
ABSTRACT
It is well-recognized that macrophages, which arise from circulating precursors, enhance tumor progression in patients and animal models. However, less is known regarding the role of tissue-resident macrophages in metastasis. Moreover, the identification of tumor factors which influence macrophage function in the metastatic niche remains incomplete. Here, we investigated one such cytokine known as thymic stromal lymphopoietin (TSLP). Our rationale to focus on TSLP was based on two non-overlapping findings; first, TSLP exacerbates asthma in part by altering the lung macrophage response and, secondly, TSLP is produced by certain mouse and human tumor systems, although its role in neoplasia remains understudied. Thus, we tested the hypothesis that tumor-derived TSLP augments lung metastasis by rendering alveolar macrophages pro-tumorigenic. To test this hypothesis, we principally employed the 4T1 tumor model, which produces high levels of TSLP and metastasizes to the lung. TSLP loss-of-function significantly reduced spontaneous lung metastasis, as well as lung colonization. Moreover, similar outcomes were observed in both wild-type and immune-deficient hosts, suggesting that TSLP acted on innate immune cells such as macrophages. To test this notion, pharmacologic depletion of alveolar macrophages significantly reduced lung tumor growth of the TSLP-expressing, but not TSLP-deficient tumor population. In contrast, depleting macrophages originating from the circulation did not impact lung tumor growth. Lastly, TSLP increased the invasive and angiogenic gene expression profile of the alveolar macrophage population. Altogether, our study identified a novel TSLP-alveolar macrophage axis in lung metastasis, which offers new insights into mechanisms of metastasis and potential therapeutic targets.
Introduction
It is now clear that host innate and adaptive immune responses against neoplastic disease are critical determinants of patient outcome.Citation1 Regarding the myeloid response in cancer, high macrophage infiltration has been tied to a poor prognosis.Citation2-5 The field of cancer biology has largely focused on the role of tumor-associated macrophages (TAMs), which are recruited to tissues as inflammatory monocytes in response to tumor- or stromal-derived chemotactic factors such as CCL26 and CSF-1.Citation7,Citation8 In cancer settings, TAM products can facilitate tumor progression to metastasisCitation9 via various mechanisms, both immune system-dependent and –independent, such as immune suppression and tumor angiogenesis, respectively.Citation10 However, an increasing number of studies suggest that TAMs are heterogeneous and can possess pro- or antitumor roles in response to the inflammatory composition of the tissue microenvironment. For example, IFN-γ stimulates macrophage production of immune-activating cytokines, such IL-12, facilitating CD8+ cytotoxic T cell differentiation and effector function.Citation11 In contrast, in response to IL-4 and/or IL-13, macrophages produce immune suppressive factors, such arginase-1, IL-10 and TGF-β, which antagonize T cell activation and function.Citation12,Citation13
In addition to macrophages derived from circulating monocytes, tissue-resident macrophages reside within tissues or organs. The role that these tissue-resident macrophages play in neoplastic progression is less understood. This is important, as common metastatic sites such as the lung, liver and brain have specific tissue-resident macrophage populations.Citation14 In contrast to TAMs, tissue-resident macrophages are yolk-sac-derived and self-populate within tissues.Citation15,Citation16 Due to their existence in tissues prior to the arrival of tumor cells, it is thought that these cells are receptive to circulating tumor-derived factors (TDFs)Citation17,Citation18 and act to condition tissues for tumor colonization and proliferation via various mechanisms.Citation19 Despite accumulating evidence for a role of tissue-resident macrophages in metastasis,Citation20,Citation21 the TDFs that influence their biology are largely unknown. Moreover, the contribution of tissue-resident macrophages to neoplastic progression and their impact on patient outcome remains unclear.
TDFs within the inflammatory microenvironment can dictate the invasive potential of tumors and may do so at least in part by engaging or mobilizing macrophages,Citation22 as noted earlier. One such TDF, thymic stromal lymphopoietin (TSLP), has recently been identified in human and mouse cancersCitation23,Citation24 including mammary carcinoma,Citation25 although its precise role in these cancer settings remains elusive. Earlier studies revealed a role for TSLP in promoting primary tumor growth, but did not investigate how TSLP influenced the metastatic process. Conversely, other work showed that TSLP plays an antitumor role, especially early in tumorigenesis,Citation26 suggesting that its action in cancer settings may be context-dependent. These divergent studies support a need for further investigation of TSLP in tumor biology.Citation27
In mouse models of lung disorders, such as allergy, TSLP enhances CD4+ Th2 cell, as well as M2-like macrophage, responses. Furthermore, TSLP is upregulated in the lung during episodes of asthma, suggesting that immune cells in the lung may be poised to respond to TSLP and play key roles in tissue pathology. The observations that TSLP has been tied to type-2 immunity in the lungCitation28 and that type-2 immunity has been broadly linked to tumor progression,Citation29 suggest that TSLP may contribute to lung disease processes, such as lung tumor growth or metastasis. Moreover, the impact of TSLP on macrophage function in cancer, and tissue-resident macrophages more precisely, has not yet been investigated. Thus, we hypothesized that TSLP enhances lung metastasis or colonization by rendering the tissue-resident alveolar macrophage (AM) population pro-tumorigenic.
Altogether, our findings demonstrate a novel TSLP/AM macrophage axis that potentiates lung tumor metastasis and colonization. The gene expression profile of TSLP-exposed AMs in vitro and in vivo is highly consistent with a phenotype supportive of increased tumor growth and metastasis. With the knowledge that the immune response is critical to patient outcome, immunotherapy is on the forefront of cancer research. Our work supports the notion that in cancers whereby TSLP is produced, TSLP blockade may serve as a unique approach to ‘rescue’ the macrophage response and improve therapies that require an intact myeloid system.
Results
Loss of tumor-derived TSLP decreases tumor progression
To test our hypothesis that TSLP alters AM function to enhance metastasis, we used the 4T1 mammary tumor model that produces high levels of TSLP and metastasizes to the lung.Citation30,Citation31 Several other mammary tumor cell lines were tested for TSLP expression including AT-3, E0771 and EMT6,Citation32-34 but only 4T1 cells expressed detectable levels of TSLP (Fig. S1A). We showed that TSLP circulates systemically following orthotopic implantation (), a prerequisite for its ability to act in distant sites. Serum TSLP concentration increased in direct correlation with tumor volume. While earlier studies showed that loss of TSLP in 4T1 cells decreased primary tumor growth,Citation30 the effect this had on metastasis remained unknown. We therefore made use of a shRNA-based loss-of-function approach to knockdown TSLP expression. Several constructs were tested and all successfully reduced TSLP expression relative to the vector control (VC) population (Fig. S1B). We randomly selected the 4T1 population that expressed construct ID v2MM_53644, which maintained low expression levels of TSLP and grew efficiently in vitro. To ensure stability of the phenotype, we generated multiple clones and confirmed TSLP knockdown by mRNA expression of two separate clones ( and Fig. S1C). We also confirmed TSLP loss in both clones at the protein level ( and Fig. S1D).
Figure 1. Knockdown of TSLP reduces primary tumor growth and spontaneous metastasis. (A) Serum TSLP concentration as measured by ELISA in 4T1 tumor-bearing BALB/c mice (n = 11) reflecting varying tumor volumes. Data analyzed by Spearman correlation. (B) TSLP expression as measured by qRT-PCR analysis. Data normalized to the housekeeping gene GAPDH and 4T1-VC group set to 1.0 to determine relative expression of the 4T1-KD group (silenced with construct ID v2MM_53644; clone 1). (C) TSLP concentration in cell-free supernatants of the indicated cell lines (1 × 106 cells/ml after 24 hours), as measured by ELISA. Results in B & C represent the mean ± SEM of 3 or 4 separate experiments, respectively. (D) Tumor growth of the indicated cell population in wild-type (WT) female BALB/c mice. Growth differences are significant at all time points measured (each line represents a single mouse (n = 16 4T1-KD, n = 17 4T1-VC; compilation of 3 independent experiments). (E) Spontaneous metastasis of the indicated cell populations at endpoint when primary tumor volumes between groups were comparable. Lung metastatic foci quantified using H&E-stained slides (each point represents an individual mouse; compilation of 3 independent experiments; n = 10 4T1-KD, n = 13 4T1-VC). (F) Survival post-surgical resection of the primary tumor in WT female BALB/c mice (n = 17 4T1-KD, n = 14 4T1-VC; compilation of 2 independent experiments). *P < 0.05, **P < 0.01.
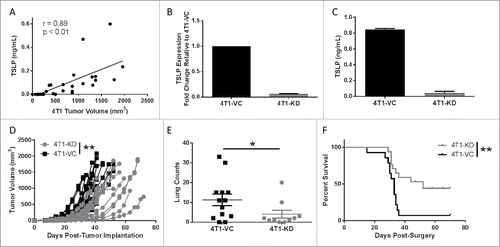
Next, we examined whether TSLP-deficiency affected tumor growth in vitro. Altogether, no significant differences in tumor cell growth were observed between the 4T1-VC and 4T1-KD (clone 1), as measured by 3H-thymidine uptake, MTS assays or colony formation (Fig. S2A-C). We confirmed that these cell populations did not differ in their ability to produce G-CSF (Fig. S2D), another cytokine abundantly produced by 4T1 cells.Citation32 To determine whether tumor-derived TSLP levels impacted tumor growth in vivo, we orthotopically implanted either 4T1-KD (clone 1 or clone 2) or 4T1-VC cells into syngeneic immune competent hosts and measured tumor growth. In both cases, 4T1-KD cells grew significantly slower compared to 4T1-VC cells ( and Fig. S1E), which is consistent with earlier findings showing that reduced TSLP levels in 4T1 cells diminished primary tumor growth rate.Citation30 We further showed that 4T1 cells did not express the TSLP receptor (Fig. S2E), making it unlikely that any difference in tumor growth rate in vivo was due to an intrinsic effect of TSLP, but rather an extrinsic effect involving host interactions.
To determine if TSLP levels impacted spontaneous metastasis, we conducted two types of analyses, either leaving intact or surgically removing the primary tumor. In regard to the former analysis, we allowed primary tumors to grow to similar volumes in both groups of mice (Fig. S3A), and evaluated metastatic burden in the lungs. Although both groups of mice bore equivalent primary tumor sizes at endpoint, we observed significantly decreased lung metastasis in mice bearing 4T1-KD cells (clone 1 or clone 2) compared to those bearing 4T1-VC cells ( and Fig. S1F). We randomly selected 4T1-KD clone 1 (henceforth termed, 4T1-KD) for all subsequent experiments. Then, we orthotopically implanted both groups of tumor cells and when primary tumor size reached 150 mmCitation3 they were surgically removed and mice were followed for survival. Mice were carefully monitored for 70 days post-surgery. Mice bearing 4T1-KD cells displayed a significant survival advantage over those bearing 4T1-VC cells (). What was even more striking was that while >90% of mice bearing 4T1-VC cells succumbed to lung metastasis, ∼50% of mice bearing 4T1-KD cells did not develop detectable metastasis (based on histologic analysis at endpoint). These data demonstrated that tumor-derived TSLP plays a significant role in spontaneous metastasis.
Extravasation alone cannot account for differences in lung metastasis
To further elucidate the basis for differences in metastasis, we focused on the process of extravasation, which may have affected the numbers of tumor cells successfully reaching the lung. To address this, we utilized an experimental lung metastasis approach, whereby tumor cells are injected into the bloodstream, bypassing steps such as extravasation from the primary tumor site. We observed significant decreases in lung tumor growth in mice receiving 4T1-KD cells compared to those receiving 4T1-VC cells ( and ), which was consistent with our earlier findings ( and Fig. S1F).
Figure 2. The pro-metastatic effects of TSLP are not due solely to differences in extravasation. In all panels, WT female BALB/c mice were injected intravenously with the indicated cell population (5 × 104cells/mouse). Mice were euthanized when the 4T1-VC group showed signs of morbidity, typically 26–30 days post-injection. (A) Images post-India ink staining of tumor-bearing lungs, with data quantified in (B) (each point represents an individual mouse; compilation of 3 independent experiments; n = 12 4T1-KD, n = 11 4T1-VC). (C) RT-PCR analysis (top) and quantification (bottom) of AH1 expression in the lungs of non-tumor-bearing (NTB; n = 2) or the indicated tumor-bearing mice 2–3 days post-tumor injection. (D) RT-PCR (top) and quantification (bottom) of TSLP expression of tumor cells propagated ex vivo from the lungs of the indicated tumor-bearing mice. In C and D, each lane represents an individual mouse (n = 4–5 mice per group). *P < 0.05; **P < 0.01; ns, P = 0.68.
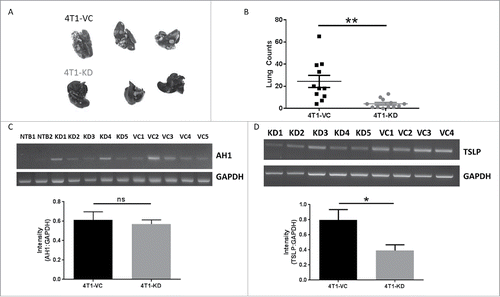
To exclude the possibility that differences in lung tumor growth reflected differences in lung infiltration, we quantified AH1 expression in the lungs of tumor-bearing mice at early time points (2–3 days) post-inoculation. AH1 (gp70) is a tumor antigen that is expressed by 4T1 and other mouse tumor cell lines, but not normal lung tissue,Citation35 thus serving as a surrogate marker of lung tumor infiltration. As expected, we observed no detectable AH1 expression in the lungs of non-tumor bearing mice. In contrast, AH1 expression was detectable in both groups of tumor-bearing mice and was similar at such early time points (). Finally, to ensure that the cells maintained their TSLP expression patterns in vivo, we collected lungs from tumor-bearing mice at endpoint and cultured single cell suspensions under selection with 6-thioguanine, an agent to which 4T1 has intrinsic resistance,Citation31 eliminating all cells in the lung except tumor cells. The resulting populations were analyzed for TSLP expression. We found that both tumor populations retained their expected TSLP expression profiles in that 4T1-KD cells expressed significantly lower levels of TSLP compared to 4T1-VC cells, suggesting that tumor expression of TSLP was stable throughout the course of tumor growth (). Overall, these data suggested that TSLP-mediated effects on lung tumor growth are likely due to interactions with the host, particularly within the lung microenvironment.
TSLP exerts its effects on metastasis independent of adaptive immunity
To better unravel the mechanisms by which TSLP influences metastasis, particularly the involvement of the adaptive immune system, we performed tumor growth experiments in immune-deficient (SCID) mice. This was based on earlier findings that implicated the CD4+ T cell response, likely the Th2 subtype, in the mechanism by which TSLP enhanced primary tumor growth.Citation24,Citation25,Citation30 Consistent with that finding, we observed that primary tumor growth rates of both TSLP-expressing and TSLP-deficient cells were comparable in SCID mice (). Thus, these data supported the notion that TSLP-mediated effects on primary tumor growth were dependent upon adaptive immune responses ( and ). We confirmed the importance of CD4+ T cells using an anti-CD4-depletion strategy, which increased the growth of 4T1-KD cells to levels similar to that seen with 4T1-VC cells (Fig. S3B).
Figure 3. TSLP-mediated effects on metastasis are independent of the adaptive immune system. (A) Primary tumor growth of the indicated cell population in SCID mice, as in (n = 15 per group). (B) Quantification of spontaneous lung metastasis of the indicated cell population at endpoint tumor volumes in A (n = 15 per group). (C) Experimental lung metastasis in SCID mice, as in (n = 9-10 per group). (D and E) Similar to A and B, except that tumor growth studies were performed in athymic mice (n = 9-10 per group). Each line or data point represents results from an individual mouse; compilation of 2 independent experiments in all panels. **P < 0.01; ***P < 0.001; ns, P > 0.1.
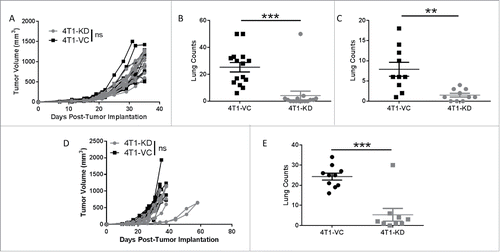
To determine whether immune status impacted metastasis, lungs were histologically analyzed at endpoint when tumor volumes remained similar. Interestingly, we still observed a significant difference in metastasis between the two tumor-bearing groups (), similar to immune competent mice (). To determine whether differences in extravasation from the primary tumor site accounted for differences in metastasis, we injected tumors intravenously in SCID mice and quantified lung tumor foci at endpoint. As with immune competent hosts (), the differences in lung tumor growth were maintained (). These experiments were then extended to a second immune-deficient mouse model; i.e., the athymic mouse model. As with SCID mice, we observed no overall significant difference in primary tumor growth between the two different tumor populations, but still observed significant differences in metastasis at endpoint ( and ). Taken together, these data suggest that tumor-derived TSLP exerts its effects on primary tumor growth via its action on CD4+ T cells, and its effects on metastasis and colonization are independent of cellular elements of the adaptive immune system but, perhaps, depend upon components of the innate immune response.
Next, we sought to extend the relationship between TSLP and metastasis using a second tumor model. To that end, we identified a second tumor cell line that produces TSLP known as KCMH-1, a squamous cell carcinoma.Citation36 As with 4T1 cells, we silenced TSLP expression in KCMH-1 cells and then evaluated differences in tumor growth using the experimental lung metastasis approach in SCID hosts (Fig. S4). First, we showed that KCMH-1 cells, silenced with the same TSLP-shRNA construct used in 4T1 cells (), did not express detectable levels of TSLP compared to the TSLP-expressing population (Fig. S4A). Second, we showed that the loss of TSLP did not alter tumor cell growth in vitro or tumor cell growth in vivo (Fig. S4B and S4C), which was similar to that seen with the 4T1 model. Third, since this model is not as well-characterized as 4T1, we sought to evaluate differences in lung tumor burden at varying input tumor cell concentrations. In doing so, we observed a consistent pattern of diminished lung tumor burden at all three inoculum concentrations in mice bearing the TSLP-deficient population compared to the TSLP-expressing population (Fig. S4D). This analysis is consistent with the notion that increased levels of TSLP enhance tumor growth in the metastatic microenvironment and strengthen our findings in the 4T1 model regarding the novel relationship between TSLP and metastasis.
TSLP mediates its effects on metastasis through an AM-dependent mechanism
Based on the notion that TSLP exerts its effects on metastasis via innate immune responses and that TSLP has been shown to alter macrophage responses in the lung in non-cancer models,Citation28 we next investigated whether TSLP acted through macrophages to enhance metastasis in our models. We therefore employed a well-established pharmacologic approach to deplete macrophages utilizing clodronate liposomes. Several groups have shown route-specific depletion effects: while intraperitoneal administration of clodronate liposomes effectively eliminates monocyte-derived inflammatory macrophages (IMs),Citation37 intranasal administration of this agent depletes the AM compartment.Citation38,Citation39 Because TSLP has been strongly linked to the lung microenvironment,Citation40,Citation41 we hypothesized that TSLP would have profound effects on AMs, but nonetheless performed depletions in both compartments. We chose the experimental lung metastasis approach to test this hypothesis in an effort to focus specifically on actions within the lung microenvironment.
We demonstrated that intranasal clodronate administration had a profound effect on lung tumor burden of mice receiving 4T-VC cells, and importantly, reduced their growth to an extent comparable to that of 4T1-KD cells (). Clodronate treatment had no significant effect on reducing lung tumor burden in mice receiving 4T1-KD cells. To confirm that this treatment selectively depleted AMs, but not IMs, we performed flow cytometric analysis in tumor-bearing mice at endpoint. Our data showed that intranasal clodronate treatment significantly reduced the frequency of AMs, as defined by the phenotype CD45+CD11blo/- CD11c+F4/80+, but had no significant effect on IMs, as defined by the phenotype CD45+CD11b+F4/80+ ( and ).
Figure 4. TSLP exerts its pro-metastatic effects via alveolar macrophages. (A) The indicated tumor cell population was injected intravenously into BALB/c mice (n = 14-15 per group; compilation of 3 independent experiments) as in , and then administered either clodronate (indicated by “+”) or control liposomes (indicated by “-”) via an intranasal route as described in the Materials and Methods. VC+ vs KD+, P = 0.06; KD+ vs. KD-, P = 0.08. (B) Quantification of AM or IMs (mean ± SEM of 3 separate mice per condition), based on the flow gating strategy (C) at endpoint. AMs were first gated on CD45+ cells, followed by gating on CD11blo/− cells and then CD11c+F4/80+ cells. IMs were first gated on CD45+ cells, followed by gating on CD11b+F4/80+ cells. ns, P > 0.07 for all comparisons. (D) Similar to A, except that clodronate or control liposomes were delivered intraperitoneally into mice (n = 9-10 mice per group; compilation of 2 independent experiments). VC+ vs. VC-, P = 0.08. (E) Quantification of macrophages in the spleen (ns, P = 0.51) or peripheral blood (mean ± SEM of 4–5 mice per condition; ns, P = 0.11) based on the flow gating strategy in Supplemental Figure 5. *P < 0.05; **P < 0.01; ns, not significant.
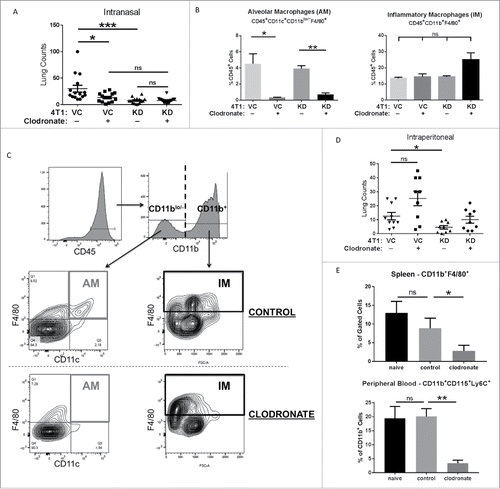
We extended these studies to intraperitoneal administration of clodronate to determine if depletion of IMs impacted lung tumor burden. In contrast to the intranasal route, intraperitoneal clodronate treatment did not reduce lung tumor burden in mice receiving 4T1-VC cells relative to administration with control liposomes (). We confirmed that this route systemically depleted macrophages, such as in the spleen (i.e., CD11b+F4/80+) and circulation (i.e., CD11b+CD115+Ly6C+) (; Fig. S5A and S5B). To further demonstrate that IMs were not involved in affecting lung tumor burden, we utilized anti-CCL2 or anti-CSF-1 antibodies, as previously reported.Citation6,Citation8 However, in our models, neither of these agents had any significant effect on reducing tumor burden (data not shown), strengthening our conclusion that TSLP does not mediate its effects via IMs. Thus, our data are consistent with the hypothesis that AMs play important roles during TSLP-mediated mechanisms of metastasis, but not necessarily the entire metastatic outcome as 4T1-KD cells still formed lung tumor foci with or without AM depletion. This specific or selective role for AMs in TSLP-mediated metastasis is supported by our observations that IMs remained present and were unaffected after clodronate treatment ().
Lastly, to further address the specific or selective involvement of AMs compared to other myeloid populations, we analyzed tumor-bearing mice for the accumulation of myeloid-derived suppressor cells (MDSCs), another pro-tumor myeloid population strongly induced in the 4T1 tumor model.Citation32,Citation42 To that end, we analyzed both the peripheral blood and lungs of tumor-bearing mice at experimental endpoint for the two major MDSC subsets, monocytic (M-MDSC) or polymorphonuclear (PMN-MDSC). Analysis was performed based on the following canonical marker phenotypes: M-MDSCs, CD11b+Ly6ChiLy6G−; and PMN-MDSCs, CD11b+Ly6Clo Ly6G+.Citation42 In lung tissue, the CD45 marker was also included to distinguish hematopoietic from non-hematopoietic cell types. We observed no significant change in M-MDSC frequency in the peripheral blood or lungs of 4T1-VC or 4T1-KD tumor-bearing mice compared to the NTB controls (Fig. S3C and S3D). The most significant change was observed by PMN-MDSCs (as we and others have reported in the 4T1 modelCitation32,Citation43) in the peripheral blood and lungs, wherein both the 4T1-VC and 4T1-KD tumor-bearing mice displayed substantial increases in the frequencies of these cells compared to the NTB controls (Fig. S3C and S3D). Importantly, there was no significant difference in PMN-MDSC accumulation in mice bearing 4T1-VC vs. 4T1-KD tumors in either the blood or lungs. These data suggest that altering tumor-derived TSLP levels did not impede MDSC production, particularly the PMN-MDSC subset, and that the differences in metastasis could not be correlated to MDSC burden. Altogether, these data strengthen the hypothesis that TSLP mediates its effects on lung tumor growth via alterations in AM biology.
TSLP alters the invasive/angiogenic gene signature of AMs
Upon discovering that depletion of AMs reduced the growth of TSLP-expressing tumor cells in the lung, we sought to address in what ways TSLP was changing the biology of these cells. We hypothesized that VEGF-A may be differentially regulated in settings of high versus low TSLP. VEGF-A has been extensively studied as a pro-metastatic cytokine produced by macrophages that acts independent of immune suppressive mechanisms.Citation19,Citation44,Citation45 In these studies, a co-culture system was established in which we combined AMs from naïve mice with tumor cells, either 4T1-KD or 4T1-VC, in a contact-independent transwell system and RNA was extracted from the AMs for qRT-PCR analysis (). This allowed for exchange of tumor-derived cytokines, including TSLP, while separating macrophages from tumor cells. Additionally, to demonstrate that any observed effect on gene expression was due specifically to TSLP levels, we added recombinant TSLP to separate wells of the AM/4T1-KD co-culture system. Our results demonstrated that VEGF-A expression in AMs increased significantly in settings of high vs. low TSLP. Moreover, the addition of recombinant TSLP to AM/4T1-KD co-cultures restored the expression of VEGF-A to the level seen with AM/4T1-VC co-cultures ().
Figure 5. VEGF-A is differentially expressed in AMs in vivo in response to TSLPhi vs. TSLPlo tumor settings. (A) Strategy shown for the experimental design and RNA sample selection for panel B. (B) VEGF-A expression as measured by qRT-PCR analysis. 100 ng/mL recombinant TSLP was added to the AM/4T1-KD co-culture system and gene expression was quantified. (C) VEGF-A expression of AMs in vivo collected from the indicated tumor-bearing mice at 7 (left), 14 (middle) or 28 (right) days post-intravenous injection. For B and C, data were normalized to the housekeeping gene GAPDH. Then one 4T1-VC sample in panel B (n = 3 biologic replicates) or one 4T1-VC sample from each time point in panel C was set to 1.0 to determine the relative expression of the other samples. Panel C represents a total of 9 separate mice per tumor-bearing group, covering the 3 distinct time points shown. Therefore, at each time point, 3 separate mice from each cohort were analyzed, and for each mouse, 3–4 technical replicates were collected. Results represent the mean ± SEM of all replicates for each tumor-bearing group at each time point. *P < 0.05; **P < 0.01; ***P < 0.001; ns, P > 0.1.
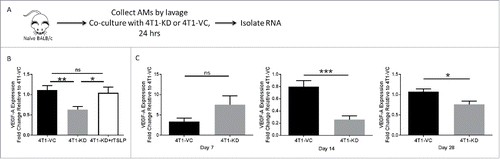
We next sought to determine if VEGF-A expression in AMs was differently regulated in vivo in response to the different TSLP-expressing tumors. We injected 4T1-VC or 4T1-KD cells intravenously to establish tumor growth in the lung and extracted AMs from these mice at 7, 14 or 28 days post injection. While VEGF-A was not differentially regulated at 7 days regardless of tumor TSLP levels, there was significantly decreased VEGF-A expression in AMs of mice that received 4T1-KD cells compared to mice that received 4T1-VC cells 14 days post-tumor injection, which was maintained at endpoint ().
Based on the finding that TSLP altered VEGF-A expression in AMs, we sought to determine whether there was a more global change in the invasive/angiogenic gene signature of these macrophages. Moreover, a major immune system-independent mechanism by which macrophages enhance metastasis is via tissue remodeling and angiogenesis.Citation44 We therefore utilized a PCR Array system focused on this particular gene profile. This experiment was conducted using the co-culture system as in to identify genes that were differentially regulated in settings of high vs. low tumor-derived TSLP. In both arrays, about 30% of the 84 genes tested were modulated in settings of high vs. low tumor-derived TSLP ( and ). Of those genes, 26 were similarly upregulated between the biologic replicates when AMs were co-cultured with 4T1-VC cells compared to 4T1-KD cells (), and five genes were highly upregulated. Interestingly, these five upregulated genes have been shown to enhance metastasis in various tissue modelsCitation45: VEGF-A, MMP9, CXCL5, PDGF-A and CTGF. VEGF-A, PDGF-A and CTGF are associated with increasing vascular supply to tumors to support their metastatic seeding and growth.Citation46-48 MMP9 is known to promote tissue remodeling, which is essential for the invasion of normal tissue by tumor cells.Citation49 CXCL5 is a chemoattractant for CXCR2-expressing cells, namely neutrophils and MDSCs, which have been shown to promote tumor progression.Citation50,Citation51
Figure 6. TSLP alters the tissue remodeling and angiogenesis pathways in AMs. AM/tumor co-cultures were utilized for all panels; strategy for the experimental design and RNA sample selection shown in . (A) Heat-map of differential gene expression between AMs co-cultured with either 4T1-VC or 4T1-KD cells in two independent biologic experiments. Data shown highlight the genes most highly upregulated (red) or downregulated (green) (by 4.66-fold) for the two biologic replicates. (B) Data summarizes the number of differentially regulated genes (>2-fold, up or down) in 4T1-VC vs. 4T1-KD cells among the 84 total genes analyzed in this format. (C, left; D) Selected genes in A were validated by qRT-PCR analysis using the same RNA employed for the expression array. Data were normalized to the housekeeping gene GAPDH, and the 4T1-VC group set to 1.0 to determine relative expression of the 4T1-KD group. (C, right) VEGF-A protein levels as measured by ELISA. For data in C and D, the results represent the mean ± SEM of all replicates (n = 4/experiment or 8 total for the ELISA; n = 3–4/experiment or 6–8 total for the qRT-PCR) covering the two independent biologic experiments performed at different times. (E) The indicated tumor cell population was implanted orthotopically into mice (n = 3–5 per group), and the lungs were collected at endpoint for analysis by immunohistochemistry for CD31 expression. Left, Representative photomicrographs of CD31-immunostained metastatic lesions of mice bearing the indicated tumor cell population (20X magnification; scale bar represents 100-μm). Right, Quantification of microvessel density, as described in the Methods section. *P < 0.05; **P < 0.01; ***P < 0.001.
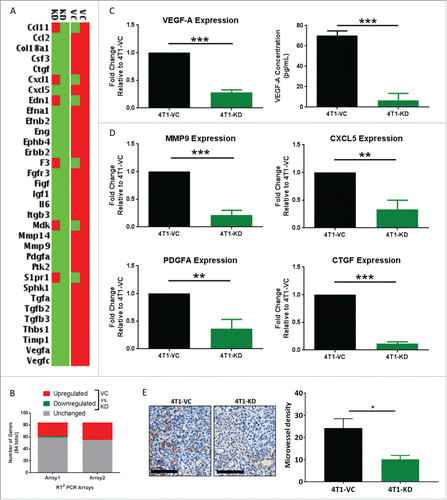
We validated the expression of these five genes, and indeed found them to be highly expressed in AMs after exposure to 4T1-VC cells compared to 4T1-KD cells (, left; ). To confirm that differences in gene expression translated to differences in protein levels, we measured VEGF-A levels and showed that AMs co-cultured with 4T1-KD cells secreted significantly less VEGF-A compared to AMs co-cultured with 4T1-VC cells (). Interestingly, such gene differences were not observed with co-cultures of tumor cells and bone marrow-derived macrophages, which are representative of IMs (Fig. S6A-D). The finding that bone marrow-derived macrophages did not respond in the same way as AMs in this in vitro co-culture system was unlikely due to the absence of the TSLP receptor, as both macrophage sources expressed it (Fig. S2E and Fig. S7). This, together with our data that depleting IMs did not impact lung tumor burden, suggests that the effect of TSLP is specific to macrophages of the lung microenvironment.
To assess functional changes in the lung, such as angiogenesis in response to tumor-derived TSLP, we sought to quantify CD31+ microvessel density in mice bearing either 4T1-VC or 4T1-KD tumor cells. We orthotopically implanted tumor cells and measured CD31 expression by immunohistochemistry within the metastatic lesions. shows photomicrographs of CD31-immunostained lung sections from such tumor-bearing mice. Lung sections from 4T1-VC tumor-bearing mice showed a large number of CD31+ endothelial cell clusters distributed within the metastases. In comparison, lung metastases from 4T1-KD tumor-bearing mice showed fewer vessels. Quantitative analysis revealed a statistically significant lower microvessel density in lung metastases of mice bearing 4T1-KD tumor cells compared to those bearing 4T1-VC tumor cells (), consistent with the pattern of VEGF-A expression. Overall, these data are consistent with the hypothesis that TSLP enhances metastasis by increasing tissue remodeling and angiogenic genes in AMs.
Discussion
Understanding mechanisms of tumor progression to metastasis are critical to the development of prognostic or therapeutic targets. Inhibition of T cell-mediated immunity and mobilization of pro-tumorigenic macrophages represent major determinants of metastatic potential.Citation45 There are two major sources of pro-tumorigenic macrophages: bone-marrow-derived, which are recruited via the circulation, or tissue-resident, which dwell within the local tissue/organ environment. The vast majority of studies devoted to macrophage-tumor biology, however, have focused on the former population, broadly termed tumor-associated macrophages (TAMs). Less is known regarding the influence of tissue-resident macrophages; yet, a few studies have documented pro-tumor TAM-like activities.Citation20,Citation21 Thus, more work is needed to fully appreciate the relevance of tissue-resident macrophages to metastasis, particularly since this population already inhabits potential metastatic microenvironments at the time of primary neoplastic growth. We hypothesize that under such circumstances, TDFs travel systemically from the primary site to distal tissues/organs, ‘conditioning’ such resident macrophage populations to support the metastatic process.
The goals of this study were two-fold: 1) to further investigate mechanisms of tumor metastasis, with emphasis on the understudied TDF, TSLP; and 2) to determine the role of tissue-resident macrophages, particularly AMs, in this mechanism. We focused on AMs based on the rationale that TSLP contributes to various lung disordersCitation28,Citation40 and that the 4T1 model proficiently metastasizes to the lung.Citation31 Our data demonstrate that: i) tumor-derived TSLP enhances lung metastasis in multiple settings, including those with or without surgical removal of the primary tumor or bypassing the primary tumor altogether using an experimental metastasis model; ii) tumor-derived TSLP mediates these pro-metastatic effects in the absence of the adaptive immune system, as determined using SCID or athymic hosts, thus precluding immune suppression as a mechanistic basis; iii) elements of the innate immune system, namely tissue-resident AMs, play important roles in mediating the pro-metastatic effects of TSLP; and iv) TSLP alters the angiogenic/tissue remodeling profile of the AMs, supporting the hypothesis that AMs modified by TSLP enhance their pro-metastatic activities. Thus, this work identifies a novel TSLP-macrophage axis in cancer biology.
Importantly, the 4T1 model was selected based on the rationale that it produces high levels of TSLP and metastasizes proficiently to the lung.Citation30,Citation31 Earlier work used this model to reveal the pro-tumorigenic actions of TSLP in primary tumor growth and the dependence of the CD4+ T cell response on that outcome.Citation24,Citation30 We confirmed this finding, showing that tumor-derived TSLP accelerates primary tumor growth in immune competent, but not in CD4+ T cell-deficient hosts. What remained unclear was the impact of tumor-derived TSLP on metastasis and the cellular basis underlying that outcome. Here, we expanded on the role of this understudied TDF in the neoplastic process and demonstrated an unrecognized influence on metastasis. Thus, this is the first description of a mechanism by which TSLP enhances metastasis and via alterations in AM biology. Why one tumor site is T cell-dependent, while another site T cell-independent is not clear, but may reflect in part the complexity and nature of the tissue-specific microenvironment.
Our data support the hypothesis that differences in metastatic phenotype reflected differences in TSLP expression and its impact on the host, based on the observations that both TSLP-expressing and -deficient 4T1 populations: i) proliferated similarly in vitro and in vivo in immune-deficient or CD4+ T cell-depleted hosts; ii) accumulated similarly in the lungs early after intravenous injection; iii) did not express detectable levels of cell surface TSLP receptor; iv) influenced AM biology in a distinctive manner, resulting in a tissue remodeling/angiogenic gene expression profile that aligned with the corresponding metastatic phenotype, including VEGF-A, a well-recognized pro-tumorigenic factor; and v) induced similar levels of VEGF-A expression upon the addition of recombinant TSLP to the AM/4T1-KD co-culture system.
To demonstrate a role for AMs in TSLP-driven metastasis, we depleted this macrophage population using clodronate liposomes delivered intranasally, which led to significantly reduced lung tumor burden. In contrast, depletion of AMs had no significant effect on lung tumor burden in settings of low tumor TSLP (i.e., 4T1-KD cells), demonstrating that TSLP influences tumor growth by engaging AMs. It is important to point out that 4T1-KD cells still formed some metastatic foci, consistent with TSLP-independent mechanisms of tumor survival, growth or colonization. In contrast, systemic administration of clodronate, which led to significantly less IMs, did not reduce 4T1-VC lung tumor burden, further supporting a role for AMs in this mechanism. It is also important to point out that these studies made use of the experimental metastasis approach to focus specifically on interactions within the lung microenvironment. Efforts to extend this analysis to a post-surgical setting, however, were met with technical challenges, as mice were unable to survive the treatment. Therefore, further studies are needed to examine the effect of AM depletion in settings of spontaneous metastasis. Moreover, we showed that MDSCs were still generated, regardless of tumor-derived TSLP levels, and did not correlate with metastatic outcome, thereby strengthening the hypothesis that TSLP influenced metastasis via an AM-dependent mechanism.
We have also identified novel changes in macrophage biology, namely AMs but not IMs in response to TSLP. Our data demonstrate that in response to tumor-derived TSLP, AMs upregulated a number of genes involved in tissue remodeling and angiogenesis. Although it remains to be investigated in detail what genes are causally responsible, it is well-appreciated that macrophage products such as VEGF-A and MMP9 play significant roles in the metastatic process.Citation48,Citation49 In accord with this concept, we showed that the CD31+ microvessel density within the metastatic lung lesions of mice bearing 4T1-KD cells was significantly lower compared to those bearing 4T1-VC cells, supporting a relationship between angiogenesis and tumor-derived TSLP levels. Additional studies are warranted, however, to further investigate in detail this potential tumor-derived TSLP-endothelial cell axis; for example, by determining the impact of AM depletion on CD31 expression in mice bearing 4T1-VC vs. 4T1-KD tumors. Moreover, while we identified TSLP as a major player in this process, it is unclear which TDFs act in concert with TSLP to exert these effects, as TSLP alone has not been shown to be a ‘polarizing’ factor for macrophage functionalityCitation52; thus, more detailed studies are needed. Additionally, it remains to be fully understood what signaling mechanisms underlie the changes in AM behavior; therefore, future studies are necessary to focus on additional elements of the TSLP-TSLP receptor axis and the downstream events ensuing TSLP receptor engagement.
Altogether, these data suggest that TSLP plays a key role in lung metastatic outcome via its action on AMs in the lung microenvironment. It is also possible that TSLP may redirect metastasis to other sites beside the lung, including metastasis to lymph nodes, which warrant further investigation. This is consistent with a study in human gastric carcinoma,Citation53 which showed a significant relationship between TSLP overexpression and metastasis to lymph nodes. Thus, from a conceptual standpoint, our work may have broader implications in cancer types, independent of their origin, that overproduce TSLP. Our findings provide a rationale to explore novel therapeutic targets and support the concept of localized administration of therapeutics to common metastatic sites such as the lung. Interestingly, anti-TSLP therapies are already being examined in clinical trials in asthma, where patients are receiving inhaled anti-TSLP blocking antibodies.Citation54 Thus, while more work is required to determine if this is a feasible approach in cancer models, our data suggest that this may be an appropriate line of investigation in cancer types that produce TSLP.Citation24,Citation25,Citation53 Additionally, several studies are focused on targeting macrophages or their products in cancer. Our data supports this concept and suggests that locally depleting or altering the activities of tissue-resident macrophages in metastatic microenvironments may also represent viable modalities. In summary, our work identifies a novel tumor TSLP-macrophage axis that contributes to metastatic potential and could shed light on novel approaches to cancer therapy.
Materials and methods
Mice: Female BALB/c (BALB/c; H-2d) and athymic (nude; nu/nu) mice were obtained from Charles River (Frederick, MD). Female SCID mice (C.B IgH-1b/Cr) were obtained from the Animal Breeding Facility at Roswell Park Cancer Institute (Buffalo, NY). All animal studies were approved and performed under Institutional Animal Care and Use Committee (IACUC) protocols 1117M and 1108M.
Tumor cell lines: The 4T1 cell line was obtained from ATCC (Manassas, VA) and maintained as described.Citation55 The KCMH-1 cell line was kindly provided by Dr. Noriyasu Hirasawa at Tohoku University in Japan and maintained as described.Citation36 To modify TSLP levels produced by 4T1 and KCMH-1 cells, we used shRNA methodology to silence TSLP expression using a pSM2 retrovirus system (GE Dharmacon, Lafayette, CO) expressing different shRNA-TSLP constructs or an empty vector control (VC) (sequences in Supplemental Table 1). Transductions were performed by the RPCI shRNA Shared Resource. The efficiency of knockdown was measured at RNA and protein levels, as described below, and stably persisted in vitro under puromycin selection (3µg/ml). Based on the efficiency of knockdown in pilot experiments, we selected 4T1 and KCMH-1 cells transduced with construct ID v2MM_53644. To ensure stable knockdown for the multiple experiments involving 4T1 cells, single cell clones were established expressing this construct, termed 4T1-KD clone 1 (henceforth, 4T1-KD) or 4T1-KD clone 2. Cell lines were maintained in vitro for one month and fresh aliquots were obtained from a cryopreserved source of similar lot number. Tumor TSLP levels were verified prior to use. Cell lines were tested for mycoplasma by PCR and mycoplasma-negative tumor cells were used in all experiments.
Tumor growth experiments: 5 × 104 4T1 cells were implanted orthotopically into mammary gland #4 of syngeneic female mice or intravenously injected via the tail vein at 8–12 weeks of age. Primary 4T1 tumor growth was measured as described.Citation55 Surgical removal of the primary tumor was performed when tumor volume reached ∼150 mm,Citation3 as described.Citation56 For the KCMH-1 model, 2 × 105 cells were implanted subcutaneously in the flank; while 5 × 104, 1 × 105 or 2 × 105 cells were injected via tail vein, as with the 4T1 model. In all tumor settings, mice were euthanized when primary tumor size reached ethical limits (2,000 mmCitation3) or when mice were moribund. Experimental design for CD4 depletion (clone GK1.5, BioXCell, West Lebanon, NH) is described in Supplemental Figure 4.
Macrophage depletion: For macrophage depletion, we made use of clodronate or control liposomes (Formumax, Sunnyvale, CA) which were delivered via two different routes, as described, Citation20,37: 100µl intraperitoneally for systemic depletion or 60 µl intranasally for local (lung) depletion twice before tumor inoculation (as a pre-depletion) and then twice weekly throughout the course of tumor growth, totaling ∼10 injections.
In vitro growth assays: Three types of assays were performed to quantify effects of TSLP knockdown on tumor survival/proliferation in vitro: 3H-thymidine incorporation, MTS colorimetric assay and colony formation. Experimental assays are described in detail in Supplemental Figure 2.
ELISA: Tumor cells (1 × 106/ml) were plated overnight and cell-free supernatants were collected. Blood was collected from mice either by retro-orbital eye bleed or cardiac puncture at endpoint. Serum was collected following centrifugation. Protein levels were measured using an ELISA kit for TSLP, VEGF-A (both eBioscience, San Diego, CA) or G-CSF (RayBiotech, Norcross, GA).
RNA analyses: Total RNA was isolated via RNA extraction kit (Qiagen, Valencia, CA). RNA was converted to cDNA by iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) and used for PCR amplification using specific primer sets. PCR products were run on a 2% agarose gel stained with Red Safe (FroggaBio, Ontario, CA), visualized under UV light using a Bio-Rad Gel-doc system and the data quantified using ImageJ 1.50i software. Quantitative RT-PCR was performed using a Bio-Rad DNA engine real-time PCR system. SYBR-Green (Life-Technologies, Carlsbad, CA) was used as the dye for quantification. Data were quantified using the formula-fold change = 2−ΔΔCt. All results were reported as the ratio of the specific mRNA signal normalized to the housekeeping gene, GAPDH. PCR Array was performed utilizing the specified protocol of the Mouse Angiogenesis Kit (Qiagen, Valencia, CA). Mouse primer sequences are listed in Supplemental Table 1.
Macrophage studies: AMs were collected via bronchioalveolar lavage in 6 ml 1% BSA in PBS with 0.1% EDTA. Generation of bone marrow-derived macrophages is described in Supplemental Figure 6. Macrophages were co-cultured with tumor cells at a 1:1 ratio in a contact-independent transwell (6.5 mm Transwell with 8.0 µm pore polycarbonate membrane insert, Corning, Corning, NY). After 24 hours, RNA was extracted, as described earlier.
Histology: 4-μm sections were cut from formalin-fixed, paraffin-embedded tissues. Specimens were stained with H&E for histologic analyses. When tumor cells were administered intravenously, postmortem lungs were inflated with 15% India ink solution and preserved in Fekete's solution, as described.Citation35 Tumors were counted using a dissection microscope.
CD31 immunohistochemistry: 4-μm sections were cut from formalin-fixed, paraffin-embedded lung tissues, placed on charged slides, and dried at 60°C for one hour. Slides were cooled to room temperature and placed onto the Dako Omnis Autostainer (Agilent Technologies, Santa Clara, CA), wherein they were de-paraffinized with Clearify (CACLEGAL; American MasterTech, Lodi, CA). Flex Target Retrieval Solution, High pH (Dako) was added for 30 minutes for target retrieval. Slides were then incubated with rat anti-mouse CD31 antibody (DIA-310, clone SZ31, which works in formalin-fixed, paraffin-embedded tissues; Dianova, Hamburg, Germany) at 1/30 dilution for 40 minutes. Rabbit anti-rat IgG (Abcam, Cambridge, MA) was applied for 30 minutes, followed by Rabbit Envision (Dako) for 30 minutes. DAB (Dako) was applied for 5 minutes for visualization, and the slides were counterstained with hematoxylin. Slides were scanned and digitized using the Scanscope XT system (Aperio Technologies, Vista, CA). Digital images were captured using ImageScope software (Aperio). For each lung section, CD31+ endothelial clusters within metastatic lesions were counted on multiple fields at 20X magnification and reported as microvessel density (i.e., number of vessels per 20X field). Three to 5 mice/group were evaluated, totaling 10–17 fields quantified/group.
Flow Cytometry: Cells were analyzed utilizing a BD LSRFortessa cytometer running Diva version 6.1.3 and FCS Express version 4. Directly conjugated antibodies included those reactive against: CD11b (M1/70, BioLegend), CD11c (N418, BioLegend), CD45.2 (104, eBioscience), F4/80 (BM8, BioLegend), TSLPR (polyclonal, R&D Systems), CD16/32 (93, eBioscience), Gr-1 (RB6-8C5, BD Biosciences), Ly6G (1A8, BD Biosciences) or Ly6C (AL-21, BD Biosciences).
Statistical analyses: For comparisons between control and experimental groups, data were expressed as the mean ± SEM for the indicated number of replicates or mice. Statistical analysis was determined using two-tailed unpaired t-tests, log-rank test for survival, linear regression for analysis where varying tumor doses were used, and Mann-Whitney for metastatic counts. P-values < 0.05 were considered significant. *P < 0.05; **P < 0.01; ***P < 0.001.
Grant support
This work was supported by R01CA172105 from the National Cancer Institute/NIH (to SIA), a pre-doctoral fellowship award F30CA200133 (to LBM) and an NIH training grant T32CA085183 (to SC). This work utilized shared resources at Roswell Park Cancer Institute (RPCI) supported by the NCI Cancer Center Support Grant P30CA016056.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental.Data_1419115.edited.docx
Download MS Word (1.6 MB)Acknowledgments
We thank Mary Lynn Hensen for excellent technical support, particularly for maintaining all mouse colonies; Dr. Irwin Gelman and Renae Holtz from the shRNA core facility (Roswell Park) for assistance with the transductions; Dr. Austin Miller from the Department of Biostatistics and Bioinformatics for statistical analysis (Roswell Park); and Dr. Paul Bogner from the Department of Pathology (Roswell Park) for assistance with the metastatic analysis during pilot experiments. We thank Dr. Noriyasu Hirasawa from Tohoku University (Japan) for kindly providing us with the KCMH-1 cell line.
Additional information
Funding
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi:10.1016/j.cell.2011.02.013. PMID: 21376230.
- Medrek C, Ponten F, Jirstrom K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer. 2012;12:306. doi:10.1186/1471-2407-12-306. PMID:22824040.
- Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, Delaney A, Jones SJ, Iqbal J, Weisenburger DD, et al. Tumor-associated macrophages and survival in classic Hodgkin's lymphoma. N Engl J Med. 2010;362(10):875–85. doi:10.1056/NEJMoa0905680. PMID:20220182.
- Hu W, Qian Y, Yu F, Liu W, Wu Y, Fang X, Hao W. Alternatively activated macrophages are associated with metastasis and poor prognosis in prostate adenocarcinoma. Oncol Lett. 2015;10(3):1390–96. doi:10.3892/ol.2015.3400. PMID:26622679
- Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117(5):1155–66. doi:10.1172/JCI31422. PMID:17476345.
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–5. doi:10.1038/nature10138. PMID:21654748.
- Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25(6):846–59. doi:10.1016/j.ccr.2014.05.016. PMID;24898549
- Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193(6):727–40. doi:10.1084/jem.193.6.727. PMID:11257139.
- Smith HA, Kang Y. The metastasis-promoting roles of tumor-associated immune cells. J Mol Med (Berl). 2013;91(4):411–29. doi:10.1007/s00109-013-1021-5. PMID:23515621.
- Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18(5):349–55. doi:10.1016/j.semcancer.2008.03.004. PMID:18467122
- Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. 2014;5:614. doi:10.3389/fimmu.2014.00614. PMID:25506346
- Sinha P, Clements VK, Ostrand-Rosenberg S. Interleukin-13-regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res. 2005;65(24):11743–51. doi:10.1158/0008-5472.CAN-05-0045. PMID:16357187.
- DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens LM. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16(2):91–102. doi:10.1016/j.ccr.2009.06.018. PMID:19647220.
- Davies LC, Taylor PR. Tissue-resident macrophages: then and now. Immunology. 2015;144(4):541–48. doi:10.1111/imm.12451. PMID:25684236.
- Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–51. doi:10.1038/nature13989. PMID:25470051.
- Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, Beaudin AE, Lum J, Low I, Forsberg EC, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. 2015;42(4):665–78. doi:10.1016/j.immuni.2015.03.011. PMID:25902481.
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159(6):1312–26. doi:10.1016/j.cell.2014.11.018. PMID:25480296.
- van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G, Hoffmann E, Beyaert R, Saeys Y, Lambrecht BN, et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity. 2016;44(4):755–68. doi:10.1016/j.immuni.2016.02.017. PMID:26992565.
- Galdiero MR, Bonavita E, Barajon I, Garlanda C, Mantovani A, Jaillon S. Tumor associated macrophages and neutrophils in cancer. Immunobiology. 2013;218(11):1402–10. doi:10.1016/j.imbio.2013.06.003. PMID:23891329.
- Sharma SK, Chintala NK, Vadrevu SK, Patel J, Karbowniczek M, Markiewski MM. Pulmonary alveolar macrophages contribute to the premetastatic niche by suppressing antitumor T cell responses in the lungs. J Immunol. 2015;194(11):5529–38. doi:10.4049/jimmunol.1403215. PMID:25911761.
- Wang DH, Lee HS, Yoon D, Berry G, Wheeler TM, Sugarbaker DJ, Kheradmand F, Engleman EG, Burt BM. Progression of EGFR mutant lung adenocarcinoma is driven by alveolar macrophages. Clin Cancer Res 2016;23(3):778–88. doi:10.1158/1078-0432.CCR-15-2597. PMID:27496865.
- Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–22. doi:10.1016/j.ccr.2012.02.022. PMID:22439926.
- Tasian SK, Doral MY, Borowitz MJ, Wood BL, Chen IM, Harvey RC, Gastier-Foster JM, Willman CL, Hunger SP, Mullighan CG, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood. 2012;120(4):833–42. doi:10.1182/blood-2011-12-389932. PMID:22685175.
- De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, Braga M, Di Carlo V, Doglioni C, Protti MP. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. 2011;208(3):469–78. doi:10.1084/jem.20101876. PMID:21339327.
- Pedroza-Gonzalez A, Xu K, Wu TC, Aspord C, Tindle S, Marches F, Gallegos M, Burton EC, Savino D, Hori T, et al. Thymic stromal lymphopoietin fosters human breast tumor growth by promoting type 2 inflammation. J Exp Med. 2011;208(3):479–90. doi:10.1084/jem.20102131. PMID:21339324.
- Demehri S, Cunningham TJ, Manivasagam S, Ngo KH, Moradi Tuchayi S, Reddy R, Meyers MA, DeNardo DG, Yokoyama WM. Thymic stromal lymphopoietin blocks early stages of breast carcinogenesis. J Clin Invest. 2016;126(4):1458–70. doi:10.1172/JCI83724. PMID:26927668.
- Burgess DJ. Tumour immunology: Context is key for TSLP. Nat Rev Cancer. 2012;12(12):796–97. doi:10.1038/nrc3405. PMID:23175117.
- Roan F, Bell BD, Stoklasek TA, Kitajima M, Han H, Ziegler SF. The multiple facets of thymic stromal lymphopoietin (TSLP) during allergic inflammation and beyond. J Leukoc Biol. 2012;91(6):877–86. doi:10.1189/jlb.1211622. PMID:22442496.
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–67. doi:10.1038/nature01322. PMID:12490959.
- Olkhanud PB, Rochman Y, Bodogai M, Malchinkhuu E, Wejksza K, Xu M, Gress RE, Hesdorffer C, Leonard WJ, Biragyn A. Thymic stromal lymphopoietin is a key mediator of breast cancer progression. J Immunol. 2011;186(10):5656–62. doi:10.4049/jimmunol.1100463. PMID:21490155.
- Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Curr Protoc Immunol. 2001; Chapter 20(Unit 20.2). doi:10.1002/0471142735.im2002s39. PMID:18432775
- Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, Bogner PN, Farren MR, Lee KP, Liu K, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. 2013;123(10):4464–78. doi:10.1172/JCI68189. PMID:24091328.
- Johnstone CN, Smith YE, Cao Y, Burrows AD, Cross RS, Ling X, Redvers RP, Doherty JP, Eckhardt BL, Natoli AL, et al. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis Model Mech. 2015;8(3):237–51. doi:10.1242/dmm.017830. PMID:25633981.
- Twentyman PR, Wright KA, Fox NE. Characterisation of a mouse tumour cell line with in vitro derived resistance to verapamil. Br J Cancer 1990;61(2):279–84. doi:10.1038/bjc.1990.52. PMID:1968761.
- Caldwell SA, Ryan MH, McDuffie E, Abrams SI. The Fas/Fas ligand pathway is important for optimal tumor regression in a mouse model of CTL adoptive immunotherapy of experimental CMS4 lung metastases. J Immunol. 2003;171(5):2402–12. doi:10.4049/jimmunol.171.5.2402. PMID:12928387.
- Segawa R, Yamashita S, Mizuno N, Shiraki M, Hatayama T, Satou N, Hiratsuka M, Hide M, Hirasawa N. Identification of a cell line producing high levels of TSLP: advantages for screening of anti-allergic drugs. J Immunol Methods. 2014;402(1–2):9–14. doi:10.1016/j.jim.2013.10.012. PMID:24252242.
- Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjallman AH, Ballmer-Hofer K, Schwendener RA. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer. 2006;95(3):272–81. doi:10.1038/sj.bjc.6603240. PMID:16832418.
- Li Z, Xu X, Feng X, Murphy PM. The Macrophage-depleting Agent Clodronate Promotes Durable Hematopoietic Chimerism and Donor-specific Skin Allograft Tolerance in Mice. Sci Rep. 2016;6:22143. doi:10.1038/srep22143. PMID:26917238.
- Park MK, Ngo V, Kwon YM, Lee YT, Yoo S, Cho YH, Hong SM, Hwang HS, Ko EJ, Jung YJ, et al. Lactobacillus plantarum DK119 as a probiotic confers protection against influenza virus by modulating innate immunity. PLoS One. 2013;8(10):e75368. doi:10.1371/journal.pone.0075368. PMID:24124485.
- Liu YJ. Thymic stromal lymphopoietin: master switch for allergic inflammation. J Exp Med. 2006;203(2):269–73. doi:10.1084/jem.20051745. PMID:16432252.
- Zhou B, Comeau MR, De Smedt T, Liggitt HD, Dahl ME, Lewis DB, Gyarmati D, Aye T, Campbell DJ, Ziegler SF. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. 2005;6(10):1047–53. doi:10.1038/ni1247. PMID:16142237.
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68. doi:10.1038/nri3175. PMID:22437938.
- Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176(1):284–90. doi:10.4049/jimmunol.176.1.284. PMID:16365420.
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi:10.1016/j.cell.2010.03.014. PMID:20371344.
- Williams CB, Yeh ES, Soloff AC. Tumor-associated macrophages: unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer. 2016;2:15025. doi:10.1038/npjbcancer.2015.25. PMID: 26998515.
- Ohgawara T, Kubota S, Kawaki H, Kurio N, Abd El Kader T, Hoshijima M, Janune D, Shimo T, Perbal B, Sasaki A, et al. Association of the metastatic phenotype with CCN family members among breast and oral cancer cells. J Cell Commun Signal. 2011;5(4):291–99. doi:10.1007/s12079-011-0133-3. PMID:21499980.
- Tripurani SK, Cook RW, Eldin KW, Pangas SA. BMP-specific SMADs function as novel repressors of PDGFA and modulate its expression in ovarian granulosa cells and tumors. Oncogene. 2013;32(33):3877–85. doi:10.1038/onc.2012.392. PMID:22964636.
- Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, Bentires-Alj M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515(7525):130–33. doi:10.1038/nature13862. PMID:25337873.
- Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget. 2014;5(9):2736–49. doi:10.18632/oncotarget.1932. PMID:24811362.
- Yu PF, Huang Y, Han YY, Lin LY, Sun WH, Rabson AB, Wang Y, Shi YF. TNFalpha-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2+ neutrophils. Oncogene. 2017;36(4):482–90. doi:10.1038/onc.2016.217. PMID:27669436.
- Waight JD, Hu Q, Miller A, Liu S, Abrams SI. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS One. 2011;6(11):e27690. doi:10.1371/journal.pone.0027690. PMID:22110722.
- Han H, Headley MB, Xu W, Comeau MR, Zhou B, Ziegler SF. Thymic stromal lymphopoietin amplifies the differentiation of alternatively activated macrophages. J Immunol. 2013;190(3):904–12. doi:10.4049/jimmunol.1201808. PMID:23275605.
- Barooei R, Mahmoudian RA, Abbaszadegan MR, Mansouri A, Gholamin M. Evaluation of thymic stromal lymphopoietin (TSLP) and its correlation with lymphatic metastasis in human gastric cancer. Med Oncol. 2015;32(8):217. doi:10.1007/s12032-015-0653-4. PMID:26175262.
- Gauvreau GM, O'Byrne PM, Boulet LP, Wang Y, Cockcroft D, Bigler J, FitzGerald JM, Boedigheimer M, Davis BE, Dias C, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med. 2014;370(22):2102–10. doi:10.1056/NEJMoa1402895. PMID:24846652.
- Stewart TJ, Abrams SI. Altered immune function during long-term host-tumor interactions can be modulated to retard autochthonous neoplastic growth. J Immunol. 2007;179(5):2851–59. doi:10.4049/jimmunol.179.5.2851. PMID:17709499.
- Danna EA, Sinha P, Gilbert M, Clements VK, Pulaski BA, Ostrand-Rosenberg S. Surgical removal of primary tumor reverses tumor-induced immunosuppression despite the presence of metastatic disease. Cancer Res. 2004;64(6):2205–11. doi:10.1158/0008-5472.CAN-03-2646. PMID:15026364.