
ABSTRACT
Pancreatic Ductal Adenocarcinoma (PDA) is characterized by a complex tumor microenvironment that supports its progression, aggressiveness and resistance to therapies. The delicate interplay between cancer and immune cells creates the conditions for PDA development, particularly due to the functional suppression of T cell anti-tumor effector activity. However, some of the mechanisms involved in this process are still poorly understood. In this study, we analyze whether the functional and epigenetic profile of T cells that infiltrate PDA is modulated by the microenvironment, and in particular by tumor-associated macrophages (TAMs). CD4 and CD8 T cells obtained from mice orthotopically injected with syngeneic PDA cells, and untreated or treated with Trabectedin, a cytotoxic drug that specifically targets TAMs, were sorted and analyzed by flow cytometry and characterized for their epigenetic profile. Assessment of cytokine production and the epigenetic profile of genes coding for IL10, T-bet and PD1 revealed that T cells that infiltrated PDA displayed activated Il10 promoter and repressed T-bet activity, in agreement with their regulatory phenotype (IL10high/IFNγlow, PD1high). By contrast, in Trabectedin-treated mice, PDA-infiltrating T cells displayed repressed Il10 and Pdcd1 and activated T-bet promoter activity, in accordance with their anti-tumor effector phenotype (IL10low/IFNγhigh), indicating a key role of TAMs in orchestrating functions of PDA-infiltrating T cells by modulating their epigenetic profile towards a pro-tumoral phenotype. These results suggest the targeting of TAMs as an efficient strategy to obtain an appropriate T cell anti-tumor immune response and open new potential combinations for PDA treatment.
Introduction
Pancreatic Ductal Adenocarcinoma (PDA) is one of the most aggressive cancers, having an incidence rate that almost coincides with the mortality rate, and represents the 5th most common cause of death due to cancer worldwide.Citation1 The 5-year survival rate is as low as 8% of diagnosed patients and most of them die in the first 6 months following diagnosis.Citation2 This is mainly due to the lack of effective markers for early detection, along with resistance to standard radio- and chemo-therapy.Citation3,4
PDA is characterized by a complex and interactive microenvironment that supports cancer initiation and progression, including stromal cells, cancer cells and immune cells.Citation5-7 For this reason, targeting components of the tumor microenvironment as a strategy to reduce cancer cell proliferation and survival may represent a promising method for overcoming resistance of pancreatic cancer cells to standard therapies.
For many tumors, it has been demonstrated that the presence of Tumor-Infiltrating Lymphocytes (TILs) is associated with better prognosis.Citation8–11 In PDA as well, the elevated number of both infiltrating CD4 and CD8 T cells was demonstrated to correlate with a better outcome.Citation12 In previous studies, we have demonstrated the presence of T cells specific for the PDA-associated antigen alpha-enolase (ENO1), both in the tumor and in the blood of PDA patients.Citation13–16 More recently, we have described that circulating ENO1-specific T cells can represent a prognostic marker due to their correlation with prolonged survival.Citation17
The status of T cells in cancer should be considered in the context of other immune cells, such as Tumor-Associated Macrophages (TAMs) or Myeloid-Derived Suppressor Cells (MDSCs).Citation18 Both these populations are known to create an immune suppressive environment through either the secretion of cytokines such as IL10 and TGFβ, or the expression of inhibitory molecules such as PDL-1.Citation19–21 This highly repressive environment inhibits the activation of CD8 T cells, and induces a switch of CD4 T cells towards T helper (Th)-2 and Treg phenotypes.Citation22,23 However, the presence of mixed stimuli in the microenvironment creates conditions for reversible changes in infiltrating cells, including TILs. These modifications derive from the activation or inhibition of signaling pathways and chromatin remodeling. The histone modification landscape is highly involved in gene transcription control and can be affected by several modifications, such as acetylation, methylation and phosphorylation.Citation24 In particular, specific histone modifications have been associated with gene transcription activation, such as tri-methylation of lysine 4 on histone 3 (H3K4me3), or repression, such as tri-methylation of lysine 27 on histone 3 (H3K27me3), and are known to rapidly change in response to environmental conditions.Citation25
Epigenetic changes in the tumor microenvironment play a central role in tumor initiation, progression and spreading.Citation26–29 However, despite the increasing number of studies investigating PDA progression and epigenetic modulation in T cells, the role of tumor microenvironment in the dynamic change of epigenetic modifications in PDA-infiltrating T cells is poorly investigated.
In this study, we compared the phenotypic and epigenetic profile of infiltrating T cells in normal pancreas and PDA, with or without perturbation of the tumor stroma by depleting macrophages. In order to do this we exploited the selective targeting of mononuclear phagocytes by Trabectedin.Citation30–32 Trabectedin (ET-743, Yondelis®) was originally derived from Ecteinascidia turbinata,Citation33 and is currently produced semi-synthetically. It is approved in Europe and USA as a second-line therapy for treating patients with advanced soft tissue sarcoma and relapsed ovarian cancers.Citation34,35 It has been already clarified that its antitumor effects are due to multiple mechanisms, namely binding to the minor groove of DNA, causing blocking of proliferation and different DNA repair pathways, through the transcription of p21, GADD45 A and G2-related cyclins, and inducing apoptosis via JNK and caspase 3 activation.Citation36–39 Trabectedin has been demonstrated to be effective against different tumor cell lines, and when combined with gemcitabine, the cytotoxic effects on PDA cells are enhanced.Citation40–42 Interestingly, Trabectedin specifically targets mononuclear phagocytes through activation of the caspase 8 cascade via TRAIL receptors, which are expressed in monocytes and TAMs.Citation43 For this reason we used Trabectedin to perturb the pancreatic tumor microenvironment.
Analyzing the immune infiltrate after Trabectedin treatment revealed changes in the cytokine production and surface molecule expression that mirrored epigenetic changes in the promoter and enhancer regions of specific genes, leading to the rescue of the anti-tumor effector phenotype of TILs.
In conclusion, our results indicate that targeting PDA microenvironment affects infiltrating T cells through epigenetic regulation, paving the way for combinatorial immunotherapeutic strategies.
Results
Characterization of PDA immune infiltrating cells
K8484 murine PDA cells were orthotopically injected into the pancreata of C57 BL/6 mice. After 28 days, mice were sacrificed and tumor masses and normal pancreata were excised. To characterize the infiltrating cells, we enzymatically dissociated the pancreata and analyzed the cells by flow cytometry (Suppl. Fig. 1). A significant decrease of the lymphocyte population in tumors was observed in all mice, with CD4 and CD8 T cells being reduced by 60% and 50%, respectively, compared to the normal pancreas (NP) ( left panel). Investigation of the macrophage population by measuring the F4/80 marker showed no differences in cell number, but an increased PDL-1 expression was detected in the resident macrophages characterized by a higher F4/80 expressionCitation44 ( right panels).
Figure 1. Phenotypic analysis of PDA immune infiltrating cells. (A) Characterization of T cells and F4/80 positive and negative populations in normal pancreas (NP) versus PDA. PDL-1 expression on F4/80hi and F4/80low cells is shown as delta Geo Mean of the fluorescence intensity after subtraction of isotype-related values. (B) Analysis of cytokine production and surface marker expression in NP or PDA-infiltrating T cells. Each graph shows the percentage of positive cells for the specific marker as indicated on the Y-axis. Data are represented as mean ± SEM of pooled cells from three independent experiments with two/three mice per experiment. Statistical analysis by unpaired Student's t test.
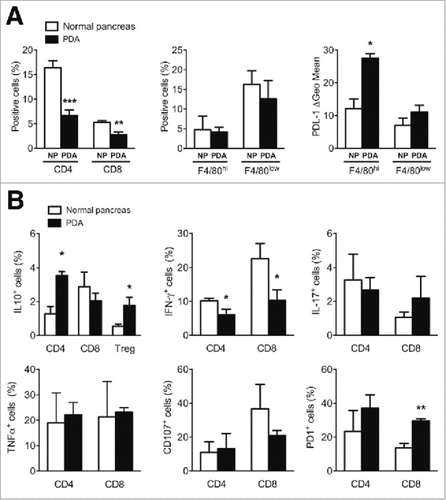
Compared to the corresponding cell subsets dispersed in the NP, CD4 T cells and Treg cells that infiltrated PDA displayed an increased percentage of both cell type producing IL-10, whereas no differences were observed in the percentage of CD8 T cells producing this cytokine from PDA or NP ( upper left panel).
Conversely, while PDA-infiltrating CD4 and CD8 T cells producing IL-17 and TNFα displayed no significant differences in their percentage, a decreased percentage of IFNγ+ cells that paralleled with an increase of PD1+ cells and a decrease of CD107+ cells was observed for PDA-infiltrating CD8 T cells only ( middle and right panels).
These results indicate that the PDA microenvironment plays an immunosuppressive role, as demonstrated by i) increased expression of PDL-1 in resident macrophages (F4/80hi); ii) the reduction of infiltrating CD4 T cells, which switched from a Th1 “effector” (IFNγhi/IL-10low, PD1low) to a suppressive (IFNγlow/IL-10high, PD1high) phenotype; iii) the reduction of infiltrating CD8 T cells, which switched from an “effector” (IFNγhi/CD107high, PD1low) to an “exhausted” (IFN-γlow/CD107low, PD1high) phenotype and iv) the higher production of IL10 by infiltrating Treg cells.
PDA microenvironment affects anti-tumor immune response through epigenetic mechanisms
Since phenotypic analysis of PDA immune cells showed a notable increase in Treg and CD4 cells producing IL10, together with a decrease in those producing IFNγ, we investigated whether the PDA microenvironment affected epigenetic marks at the promoter regions of Il10 and T-bet loci; T-bet being the main transcription factor that induces IFN-γ expression in T cells.Citation45 We sorted CD4, CD8 and Treg cells from the NP and PDA and measured H3K4me3, an active gene histone mark, and H3K27me3, a repressive mark, at the promoters of Il10 and T-bet, using chromatin immunoprecipitation (ChIP). In CD4 T cells from PDA, the active H3K4me3 mark was highly enriched at the IL10 promoter compared to the repressive H3K27me3 mark, while equal enrichment of both marks was observed in CD4 T cells purified from NP ( left panel). In CD8 T cells, no significant differences were found between NP and PDA ( middle panel), while PDA-infiltrating Treg cells showed a significant decrease in the repressive mark H3K27me3 compared to the NP-purified ones ( right panel). This data corroborated the observation that IL10 was highly expressed by PDA-infiltrating CD4 and Treg cells, while no differences were observed for CD8 T cells ( upper right panel).
Figure 2. Epigenetic changes of Il10 and T-bet promoters in PDA-infiltrating T cells. (A) ChIP analysis targeting H3K4me3 and H3K27me3 at Il10 promoter in CD4, CD8 and Treg cells sorted from NP and PDA. (B) ChIP analysis targeting H3K4me3 and H3K27me3 at T-bet promoter in CD4 and CD8 T cells sorted from NP and PDA. Columns represent the percentage of input chromatin. Data are represented as mean ± SEM of pooled cells from two independent experiments with three mice per experiment, in which tumor cell suspensions were pooled together for the sorting. Statistical analysis by unpaired Student's t test. #, ##, ### P values statistically different between NP and PDA; §§§ P values statistically different between permissive and repressive marks in the NP group.
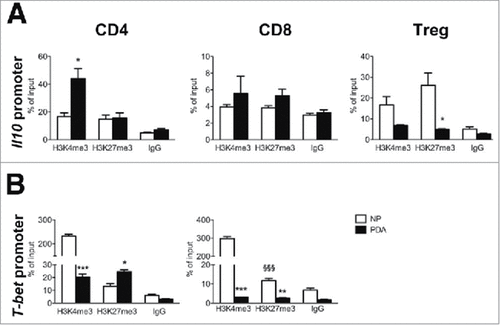
Conversely T-bet promoter was marked by high levels of permissive H3K4me3 and very low levels of repressive H3K27me3 in both CD4 and CD8 T cells from NP, while no differences were observed for the two marks in PDA-infiltrating CD4 and CD8 T cells (), suggesting that T-bet promoter activity is modulated when T cells move from NP to the tumor microenvironment, affecting IFNγ production. These results corroborated the flow cytometry data, showing that CD4 and CD8 T cells from NP had increased IFNγ production compared to the corresponding subsets in PDA ( middle panel), and strongly suggests a role for the tumor microenvironment in orchestrating the immune response by an epigenetic-mediated suppression.
Depletion of macrophages affects the tumor microenvironment and T cell activation
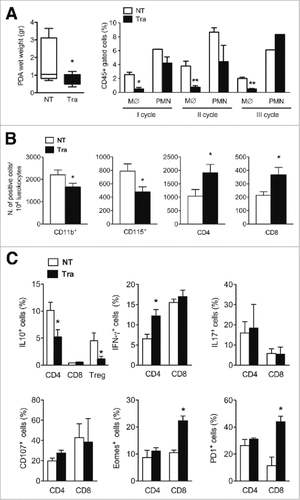
To interfere with the tumor microenvironment, we exploited the DNA-binding anti-tumor drug Trabectedin, which selectively induces monocyte apoptosis as a secondary effect.Citation30 Mice injected with K8484 tumor cells in the pancreata were untreated (NT) or treated intravenously on a weekly basis with Trabectedin (Tra), for three weeks, starting at day 7. Tumor masses were excised after 28 days and weighed before dissociation. Notably, Trabectedin significantly reduced tumor size ( left panel). To ascertain the efficacy of Trabectedin in depleting the monocyte population, we collected and analyzed blood from mice the day after every drug administration. Flow cytometry showed a strong decrease in circulating monocytes after each Trabectedin administration, while polymorphonuclear cells (PMNs) were not significantly affected ( right panel and Suppl. ). After enzymatic dissociation, we analyzed the tumor-infiltrating immune cells by flow cytometry. Trabectedin-treated mice showed a notable decrease in CD11b+ and CD115+ populations ( left panels) and a concomitant significant increase in the number of infiltrating CD4 and CD8 T cells ( right panels). Compared with untreated tumor-bearing mice, the percentage of IL-10-producing CD4 T cells and Treg cells was sharply decreased (). A significant increase in the percentage of IFNγ-producing cells was observed in CD4 T cells from Trabectedin-treated mice, while no substantial differences were observed in the percentage of IL17-producing or CD107+ cells in either of the T cell subsets (). Notably, a sharp parallel increase in the percentage of Eomesodermin/Tbr2 (Eomes)+ and PD1+ cells, consistent with a T cell “memory” population,Citation46 was observed in CD8 T cells, but not in CD4 T cells, from tumor-bearing mice treated with Trabectedin (). These results show that Trabectedin efficiently depleted monocytes, modified the tumor microenvironment and affected infiltrating T cells, by reducing IL10 production and inducing activation of CD8 T cells, as demonstrated by their “effector-memory” phenotype.
Figure 3. Phenotypic analysis of the PDA microenvironment after Trabectedin treatment. (A) Left panel: PDA wet weight from untreated (NT) and Trabectedin-treated (Tra) mice. Data are represented as box and whiskers of Tukey's method (six mice per group; statistical analysis by unpaired Student's t test). Right panel: Flow cytometry analysis of Monocytes (M∅) and Polymorphonuclear cells (PMN) in blood collected 24 h after each Trabectedin treatment from NT and Trabectedin-treated mice. (B) Absolute number of infiltrating immune cells positive for specific markers, expressed per 104 leukocytes from NT and Trabectedin-treated mice. (C) Analysis of PDA-infiltrating T cell cytokine production and surface markers from NT or Trabectedin-treated mice. Each plot shows the percentage of positive cells for the specific marker, as indicated on the Y-axis. Data are represented as means ± SEM of pooled cells from two independent experiments with three mice per condition. Statistical analysis by unpaired Student's t test.
Changes in the tumor microenvironment affect the epigenetic profile of infiltrating T cells
To explore whether Trabectedin-induced changes in IL10 and IFNγ production were caused by modifications in the epigenetic states of T cells, we sorted CD4+ and CD8+ T cells to analyze Il10 and T-bet promoters. We tested the permissive H3K4me3 mark and the repressive H3K27me3 mark through ChIP analysis in untreated and Trabectedin-treated mice. We observed that CD4 T cells from NT mice were highly enriched in the permissive H3K4me3 mark, while they showed a low enrichment of the repressive H3K27me3 mark at the Il10 promoter. By contrast, CD4 T cells from Trabectedin-treated mice displayed an opposite trend, with a significant decrease in H3K4me3 and an increase in H3K27me3 compared to NT CD4 T cells ( left panel). These data indicate that Trabectedin changed the epigenetic state of the Il10 promoter from an active to a silent state, corresponding to the decreased IL10 production by CD4 T cells ( left panel). Conversely, both marks were weakly enriched in CD8 T cells from untreated mice, and enrichment of both marks significantly increased in CD8 T cells from Trabectedin-treated mice ( right panel). These data indicate that Trabectedin modifies the epigenetic profile of the Il10 promoter in CD8 T cells, although the production of IL-10 was not detectable in CD8 T cells from untreated mice or those treated with Trabectedin ().
Figure 4. Epigenetic changes of Il10 and T-bet promoters in PDA-infiltrating T cells after Trabectedin treatment. ChIP analysis targeting H3K4me3 and H3K27me3 of (A) Il10 and (B) T-bet promoters in PDA-infiltrating CD4 and CD8 T cells sorted from NT and Trabectedin-treated mice. Columns represent percentage of input chromatin. Data are represented as means ± SEM of pooled cells from two independent experiments, in which tumor cell suspensions from three mice were pooled together for the sorting. Statistical analysis by unpaired Student's t test. #, ## P values statistically different between NT and Tra; §, §§ P values statistically different between permissive and repressive marks in the NT or Tra group.
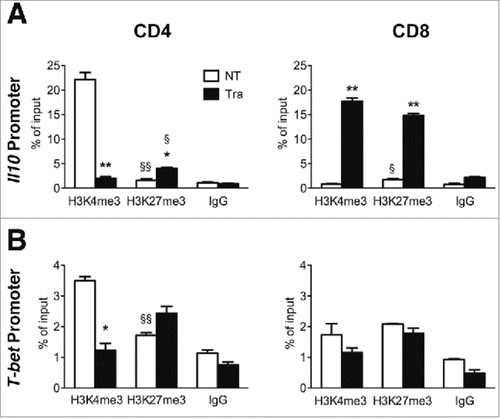
Analysis of the T-bet promoter showed an enrichment of the H3K4me3 permissive mark and a lower enrichment of the repressive mark in CD4 T cells from untreated mice, whereas in mice treated with Trabectedin, CD4 T cells displayed a significant reduction of the permissive H3K4me3 mark without any significant changes in the H3K27me3 repressive mark (). These variations did not predict the increased production of IFNγ by CD4 T cells assayed by flow cytometry (). No significant differences were observed in CD8 T cells from mice treated with Trabectedin compared to untreated mice, with no evident modifications in the H3K4me3 permissive and the H3K27me3 repressive marks of the T-bet promoter (). This did not substantially impair their ability to produce IFNγ () and the non-permissive regulation of T-bet could reflect a T cell phenotype skewed towards a “memory” status, as observed by flow cytometry analysis.
As PD1 expression significantly increased in CD8 TILs after Trabectedin treatment (), we investigated the methylation and acetylation states of regulatory sites on the Pdcd1 locus acting as enhancers located at +3.7 and −17.1kb from the transcription start siteCitation47 (Suppl. Fig. 3A). In particular, we tested the active marks H3K4me1 and acetylation on lysine 27 of H3 (H3K27 Ac), in sorted PDA-infiltrating CD4 and CD8 T cells from untreated or Trabectedin-treated mice through ChIP. Purified PDA-infiltrating CD4 T cells from Trabectedin-treated mice showed enrichment in H3K4me1 at both enhancers, while a slight decrease was observed for H3K27 Ac enrichment compared to PDA-infiltrating CD4 T cells from NT mice (Suppl. Fig. 3B). By contrast, no changes in H3K4me1 enrichment, but a significant decrease in H3K27 Ac enrichment for both +3.7 and −17.1 enhancers was observed in PDA-infiltrating CD8 T cells from Trabectedin-treated mice compared to PDA-infiltrating CD8 T cells from NT mice. These results do not fully correspond with the flow cytometry results, and suggest that PD1 up-regulation on CD8 TILs after Trabectedin treatment is independent from these two enhancers. Therefore, other enhancers may play a role in the regulation of PD1 expression, or Trabectedin may affect different pathways inducing PD1 expression.
Effect of Trabectedin on epigenetics of CD4 T cells is both direct and macrophage-mediated
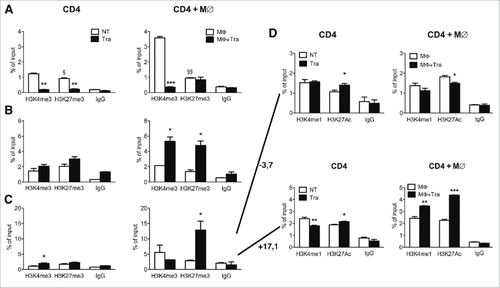
To confirm whether the effect of Trabectedin on PDA-infiltrating CD4 T cells was direct or mediated by the depletion of macrophages, we sorted CD4 T cells from normal mice and cultured them in the presence or absence of Trabectedin and in the presence or absence of supernatants from bone marrow-derived macrophages previously untreated or treated with Trabectedin. We chose CD4 T cells, as they were the most regulated in all three promoters. ChIP analysis of the Il10 promoter revealed a consistent decrease in H3K4me3 upon Trabectedin treatment in CD4 T cells cultured in medium only, and to a much greater extent in those cultured in the presence of macrophage supernatants (). This suggests that Trabectedin directly and negatively regulates IL10 production, which is amplified by the presence of macrophage-secreted factors. By contrast, ChIP analysis of the T-bet promoter showed a significant increase of both epigenetic marks only in stimulated CD4 T cells with the supernatants of macrophages cultured in the presence of Trabectedin (). Enrichment of the repressive H3K27me3 mark confirms the ex-vivo epigenetic data shown in , but the specific increase in H3K4me3 in CD4 cells cultured with the supernatants of Trabectedin-treated macrophages suggests a role of the latter in mediating the epigenetic switch in T cells. Finally, analysis of the Pdcd1 promoter revealed enrichment in H3K27me3 only when CD4 T cells were cultured in the presence of Trabectedin-treated macrophage supernatants (). Further analysis of histone modifications associated with active enhancers, H3K4me1 and H3K27 Ac, in the −3.7kb enhancer region of Pdcd1 locus, demonstrated a decrease induced by Trabectedin in both the epigenetic marks when T cells were treated in the presence of macrophage supernatants, while the opposite was observed for T cells alone ( upper panels). Analysis of the +17.1kb genomic region revealed a strong enrichment for both marks in CD4 T cells stimulated with Trabectedin-treated macrophage supernatants ( lower panels). These results suggest that Trabectedin may have a direct effect on T cells, but the presence of macrophages enhances this effect, promoting an epigenetic switch that changes their functions.
Figure 5. Effects of Trabectedin on CD4 T cells. ChIP analysis of H3K4me3 and H3K27me3 of Il10 (A) and T-bet (B) and Pdcd1 promoter (C) and associated enhancers at −3.7kb and +17.1kb from the transcription start site (D) in sorted CD4 T cells from naïve mice alone or in the presence of Trabectedin and stimulated with supernatants from M∅ in the presence or absence of Trabectedin. Columns represent percentage of input chromatin. Data are represented as means ± SEM of pooled cells from two independent experiments. Statistical analysis by unpaired Student's t test. #, ##, ### P values statistically different between NT and Tra; §, §§ P values statistically different between permissive and repressive marks in the NT or Tra group.
Trabectedin changes the macrophage secretome
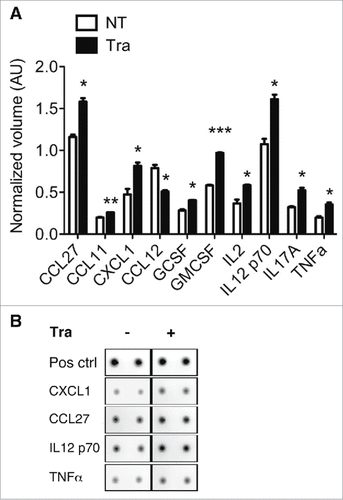
To further investigate the effect of Trabectedin on macrophages and the indirect effect on TILs, we performed a cytokine array with the conditioned supernatant used in (). Trabectedin significantly up-regulated 17 cytokines/chemokines, and down-regulated CCL12 only. Among the statistically significant up-regulated there are IL2, IL12, IL17 and TNFα, which are involved in T cell activation. Moreover, Trabectedin increased the expression of granulocyte- and granulocyte and monocyte colony stimulator factors (G-CSF and GM-CSF) and some chemokines as CCL27, CCL11 and CXCL1, which elicit the recruitment of memory T cells to inflammatory sites and neutrophil activation, respectively.Citation48,49 Overall, these results suggest that Trabectedin induces an increase in inflammatory cytokine and chemokine production by macrophages, which shapes and regulates the epigenetic landscape of specific promoters related to the activation and phenotype of T cells.
Figure 6. Evaluation of chemokine/cytokine secretion by macrophages after Trabectedin treatment. (A) Quantification of cytokines and chemokines in the supernatants of macrophages treated with Trabectedin for 24h, or untreated. Data are represented as means of normalized volumes (arbitrary units – AU) ± SEM from two independent experiments. Statistical analysis by unpaired Student's t test. (B) Representative spots from the recorded images of one array.
Discussion
Despite the continuous advances in cancer research, PDA remains one of the most challenging tumors to diagnose and treat.Citation2 In this study, we focused on the complex interplay occurring in the tumor microenvironment in order to clarify mechanisms involved in the suppression of the immune response, with particular attention to the epigenetic profile of infiltrating T cells.
Tumors engage the immune system from their inception. Initially, cells of the innate system such as macrophages and mast cells are mainly involved. However, even at early stages, cells of the acquired system are engaged, particularly T cells.Citation50 Recent data have challenged the original idea that PDA is poorly immunogenic. Above all, higher levels of tumor-infiltrating CD4 and CD8 lymphocytes and dendritic cells positively correlate with longer survival after surgical resection.Citation12 We have also described that PDA-infiltrating T cells specifically recognize alpha-enolase (ENO1), a PDA-associated antigen,Citation14 but are often frustrated in their effector functions by the presence of Tregs.Citation15 Of note, peripheral ENO1-specific T cells mirror the intra-tumoral immune response and represent a promising candidate as a prognostic marker of survival.Citation17
Here, we analyzed PDA-infiltrating T cells to define their functional state and how they were reflected in their epigenetic landscape. Although some studies have addressed the question of the number of PDA-infiltrating T cells,Citation12 to our knowledge this is the first study that characterizes the epigenetic states of PDA-TILs. Here, we demonstrated that CD4 and CD8 T cells accumulated to a lesser extent in the PDA compared to the normal pancreata, while no differences were observed in either CD11b/F4/80hi or CD11b/F4/80low myeloid cells. Of note, PDL-1 expression was increased in both CD11b/F4/80low and CD11b/F4/80hi myeloid cells, but especially in the latter. PDA-infiltrating T cells, especially CD4 and Tregs, highly produced IL-10 but not IFNγ, while there were no differences in those producing IL-17 or TNFα. Even PD1 was substantially increased on CD8 T cells, confirming that inflammation that was strictly associated with PDA growth led to activation of both types of T cells, but they were eventually restrained by the regulatory and suppressor immune cells. In addition, our results showed that the PDA microenvironment also induced alterations in the epigenome of the TILs, probably by means of soluble and cell-contact factors. These effects can be generally promoted by different, extremely heterogeneous cell types in the microenvironment, and we focused on TAMs, which are well-known pro-tumor cells that suppress anti-tumor responses through the secretion of cytokines, chemokines and exposure of inhibitory ligands.Citation51,52 Targeting myeloid cells to limit T cell suppression and induce efficient anti-tumor responses has been used as a strategy in many tumors, including PDA,Citation53,54 employing different drugs.Citation55–57 Depletion of GM-CSF or Gr1+CD11b+ myeloid cells has been described as being sufficient to inhibit implanted PDA cells and efficiently recruit CD8 T cells, which are responsible for tumor growth impairment, as their depletion can restore tumor growth.Citation39,54
We exploited Trabectedin, a drug known to affect both tumor cells and TAMs.Citation39 Trabectedin has been demonstrated to be active in inducing cell cycle arrest or cytotoxicity on a variety of tumor cells and can selectively inhibit the transcription of different pro-inflammatory factors and angiogenic mediators by macrophages.Citation42 After Trabectedin-dependent TAM deprivation, TILs showed a reactivation both at the epigenetic and functional levels, with a switch from IL10-secreting T cells towards an effector/memory phenotype, as demonstrated by the increased percentage of IFNγ+ and Eomes+ T cells. The changes in the secretome of TAMs after Trabectedin treatment suggests that this switch could be partly mediated by inflammatory cytokines and chemokines secreted in response to the treatment. PD1+ CD8 T cells are increased in Trabectedin-treated mice compared to PDA-infiltrating CD8 T cells from untreated mice, and to a lesser extent in CD4 T cells. This confirms their activation status, as PD1 is increased by T cell activation.Citation58 After activation of T cell receptors, T cells start to actively secrete cytokines (such as IFN-γ in our study) and express PD1, together with the costimulatory receptor CD28, its early and preferential target.Citation59,60 After prolonged antigen stimulation, the T cells become “exhausted” but still express PD1. Of note, the epigenetic results did not demonstrate an active transcription, as shown by the protein expression after Trabectedin treatment. Thus, other regulatory regions or signaling pathways seem to be involved in the complex expression of PD1. However, Trabectedin acts on both sides, i.e. tumor cells and immune cells, by inducing cell death and triggering an efficient anti-tumor response, respectively. These results demonstrate that modifying the microenvironment composition also affects immune infiltrating cells, not only in terms of numbers, but also by inducing a shift in their activation profiles. However, additional studies are needed to provide further evidence of the role of macrophages, by exploiting a genetic macrophage-depleted mouse model. In addition, in vitro experiments suggest a direct effect of Trabectedin on T cells, proposing a new target for this drug that deserves further investigation.
Overall, this study demonstrates that PDA-infiltrating T cells are epigenetically shaped by the microenvironment, and particularly by TAMs. The use of Trabectedin to perturb the composition of the microenvironment by depleting TAMs, and their anti-tumoral effects, opens new options for novel combinatorial therapies. Among treatments involving active immunotherapy, Trabectedin has been successfully tested in combination with anti-PD1 antibody in ovarian cancer,Citation61 and its combination with doxorubicin has been shown to be effective in a phase 2 clinical trial in leyomiosarcoma.Citation35 Regarding the combination with PD1 treatment, our results suggest that the timing of this combination can be essential. Indeed, Trabectedin treatment is able to modulate chromatin remodeling in specific genomic regulatory regions in the Pdcd1 locus, and consequently PD1 expression on T cells.
Taking into account the major role played by chromatin remodeling in the PDA microenvironment, new therapeutic strategies deploying epigenetic modulating agents also need to be considered for PDA. Some epigenetic drugs have been already tested in PDA with promising results,Citation62 but no effects on immune infiltrating cells have been described. In particular, inhibition of histone methyltransferases, by Enhancer Of Zeste Homolog (EZH) 2 or histone deacetylases (HDACs), significantly impaired PDA progression, with EZH2 interfering with the oncogenic activity of NFATC1 and HDACs promoting epithelial gene expression,Citation63-66 but there is no mention about their effect on TILs.
The most common opinion is that a successful anti-cancer therapy should exploit the combinatorial treatment of several targets in the microenvironment, from cancer cells to immune or stromal cells. Due to the strong interconnection between cell-type specific responses to the environment and the epigenetic profile, as well as the increasing number of drugs affecting epigenetic regulators, new molecular targets are constantly being identified. Perturbing the right components with combinatorial therapies may open new effective ways to fight PDA.
Materials and methods
Cell Culture
The murine pancreatic cancer cell line K8484 was obtained from mice carrying mutated KrasG12D and Trp53R172H (kindly provided by Dr. K. Olive, Columbia University). Cell lines were cultured in DMEM (Lonza, 12–719F) supplemented with 10% FBS (Gibco, 10500056), L-Glutamine (GE Healthcare, SH30034.01) and 50μg/ml of gentamicin (Gentalyn 40mg/ml, Essex Italia) at 37°C in a 5% CO2 atmosphere. Cells were detached using a solution of PBS (Lonza, 17–516F) containing 0.25% Trypsin-EDTA (Gibco, 15400054). Before injection, cells were washed twice with PBS and re-suspended in DMEM without FBS.
In vivo experiments
All animal experiments were approved by the University of Torino, Italian Ministry of Health and performed in accordance with EU laws in the animal facility of the Molecular Biotechnology Center (MBC).
C57 BL/6 mice were anesthetized and orthotopically injected into the pancreas with 1 × 105 K8484 cells resuspended in DMEM with Matrigel (BD Bioscience, 354234) at a dilution of 1:4. After 28 days, mice were euthanized; PDA were weighed and the tumor masses and normal pancreata were separated. To avoid the presence of peripheral blood cells in the pancreas, mice were intracardially perfused with 10 ml PBS. Trabectedin was administered every 7 days intravenously into the tail vein of the mice at 0.015mg/kg in physiologic solution. Age-matched mice were randomly assigned to the control group, in which mice were injected with the same volume of PBS, or the treated group. The day following drug administration, 30µl of peripheral blood was collected to analyze monocyte and polymorphonuclear cells by flow cytometry.
Trabectedin was provided as a lyophilized formulation by PharmaMar (S.A. Colmenar Viejo), dissolved in DMSO and stored at −20°C.
Tissue dissociation
Tumors and normal pancreata were enzymatically dissociated with a cocktail containing Collagenase IV (Sigma-Aldrich, C5138), Hyaluronidase V (Sigma-Aldrich, H5264), Dispase II (Roche, 04942078001), DNAse I (Millipore, 200913) and Soybean Trypsin Inhibitor (Worthington Biochemical, LS003587). Tumors and normal pancreata from three mice were pooled for analysis. Cells were filtered through 70 µm and 40 µm cell strainers to obtain a cell mono-suspension. Cells were analyzed immediately or after 12h of stimulation with PMA (Sigma-Aldrich, P-1585), Ionomycin (Sigma-Aldrich, I3909) and Brefeldin A (BioLegend, 420601), by flow cytometry.
Flow cytometry analysis and cell sorting
A total of 1 × 105 dissociated cells per tube were washed twice with PBS-0.2% BSA-0.01% NaN3. After blocking nonspecific sites with anti-CD16/CD32 mAb (MiltenyiBiotec, 130-092-574), cells were incubated with mAbs for 30min at 4°C: FITC anti-mouse CD4 (MiltenyiBiotec, 130-102-779), PerCP anti-mouse CD8 a (MiltenyiBiotec, 130-102-811), APC anti-mouse CD8 a (eBioscience, 17-0081-81), PE anti-mouse CD19 (Biolegend, 151407), PE anti-mouse CD25 (Biolegend, 101903), PE anti-mouse PDL-1 (Biolegend, 124307), PerCP anti-mouse F4/80 (Biolegend, 123125), APC anti-mouse CD127 (MiltenyiBiotec, 130-102-551), PE anti-mouse IL-10 (MiltenyiBiotec, 130-103-014), PE anti-mouse PD-1 (MiltenyiBiotec, 130-11-953), PE anti-mouse CD107 (MiltenyiBiotec, 130-102-219), APC anti-mouse IFNγ (MiltenyiBiotec, 130-102-340), FITC anti-mouse CD11b (eBioscience, 11-0112-82). Intracellular staining was performed with Foxp3/Transcription Factor Staining Buffer Set (eBioscience, 00-5523-00) using APC anti-mouse FoxP3 antibody (eBioscience, 17-5773-80). Cells were washed, re-suspended in PBS and acquired with a BD AccuriC6 Flow Cytometer (BD). Analysis was performed using FlowJo 7.5 software for tissue analysis and using BD Accuri C6 software for blood analysis.
For cell sorting, cells were blocked with a short incubation with anti-CD16/CD32 mAb and stained with FITC anti-mouse CD4, PerCP anti-mouse CD8 a and APC anti-mouse CD127 for 30min at 4°C. After washing, cells were resuspended with PBS-2mM EDTA-2% BSA and sorted with a FACSAria III and data were analyzed through BD FACSDiva software 7.0 (both by BD).
Chromatin Immunoprecipitation Assay (ChIP)
Lymphocytes purified from tissues were incubated with 1% formaldehyde in PBS for 10min at 37°C. The crosslinking reaction was stopped by adding glycine at a final concentration of 125 mM, and samples were incubated for 5min at Room Temperature (RT). Crosslinked cells were then resuspended in 25 µl of Lysis buffer (50mM Tris-HCl pH 8,10mM EDTA and 1% SDS). Cell lysates were incubated on ice for 10min, diluted with 75µl Immuno-Precipitation (IP) buffer (16.7 mMTrisHCl pH 8, 167 mMNaCl, 1.2 mM EDTA, 0.01% SDS and 1.1% Triton X-100), and then sonicated for 30 pulse 30″ON/30″OFF high with Bioruptor Twin (Diagenode). The protocol required the use of a small amount of chromatin and was modified from.Citation67 Sonicated chromatin was diluted in IP buffer to a final volume of 120 µl for each immunoprecipitation, and incubated with 0.5µg of antibodies against human H3K4me3, H3K27me3, H3K4me1 or H3K27ac (ActiveMotif; 39915, 39155, 61633 or 39133) in a BSA pre-treated 96-well dish. Lysates with Ab were incubated at 4°C overnight on an orbital shaker. Samples with IgG antibody (Santa Cruz Biotechnology, sc-2027) were run in parallel as negative controls. The following day, 30 µl of 50% Protein A Sepharose™ 4 Fast Flow slurry (GE Healthcare, 17-5280-01) was added and incubated for 2h at 4°C to purify the immune complexes. Proteins and DNA complexes not specifically associated with beads were removed by sequential washes with low-salt buffer (0.1%SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH8 and 150mM NaCl), high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20mM Tris-HCl, pH8.0 and 500 mM NaCl), LiCl washing buffer (0.25M LiCl; 1% deoxycholate sodium salt, 1 mM EDTA, 10 mMTris-HCl pH8.0 and 1% NP-40) and twice with Tris-EDTA buffer (10 mMTris-HCl pH8, 1 mM EDTA). Samples were maintained at 4°C for 5min per wash. The immunoprecipitated DNA-protein complexes were purified using 10% Chelex® 100 Resin (BioRad, 142–2822) for 10min at 95°C. Proteins were digested by incubating each sample with 20µg of Proteinase K (Thermo Fisher Scientific, AM2546) for 30min at 55°C and then 10min at 95°C to obtain Proteinase K inactivation, thus achieving DNA purification.
Oligonucleotide primer sequences used for quantification of enriched DNA after chromatin immunoprecipitation are listed in Supplementary Materials & Methods (Suppl. Table 1).
T cell conditioning with macrophage-derived supernatants
CD11b cells were magnetically purified from spleens from tumor-bearing mice following the manufacturer's instructions (MiltenyiBiotec, 130-047-142), and 2 × 106 cells/ml were cultured in the presence of 25ng/ml of murine M-CSF (MiltenyiBiotec, 130-101-706) in a 6-well plate for 4 days at 37°C. After washing with RPMI only, macrophages were incubated in the presence or absence of Trabectedin (0.05µg/ml) for 24h. Supernatants were collected and frozen at −20°C until use.
CD4 T cells were sorted from splenocytes from naïve C57BL/6 mice, as described above, and cultured in the presence or absence of supernatants from untreated or treated macrophages, and in the presence or absence of Trabectedin (0.05µg/ml) for 24h at 37°C. After two washes, cells were harvested and processed as described for ChIP.
Cytokine array
Supernatants from macrophages treated with Trabectedin (0.05µg/ml) for 24h, or untreated, were evaluated for the presence of chemokines/cytokines with the Mouse Cytokine array C2 (RayBiotech, AAM-CYT-2-2). The arrays were performed according to the manufacturer's instructions. Pictures were acquired with the ProXpress 2D Proteomic Imaging System and saved as .tiff images (PerkinElmer, Milan, Italy). Spot volumes were analyzed using the ProFinder software (PerkinElmer). To determine the relative presence of cytokines/chemokines induced by the presence of Trabectedin, each sample was normalized according to values obtained for positive spots from untreated cells.
Statistical analysis
The Student's t test (GraphPad Prism 5 Software) was used to evaluate statistically significant differences in in vitro and in vivo experiments. Values are expressed as means ± SEM. # p<0.05, ## p<0.01, ### p<0.001
Disclosure of potential conflicts of interest
The authors report no conflict of interest
KONI_A_1393596.pdf
Download PDF (10.9 MB)Acknowledgments
We thank Dr. Radhika Srinivasan for critical reading of the manuscript and Roberta Curto for technical support.
Funding
This work was supported by the Associazione Italiana Ricerca sul Cancro (5 x mille no. 12182) and (IG no. 15257); the University of Turin-Progetti Ateneo 2014-Compagnia di San Paolo (PANTHER for P.C.) (PC-METAIMMUNOTHER for F.N.); the Fondazione Ricerca Molinette; Associazione Nadia Valsecchi; and the Local University of Torino Research Funding.
References
- Fang Y, Yao Q, Chen Z, Xiang J, William FE, Gibbs RA, Chen C. Genetic and molecular alterations in pancreatic cancer: implications for personalized medicine. Med Sci Monit. 2013;19:916–26.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30.
- Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1252–61.
- Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7:163–72.
- Pergamo M, Miller G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017;24:100–5.
- Tlsty TD, Hein PW. Know thy neighbor: stromal cells can contribute oncogenic signals. Curr Opin Genet Dev. 2001;11:54–9.
- van den Hooff A. Stromal involvement in malignant growth. Adv Cancer Res. 1988;50:159–96.
- Adams S, Goldstein LJ, Sparano JA, Demaria S, Badve SS. Tumor infiltrating lymphocytes (TILs) improve prognosis in patients with triple negative breast cancer (TNBC). Oncoimmunology. 2015;4:e985930.
- Ohtani H. Focus on TILs. Cancer Immun. 2007;7:4.
- Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13.
- Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306.
- Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, Oshikiri T, Kato K, Kurokawa T, Suzuoki M, Nakakubo Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28:e26–31.
- Cappello P, Tomaino B, Chiarle R, Ceruti P, Novarino A, Castagnoli C, Migliorini P, Perconti G, Giallongo A, Milella M, et al. An integrated humoral and cellular response is elicited in pancreatic cancer by α-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. Int J Cancer. 2009;125:639–48.
- Cappello P, Principe M, Bulfamante S, Novelli F. Alpha-Enolase (ENO1), a potential target in novel immunotherapies. Front Biosci (Landmark Ed). 2017;22:944–59.
- Amedei A, Niccolai E, Benagiano M, Della Bella C, Cianchi F, Bechi P, Taddei A, Bencini L, Farsi M, Cappello P, et al. Ex vivo analysis of pancreatic cancer-infiltrating T lymphocytes reveals that ENO-specific Tregs accumulate in tumor tissue and inhibit Th1/Th17 effector cell functions. Cancer Immunol Immunother. 2013;62:1249–60.
- Cappello P, Rolla S, Chiarle R, Principe M, Cavallo F, Perconti G, Feo S, Giovarelli M, Novelli F. Vaccination with ENO1 DNA prolongs survival of genetically engineered mice with pancreatic cancer. Gastroenterology. 2013;144:1098–106.
- Niccolai E, Cappello P, Taddei A, Ricci F, D'Elios M, Benagiano M, Bechi P, Bencini L, Ringressi M, Coratti A, et al. Peripheral ENO1-specific T cells mirror the intratumoral immune response and their presence is a potential prognostic factor for pancreatic adenocarcinoma. Int J Oncol. 2016;49:393–401.
- Chew V, Toh HC, Abastado J-P. Immune microenvironment in tumor progression: characteristics and challenges for therapy. J Oncol. 2012;2012:1–10.
- Ben-Baruch A. Inflammation-associated immune suppression in cancer: the roles played by cytokines, chemokines and additional mediators. Semin Cancer Biol. 2006;16:38–52.
- Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–7.
- Pinton L, Solito S, Damuzzo V, Francescato S, Pozzuoli A, Berizzi A, Mocellin S, Rossi CR, Bronte V, Mandruzzato S, et al. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget. 2016;7:1168–84.
- Seo YD, Pillarisetty VG. T-cell programming in pancreatic adenocarcinoma: a review. Cancer Gene Ther. 2017;24:106–13.
- De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, Braga M, Di Carlo V, Doglioni C, Protti MP. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. 2011;208:469–78.
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–95.
- Barth TK, Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem Sci. 2010;35:618–26.
- Hu M, Yao J, Cai L, Bachman KE, van den Brûle F, Velculescu V, Polyak K. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37:899–905.
- Rodriguez-Canales J, Hanson JC, Tangrea MA, Erickson HS, Albert PS, Wallis BS, Richardson AM, Pinto PA, Linehan WM, Gillespie JW, et al. Identification of a unique epigenetic sub-microenvironment in prostate cancer. J Pathol. 2007;211:410–9.
- Hanson JA, Gillespie JW, Grover A, Tangrea MA, Chuaqui RF, Emmert-Buck MR, Tangrea JA, Libutti SK, Linehan WM, Woodson KG. Gene promoter methylation in prostate tumor-associated stromal cells. JNCI J Natl Cancer Inst. 2006;98:255–61.
- Dey P. Epigenetics meets the tumor microenvironment. Med Epigenetics. 2013;1:31–6.
- Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249–62.
- Sinder BP, Zweifler L, Koh AJ, Michalski MN, Hofbauer LC, Aguirre JI, Roca H, McCauley LK. Bone mass is compromised by the chemotherapeutic trabectedin in association with effects on osteoblasts and macrophage efferocytosis. J Bone Miner Res. 2017;32(10):2116–2127
- Allavena P, Signorelli M, Chieppa M, Erba E, Bianchi G, Marchesi F, Olimpio CO, Bonardi C, Garbi A, Lissoni A, et al. Anti-inflammatory properties of the novel Antitumor Agent Yondelis (Trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res 2005;65:2964–71.
- Rinehart KL, Holt TG, Fregeau NL, Keifer PA, Wilson GR, Perun TJ, Sakai R, Thompson AG, Stroh JG, Shield LS. Bioactive compounds from aquatic and terrestrial sources. J Nat Prod. 1990;53:771–92.
- López-Guerrero JA, Romero I, Poveda A. Trabectedin therapy as an emerging treatment strategy for recurrent platinum-sensitive ovarian cancer. Chin J Cancer. 2015;34:41–9.
- Pautier P, Floquet A, Chevreau C, Penel N, Guillemet C, Delcambre C, Cupissol D, Selle F, Isambert N, Piperno-Neumann S, et al. Trabectedin in combination with doxorubicin for first-line treatment of advanced uterine or soft-tissue leiomyosarcoma (LMS-02): a non-randomised, multicentre, phase 2 trial. Lancet Oncol. 2015;16:457–64.
- Simoens C, Korst AEC, De Pooter CMJ, Lambrechts HAJ, Pattyn GGO, Faircloth GT, Lardon F, Vermorken JB. In vitro interaction between ecteinascidin 743 (ET-743) and radiation, in relation to its cell cycle effects. Br J Cancer. 2003;89:2305–11.
- Gajate C, An F, Mollinedo F. Differential cytostatic and apoptotic effects of ecteinascidin-743 in cancer cells: Transcription-dependent cell cycle arrest and transcription-independent JNK and mitochondrial mediated apoptosis. J Biol Chem. 2002;277:41580–9.
- Larsen AK, Galmarini CM, D'Incalci M. Unique features of trabectedin mechanism of action. Cancer Chemother Pharmacol. 2016;77:663–71.
- D'Incalci M, Badri N, Galmarini CM, Allavena P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br J Cancer. 2014;111:646–50.
- Miao X, Koch G, Straubinger RM, Jusko WJ. Pharmacodynamic modeling of combined chemotherapeutic effects predicts synergistic activity of gemcitabine and trabectedin in pancreatic cancer cells. Cancer Chemother Pharmacol. 2016;77:181–93.
- Miao X, Koch G, Ait-Oudhia S, Straubinger RM, Jusko WJ. Pharmacodynamic modeling of cell cycle effects for gemcitabine and trabectedin combinations in pancreatic cancer cells. Front Pharmacol. 2016;7:421.
- D'Incalci M, Zambelli A. Trabectedin for the treatment of breast cancer. Expert Opin Investig Drugs. 2016;25:105–15.
- Liguori M, Buracchi C, Pasqualini F, Bergomas F, Pesce S, Sironi M, Grizzi F, Mantovani A, Belgiovine C, Allavena P. Functional TRAIL receptors in monocytes and tumor-associated macrophages: a possible targeting pathway in the tumor microenvironment. Oncotarget. 2016;7:41662–76.
- Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: Phenotypical vs. functional differentiation. Front Immunol. 2014;5:514.
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–69.
- Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ, Reiner SL. Cutting edge: IL-12 inversely regulates T-bet and Eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J Immunol. 2006;177:7515–9.
- Bally APR, Lu P, Tang Y, Austin JW, Scharer CD, Ahmed R, Boss JM. NF-κB regulates PD-1 expression in macrophages. J Immunol. 2015;194:4545–54.
- Homey B, Alenius H, Müller A, Soto H, Bowman EP, Yuan W, McEvoy L, Lauerma AI, Assmann T, Bünemann E, et al. CCL27–CCR10 interactions regulate T cell–mediated skin inflammation. Nat Med. 2002;8:157–65.
- De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, Gunzer M, Roers A, Hogg N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. 2013;121:4930–7.
- Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–22.
- Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61.
- Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–65.
- Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–35.
- Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, Greenberg PD, Hingorani SR. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63:1769–81.
- Shen K-Y, Song Y-C, Chen I-H, Chong P, Liu S-J. Depletion of tumor-associated macrophages enhances the anti-tumor immunity induced by a Toll-like receptor agonist-conjugated peptide. Hum Vaccin Immunother. 2014;10:3241–50.
- Bader JE, Enos RT, Velazquez KT, Carson MS, Nagarkatti M, Nagarkatti PS, Chatzistamou I, Davis JM, Carson JA, Robinson C, et al. Macrophage depletion using clodronate liposomes decreases tumorigenesis and alters gut microbiota in the AOM/DSS mouse model of colon cancer. Am J Physiol - Gastrointest Liver Physiol. 2017 Oct 12:ajpgi.00229.2017. doi: 10.1152/ajpgi.00229.2017. [Epub ahead of print]
- Williams CB, Yeh ES, Soloff AC. Tumor-associated macrophages: unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer. 2016;2:15025.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and Its Ligands in Tolerance and Immunity. Annu Rev Immunol. 2008;26:677–704.
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science. 2017;355:1428–1433.
- Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–7.
- Guo Z, Wang H, Meng F, Li J, Zhang S. Combined Trabectedin and anti-PD1 antibody produces a synergistic antitumor effect in a murine model of ovarian cancer. J Transl Med. 2015;13:247.
- Lomberk GA, Iovanna J, Urrutia R. The promise of epigenomic therapeutics in pancreatic cancer. Epigenomics. 2016;8:831–42.
- Blasco F, Peñuelas S, Cascalló M, Hernández JL, Alemany C, Masa M, Calbó J, Soler M, Nicolás M, Pérez-Torras S, et al. Expression profiles of a human pancreatic cancer cell line upon induction of apoptosis search for modulators in cancer therapy. Oncology. 2004;67:277–90.
- Avan A, Crea F, Paolicchi E, Funel N, Galvani E, Marquez VE, Honeywell RJ, Danesi R, Peters GJ, Giovannetti E. Molecular mechanisms involved in the synergistic interaction of the EZH2 inhibitor 3-Deazaneplanocin a with gemcitabine in pancreatic cancer cells. Mol Cancer Ther. 2012;11:1735–46.
- Chen N-M, Neesse A, Dyck ML, Steuber B, Koenig AO, Lubeseder-Martellato C, Winter T, Forster T, Bohnenberger H, Kitz J, et al. Context-dependent epigenetic regulation of nuclear factor of activated T Cells 1 in pancreatic plasticity. Gastroenterology. 2017;152:1507–1520. e15.
- Mishra VK, Wegwitz F, Kosinsky RL, Sen M, Baumgartner R, Wulff T, Siveke JT, Schildhaus H-U, Najafova Z, Kari V, et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res 2017;45(11):6334–6349
- Dahl JA, Collas P. A rapid micro chromatin immunoprecipitation assay (ChIP). Nat Protoc. 2008;3:1032–45.