Citation21 and also other factors which might directly affect NK cell reactivity were excluded in our experimental system. Moreover, when tumor cells, NK cells or both were exposed to medium generated in a manner to mirror the setting of our functional analyses, no alterations of the expression of NKG2DL (tumor cells), NKG2D (NK cells) or reduction of lysis rates were observed (Supplementary Figures 4+5). TGF-β not only causes downregulation of NKG2D expression on NK cells, which we identified as a mechanism by which platelets affect NK immune-surveillance; others reported that this cytokine also can downregulate NKG2DL expression.Citation13,37 Other soluble factors secreted by platelets like CCL5/7 that could influence NK cell reactivity independently of tumors cells and/or the NKG2D molecule system may also play a role.Citation38 Thus, the experimental system used in these experiments is particularly suited to demonstrate the relevance of altered NKG2DL expression, thereby substantiating our notion that dampening of NK reactivity after tumor-platelet interaction is caused by impaired NKG2D-mediated immune-surveillance.
While our study provides evidence that platelets can impair NK cell antitumor reactivity by mediating NKG2DL shedding, it should be noted that direct proof for the involvement of ADAM10/17 is not provided by our study. Efforts like depletion-experiments using immunoprecipitation of releasate with ADAM antibodies were not successful for technical reasons. Thus, it remains somewhat unclear to what extent sheddases directly contained in the releasate and/or other factors like platelet-derived TGF-β that reportedly induces sheddases which in turn affect NKG2DL expression, contributed to the observed effects. In addition, we did not study whether and how elevated levels of soluble MIC molecules directly affect NK reactivity. Even if direct effects of soluble NKG2DL are excluded in our study due to the chosen experimental conditions, this issue is of importance as recently the general notion that soluble NKG2DL suppress NK reactivity was challenged by a report of Raulet and colleagues in a mouse study where soluble MULT1 activated NK cells.Citation39 It will be important to determine whether this also holds true for human NKG2DL, in particular as the latter reportedly display lower affinities for NKG2D compared with MULT1.Citation9
While these questions require further elucidation, our results unravel shedding of NKG2DL as a novel mechanism beyond downregulation of NKG2D expression on NK cells, by which platelets can systemically impair NKG2D-mediated immune-surveillance. Overall, our data thereby provide further insight into the molecular mechanisms underlying the prometastatic effects of tumor-platelet-NK cell interaction.
Material and methods
Reagents
The mAb AMO1 (anti-MICA), BMO1 (anti-MICB), AUMO3 (anti-ULBP1), BUMO-1 (anti-ULBP2), CUMO3 (anti-ULBP3), 6H7 (anti-NKG2D), MAB139 (anti-NKG2D) and the respective F(ab´)2 fragments, were described previously.Citation40,41 ADAM10 and ADAM17 ectodomain antibodies for FACS analysis with confirmed specificityCitation10 were from R&D (clone 163003 and 111633, respectively), blocking anti-ADAM17 (clone D1(A12)) was from AdipoGen. Anti-EGFR (cetuximab) was from Bristol Myers Squibb and anti-CD133 was generated as described previously.Citation36 CD41a-PeCy5 was from Becton Dickinson. Species-specific PE-conjugates were from Jackson ImmunoResearch. Thrombin was from Sigma, ADP was from Chrono-Log, collagen was from Mascia Brunelli. TRAP-6 was from Tocris. GI254023X was from Tocris and BB-94 was from Calbiochem.
Cell lines
HCT-116, COLO-678, MDA-MB-231 and T-47D cells were from German Collection of Microorganisms and Cell Cultures. A549, HOP-62, HOP-92, EKVX, NCI-H226, NCI-H522, NCI-H23 and NCI-H322M lung carcinoma cell lines were from the NCI-DCTD repository and internally provided from the Sub-repository of the Eberhard Karls University Tuebingen. Authenticity was determined by validating the respective immunophenotype described by the provider using FACS every 6 month and specifically before use in experiments.
Preparation of NK cells, megakaryocytes and platelets
Polyclonal NK cells were generated from non-plastic-adherent PBMC as described previously.Citation36 Functional experiments were performed when purity was above 90%. Maturation of megakaryocytes from CD34+ cells of healthy donors using thrombopoietin (50 ng/ml) was performed as described previously.Citation23 Platelets from healthy volunteers were obtained and activated using 0.1IU/mL thrombin, 2.5 µM ADP, 5 µM TRAP-6, 10μg/ml collagen or 3 × 105 tumor cells/mL with 5% platelet-poor plasma (TCIPR) as described previously.Citation21 The respective supernatants were used as platelet-releasate.
Treatment of tumor cells with platelets
Tumor cells were coated with platelets as described previously with slight modifications.Citation16,22 In brief, tumor cells were incubated with washed platelets at a total of 150,000 platelets/μl at a tumor cell-platelet ratio of 1:150 or equivalent volumes of releasate were added. For further investigation, tumor cells were washed afterwards to remove surplus platelets and soluble factors.
Patients
Blood samples from patients with metastasized non-small cell lung cancer were obtained after written informed consent in accordance with the Helsinki protocol, and the study was performed according to the guidelines of the local Ethics Committee. Patients comprised 9 men and 15 women (male-female ratio 0.6); the median age was 61 y with a range from 52 to 83 y.
Determination of soluble NKG2DL levels
Levels of sMICA, sMICB, sULBP1, sULBP2 and sULBP3 in culture supernatants were determined by ELISA as described previously.Citation7,40,42 All indicated concentrations represent means of triplicate measurements.
Flow cytometry
Flow cytometry was performed using fluorescence-conjugates or specific mAb (10 µg/ml) and their controls followed by species-specific (1:100) PE-conjugate using a FC500 flow cytometer (Beckman Coulter) or a LSRFortessa (Becton Dickinson). Specific fluorescence indices (SFI) were calculated by dividing median fluorescences obtained with specific mAb by median fluorescences obtained with control.
Cytotoxicity assay
Tumor cell lysis was analyzed by chromium release assays as described previously.Citation43 Percentage of lysis was calculated as follows: 100 × (experimental release − spontaneous release) / (maximum release − spontaneous release).
Quantitative PCR
Total RNA was isolated using the High Pure RNA Isolation Kit and transcribed into cDNA using Transcriptor First Strand cDNA Synthesis Kit (both Roche) according to manufacturer's instructions. Amplification was performed using qPCR Mastermix Plus for SYBR Green I (Eurogentec) on a LightCycler 480 instrument. The following primer sets were used: GAPDH (5′-GAG TCA ACG GAT TTG GTC GT-3′; 5′-TTG ATT TTG GAG GGA TCT CG-5′), and ADAM10 as well as ADAM17 kits (QuantiTect primer assay, Qiagen). Expression levels relative to GAPDH were calculated using the ΔΔCT method.
Western blot
Protein from platelets was isolated with cell lysis buffer (New England Biolabs), and protein concentration was determined by a Bradford assay. Equal amounts of protein (20 μg) from each sample were resolved on a precast 4–12% NuPAGE gel and transferred on a polyvinylidene difluoride membrane (GE Healthcare). Membrane was blocked for 1 h at room temperature with Roti-Block, followed by overnight incubation at 4°C with anti-ADAM10 or anti-ADAM17 antibody (1:1000 each; Cell Signaling Technologies and Triple Point Biologics, respectively). After 1 h incubation with HRP-conjugated anti-rabbit antibody (1:5000; DAKO) the proteins were detected by using ECL reagents (GE Healthcare).
Statistics
If not indicated otherwise, values are depicting means of technical triplicates with standard deviation, and paired t-test was used to calculate statistically significant differences between groups. A p-value of < 0.05 was considered statistically significant.
Abbreviations
| ADAM | = | a disintegrin and metalloproteinase domain-containing protein |
| ADCC | = | antibody dependent cellular cytotoxicity |
| MPI | = | matrix metalloproteinase inhibitor |
| NK | = | natural killer |
| NKG2D | = | natural-killer group 2 member D |
| NKG2DL | = | NKG2D ligands |
| NSCLC | = | non-small cell lung cancer |
| SFI | = | specific fluorescence intensity |
| sMICA/B | = | soluble MICA/B |
| sNKG2DL | = | soluble NKG2DL |
| sULBP | = | soluble ULBP |
| pNKC | = | polyclonal NK cells |
| TCIPR | = | tumor cell-induced platelet-releasate |
Disclosure of potential conflicts of interest
No authors have any proprietary interest in this report.
supplimental_data.zip
Download Zip (1.2 MB)Acknowledgments
The authors thank Ilona Hagelstein, Stefanie Mueller and Elke Malenke for excellent technical assistance and Ulrich Lauer for providing tumor cells from the Sub-Repository of the Eberhard Karls University.
Funding
This work was supported by DFG (SA1360/7–3, SFB685-A07), and Deutsche Krebshilfe (111828, 111134).
References
- Gasser S, Raulet DH. Activation and self-tolerance of natural killer cells. Immunol Rev. 2006;214:130-42. doi:10.1111/j.1600-065X.2006.00460.x. PMID:17100881
- Caligiuri MA. Human natural killer cells. Blood. 2008;112:461-9. doi:10.1182/blood-2007-09-077438. PMID:18650461
- Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell. 2010;142:847-56. doi:10.1016/j.cell.2010.08.031. PMID:20850008
- Ullrich E, Koch J, Cerwenka A, Steinle A. New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunology. 2013;2:e26097. doi:10.4161/onci.26097. PMID:24353908
- Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci U S A. 2001;98:11521-6. doi:10.1073/pnas.201238598. PMID:11562472
- Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413:165-71. doi:10.1038/35093109. PMID:11557981
- Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol. 2002;169:4098-102. doi:10.4049/jimmunol.169.8.4098. PMID:12370336
- Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419:734-8. doi:10.1038/nature01112. PMID:12384702
- Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31:413-41. doi:10.1146/annurev-immunol-032712-095951. PMID:23298206
- Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, Stevanovic S, Rammensee HG, Steinle A. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 2008;68:6368-76. doi:10.1158/0008-5472.CAN-07-6768. PMID:18676862
- Boutet P, Aguera-Gonzalez S, Atkinson S, Pennington CJ, Edwards DR, Murphy G, Reyburn HT, Vales-Gomez M. Cutting edge: the metalloproteinase ADAM17/TNF-alpha-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein. J Immunol. 2009;182:49-53. doi:10.4049/jimmunol.182.1.49. PMID:19109134
- Chitadze G, Lettau M, Bhat J, Wesch D, Steinle A, Furst D, Mytilineos J, Kalthoff H, Janssen O, Oberg HH, et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int J Cancer. 2013;133:1557-66. doi:10.1002/ijc.28174. PMID:23526433
- Wolpert F, Tritschler I, Steinle A, Weller M, Eisele G. A disintegrin and metalloproteinases 10 and 17 modulate the immunogenicity of glioblastoma-initiating cells. Neuro Oncol. 2014;16:382-91. doi:10.1093/neuonc/not232. PMID:24327582
- Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6:32-43. doi:10.1038/nrm1548. PMID:15688065
- Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11:123-34. doi:10.1038/nrc3004. PMID:21258396
- Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20:576-90. doi:10.1016/j.ccr.2011.09.009. PMID:22094253
- Placke T, Kopp HG, Salih HR. Modulation of natural killer cell anti-tumor reactivity by platelets. J Innate Immun. 2011;3:374-82. doi:10.1159/000323936. PMID:21411974
- Gasic GJ, Gasic TB, Stewart CC. Antimetastatic effects associated with platelet reduction. Proc Natl Acad Sci U S A 1968;61:46-52. doi:10.1073/pnas.61.1.46. PMID:5246932
- Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood. 2004;104:397-401. doi:10.1182/blood-2004-02-0434. PMID:15031212
- Nieswandt B, Hafner M, Echtenacher B, Mannel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res 1999;59:1295-300. PMID:10096562
- Kopp HG, Placke T, Salih HR. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009;69:7775-83. doi:10.1158/0008-5472.CAN-09-2123. PMID:19738039
- Placke T, Orgel M, Schaller M, Jung G, Rammensee HG, Kopp HG, Salih HR. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012;72:440-8. doi:10.1158/0008-5472.CAN-11-1872. PMID:22127925
- Placke T, Salih HR, Kopp HG. GITR ligand provided by thrombopoietic cells inhibits NK cell antitumor activity. J Immunol. 2012;189:154-60. doi:10.4049/jimmunol.1103194. PMID:22649191
- Nausch N, Cerwenka A. NKG2D ligands in tumor immunity. Oncogene. 2008;27:5944-58. doi:10.1038/onc.2008.272. PMID:18836475
- Huergo-Zapico L, Acebes-Huerta A, Lopez-Soto A, Villa-Alvarez M, Gonzalez-Rodriguez AP, Gonzalez S. Molecular Bases for the Regulation of NKG2D Ligands in Cancer. Front Immunol. 2014;5:106. doi:10.3389/fimmu.2014.00106. PMID:24711808
- Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. J Exp Med. 2012;209:2409-22. doi:10.1084/jem.20120565. PMID:23166357
- Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998;391:591-4. doi:10.1038/35393. PMID:9468137
- Colciaghi F, Marcello E, Borroni B, Zimmermann M, Caltagirone C, Cattabeni F, Padovani A, Di Luca M. Platelet APP, ADAM 10 and BACE alterations in the early stages of Alzheimer disease. Neurology. 2004;62:498-501. doi:10.1212/01.WNL.0000106953.49802.9C. PMID:14872043
- Bergmeier W, Piffath CL, Cheng G, Dole VS, Zhang Y, von Andrian UH, Wagner DD. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates GPIbalpha shedding from platelets in vitro and in vivo. Circ Res. 2004;95:677-83. doi:10.1161/01.RES.0000143899.73453.11. PMID:15345652
- Fong KP, Barry C, Tran AN, Traxler EA, Wannemacher KM, Tang HY, Speicher KD, Blair IA, Speicher DW, Grosser T, et al. Deciphering the human platelet sheddome. Blood. 2011;117:e15−26. doi:10.1182/blood-2010-05-283838. PMID:20962327
- Colciaghi F, Borroni B, Pastorino L, Marcello E, Zimmermann M, Cattabeni F, Padovani A, Di Luca M. [alpha]-Secretase ADAM10 as well as [alpha]APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol Med. 2002;8:67-74. PMID: 12080182. PMID:12080182
- Saftig P, Reiss K. The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: novel drug targets with therapeutic potential? Eur J Cell Biol. 2011;90:527-35. doi:10.1016/j.ejcb.2010.11.005. PMID:21194787
- Wiesner T, Bugl S, Mayer F, Hartmann JT, Kopp HG. Differential changes in platelet VEGF, Tsp, CXCL12, and CXCL4 in patients with metastatic cancer. Clin Exp Metastasis. 2010;27:141-9. doi:10.1007/s10585-010-9311-6. PMID:20182908
- Lin RJ, Afshar-Kharghan V, Schafer AI. Paraneoplastic thrombocytosis: the secrets of tumor self-promotion. Blood. 2014;124:184-7. doi:10.1182/blood-2014-03-562538. PMID:24868077
- Meyer T, Amaya M, Desai H, Robles-Carrillo L, Hatfield M, Francis JL, Amirkhosravi A. Human platelets contain and release TWEAK. Platelets. 2010;21:571-4. doi:10.3109/09537104.2010.512403. PMID:20849210
- Koerner SP, Andre MC, Leibold JS, Kousis PC, Kubler A, Pal M, Haen SP, Buhring HJ, Grosse-Hovest L, Jung G, et al. An Fc-optimized CD133 antibody for induction of NK cell reactivity against myeloid leukemia. Leukemia. 2017;31:459-69. doi:10.1038/leu.2016.194. PMID:27435001
- Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, Weller M, Friese MA. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain. 2006;129:2416-25. doi:10.1093/brain/awl205. PMID:16891318
- Weyrich AS, Zimmerman GA. Platelets: signaling cells in the immune continuum. Trends Immunol. 2004;25:489-95. doi:10.1016/j.it.2004.07.003. PMID:15324742
- Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, Xu J, Rovis TL, Xiong N, Raulet DH. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science. 2015;348:136-9. doi:10.1126/science.1258867
- Hilpert J, Grosse-Hovest L, Grunebach F, Buechele C, Nuebling T, Raum T, Steinle A, Salih HR. Comprehensive analysis of NKG2D ligand expression and release in leukemia: implications for NKG2D-mediated NK cell responses. J Immunol. 2012;189:1360-71. doi:10.4049/jimmunol.1200796. PMID:22730533
- Salih HR, Goehlsdorf D, Steinle A. Release of MICB molecules by tumor cells: mechanism and soluble MICB in sera of cancer patients. Hum Immunol. 2006;67:188-95. doi:10.1016/j.humimm.2006.02.008. PMID:16698441
- Salih HR, Antropius H, Gieseke F, Lutz SZ, Kanz L, Rammensee HG, Steinle A. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood. 2003;102:1389-96. doi:10.1182/blood-2003-01-0019. PMID:12714493
- Raab S, Steinbacher J, Schmiedel BJ, Kousis PC, Steinle A, Jung G, Grosse-Hovest L, Salih HR. Fc-optimized NKG2D-Fc constructs induce NK cell antibody-dependent cellular cytotoxicity against breast cancer cells independently of HER2/neu expression status. J Immunol. 2014;193:4261-72. doi:10.4049/jimmunol.1400872. PMID:25217158