
ABSTRACT
Diminished overall survival rate of non-Hodgkin lymphoma (NHL) patients treated with a combination regimen of rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) has been recently linked to recurrent somatic mutations activating FOXO1. Despite of the clinical relevance of this finding, the molecular mechanism driving resistance to R-CHOP therapy remains largely unknown. Herein, we investigated the potential role of FOXO1 in the therapeutic efficacy of rituximab, the only targeted therapy included in the R-CHOP regimen. We found CD20 transcription is negatively regulated by FOXO1 in NHL cell lines and in human lymphoma specimens carrying activating mutations of FOXO1. Furthermore, both the expression of exogenous mutants of FOXO1 and the inhibition of AKT led to FOXO1 activation in lymphoma cells, increased binding to MS4A1 promoter and diminished CD20 expression levels. In contrast, a disruption of FOXO1 with CRISPR/Cas9 genome-editing (sgFOXO1) resulted in CD20 upregulation, improved the cytotoxicity induced by rituximab and the survival of mice with sgFOXO1 tumors. Accordingly, pharmacological inhibition of FOXO1 activity in primary samples upregulated surface CD20 levels. Importantly, FOXO1 was required for the downregulation of CD20 levels by the clinically tested inhibitors of BTK, SYK, PI3K and AKT. Taken together, these results indicate for the first time that the AKT-unresponsive mutants of FOXO1 are important determinant of cell response to rituximab-induced cytotoxicity, and suggest that the genetic status of FOXO1 together with its transcriptional activity need further attention while designing anti-CD20 antibodies based regimens for the therapy of pre-selected lymphomas.
Introduction
B-cell non-Hodgkin lymphomas (B-NHL) originate mainly from the germinal center (GC) B cells as a result of genetic alterations of pathways involved in GC reaction as well as recycling of B cells between dark zone (DZ) and light zone (LZ).Citation1 FOXO1 plays a critical role in cell fate decisions in mature B-cells.Citation2-4 Within the GC compartment, expression of this transcription factor, restricted to DZ, is required for DZ formation and contributes to the LZ-DZ transition.Citation2,Citation3 The constitutive activity of FOXO1 might contribute to malignant transformation by supporting DZ germinal center B-cell program involved in cell proliferation and impaired DNA repair.Citation2 Consistently, frequent mutations in FOXO1 gene have been reported in Burkitt lymphomaCitation5 and follicular lymphoma,Citation6 indicating their potential role in the pathogenesis of B-NHL. Moreover, recent reports have identified FOXO1 mutations in diffuse large B-cell lymphoma (DLBCL), the most common type of B-NHL, particularly in patients relapsing or refractory to standard treatment with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone (R-CHOP).Citation7 FOXO1 mutations, the majority of them predicted to be activating, were also found to correlate with a decreased overall survival in DLBCL patients uniformly treated with R-CHOP.Citation8 Although the contribution of FOXO1 mutations to the therapeutic resistance of B-NHLs becomes apparent, the molecular mechanisms underlying the R-CHOP's low efficacy have not been explained so far. Roughly 30–40% of patients develop resistance to rituximab-based immunochemotherapies (reviewed inCitation9,Citation10). The diminished levels of CD20 on the cell surface of tumor cells are among several potential mechanisms underlying the resistance to anti-CD20 monoclonal antibodies (mAbs). This holds especially true for rituximab and ofatumumab, type I mAbs that eliminate tumor cells by the activation of complement cascade.Citation11 CD20 has been reported to be down-modulated epigenetically by DNA methyltransferasesCitation12 and by histone deacetylases,Citation13 as well as at the transcriptional level.Citation14 Resistance to rituximab has been also linked to CD20 posttranscriptional regulation, associated with internalization,Citation15 shedding,Citation16 trogocytosis,Citation17 translational regulationCitation18 or conformational changes of CD20 antigen.Citation19 Moreover, treatment with anti-CD20 monoclonal antibodies may exhaust effector mechanisms (complement componentsCitation20 or CD16 expression on NK cellsCitation21) responsible for elimination of tumor cells. Recently, we reported that the block of tonic BCR (B-cell receptor) signaling activates FOXO122, and that inhibitors of the downstream BCR signaling pathway decrease CD20 expression.Citation23 In the present study, we show that FOXO1 regulates the abundance of CD20 on the surface of tumor cells, thus influencing the response to rituximab-based therapies. Our results provide strong evidence confirming FOXO1's role as a suppressor of CD20 transcription and establish the importance of FOXO1 signaling in determining the response of B-cell lymphomas to anti-CD20 based therapies. Our findings are further supported by the recent observation showing higher CD20 expression in GCB centrocyte (LZ-derived) subtype of DLBCL patients, characterized by superior prognosis after R-CHOP, as compared with the GCB centroblast (DZ-derived) subtype.Citation24
Results
Ablation of FOXO1 gene results in upregulation of CD20 levels and improved rituximab efficacy both in vitro and in vivo
To determine the potential role of FOXO transcription factors in CD20 regulation, we disrupted FOXO1 and FOXO3 loci () using the CRISPR/Cas9 genome-editing technology in Raji cells (FOXO4 expression was undetectable – data not shown). As controls we transduced Raji cells with either empty vector or sgEGFP. Only clones with sgFOXO1 exhibited a very strong, over 3-fold upregulation of surface CD20 levels, raising the possibility that inhibition of FOXO1 (but not of FOXO3) expression may lead to improvement in the rituximab efficacy due to the higher surface abundance of its target, CD20 antigen. In in vitro complement-dependent cytotoxicity (CDC) assay the survival of Raji cells, incubated with different concentrations of rituximab (0.3 – 3 µg/ml) in the presence of human complement, decreased by 20–40% in control clones with empty vector or sgEGFP, as well as in clones with sgFOXO3. The clones with sgFOXO1 were more sensitive to rituximab and their survival decreased by about 80% at the highest concentration (3 µg/ml) of rituximab ().
Figure 1. Ablation of FOXO genes and its effects on CD20 levels and rituximab efficacy in vitro and in vivo. (A) Western blotting showing FOXO1, FOXO3 and CD20 proteins levels in Raji cell clones that were previously transduced with lentiviruses encoding sgRNA (CRISPR/Cas9 technology) targeting either FOXO1 or FOXO3 loci. Clones with empty vector or sgEGFP were used as controls. β-actin level was used as loading control. (B) FACS analysis of cell surface levels of CD20 (left panel, example of graph from FlowJo software; right panel, quantification of MFI values) in clones of Raji cells characterized in panel A. (C) CDC (complement-dependent cytotoxicity) assay showing improved response of sgFOXO1 cell clones to low concentrations of rituximab in the presence of human serum. (D) Kaplan-Meier survival plot of mice inoculated intravenously with Raji cells (either mix of 3 control clones or mix of 3 clones with sgFOXO1) expressing Red Firefly luciferase. Mice (n = 7) were then injected intraperitoneally with rituximab (10 mg/kg) three times a week.
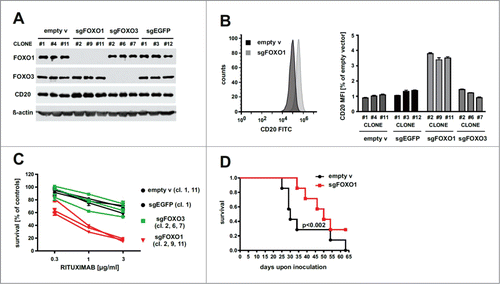
To determine whether the up-regulation of CD20, resulting from FOXO1 ablation, translates into improved antitumor efficacy of rituximab we have used SCID Fox Chase mice intravenously inoculated with control (empty vector) or sgFOXO1-transduced mixtures of Raji clones expressing Red Firefly Luciferase (). Our results show that mice treated systemically with rituximab, administered at a dose of 10 mg/kg, survived longer when inoculated with sgFOXO1-transduced Raji cells as compared with mice inoculated with control Raji cells (median survival 49 days versus 29 days, respectively) ().
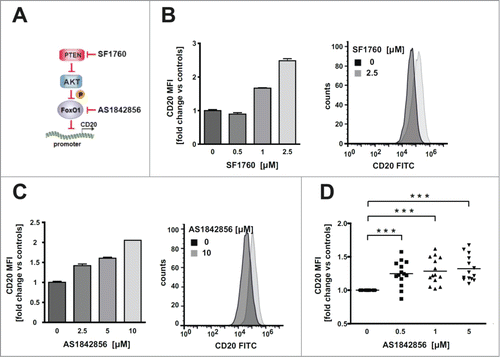
Next, we sought to determine whether the pharmacological inhibition of both FOXO1 and its upstream regulator PTEN (by AS1842856 and SF1760, respectively) modulate CD20 expression (). SF1760 and AS1842856 significantly increased CD20 levels in Raji cells at a concentrations range of 1–2.5 µM and 2.5-10 µM, respectively (). In a panel (n = 14) of primary CLL cells AS1842856 induced a significant increase in CD20 levels at a concentration range of 0.5-5 µM (), further confirming that inhibition of FOXO1 activity is able to increase the surface levels of CD20 antigen.
Figure 2. Pharmacological inhibition of FOXO1 activity elevates CD20 levels in Raji cell line and tumor cells from CLL patients ex vivo. (A) Schema illustrating signaling of PTEN/AKT/FOXO pathway, combined with PTEN inhibitor (SF1760), FOXO1 inhibitor (AS1842856) and our hypothesis concerning negative regulation of CD20 expression by FOXO1. (B-C) FACS analysis showing increased levels of cell surface CD20 antigen 48 h upon incubation of Raji cells with different concentrations of either SF1760 (panel B) or AS1842856 (panel C). (D) FACS analysis of cell surface CD20 antigen in tumor cells (CD19-positive) isolated from blood of CLL patients and treated for 48 h ex vivo with different concentrations of AS1842856. Statistical significance was determined with 1-way Anova, *** p < 0.001 vs controls.
Activation of FOXO1 downregulates CD20 expression and impairs the efficacy of rituximab in vitro
It has been previously shown that the BCR signaling inhibitors are able to activate FOXO1 in tonic BCR signal-dependent DLBCLs.Citation22 Since PI3K/AKT is one of the downstream pathways of BCR signaling, we studied the effect of clinically tested PI3K-AKT inhibitors on the activity of FOXOs. Both inhibitors, MK-2206 (allosteric inhibitor of AKT) and GDC-0068 (ATP-competitive AKT inhibitor), at nontoxic concentrations, led to the dephosphorylation and nuclear accumulation of FOXO1 in Raji and SU-DHL4 cell lines ( and Suppl. ) accompanied by a decrease of total (Suppl. ) and surface CD20 levels (, left panel and Suppl. , left panel). Likewise, the clinically used PI3K inhibitor (idelalisib) downregulated CD20 levels in both cell lines ( and Suppl. ).
Figure 3. AKT inhibitors activate FOXO1, affect CD20 levels and impair rituximab efficacy in vitro. (A) The levels of FOXO1, FOXO3 and AKT (either total or phosphorylated) in Raji cells incubated for 6–24 hours with MK-2206 or GDC-0068 (left panel) followed by nuclear/cytoplasmic fractionation. Oct-2 and EEA1 were used as nuclear and cytoplasmic markers, respectively. Quantification of nuclear FOXO1 upon incubation with either MK-2206 (1 μM) or GDC-0068 (2.5 μM) was performed, normalized to Oct-2 using the ImageStudio software and depicted on graphs (right panel). (B) FACS analysis showing decreased levels of cell surface CD20 antigen 48 h upon incubation of Raji cells with inhibitors of PI3K (idelalisib) and AKT (MK-2206 and GDC-0068) (left panel) and CDC assay showing impaired response of Raji cells to different concentrations of rituximab upon pre-incubation with 1 μM idelalisib, 1 μM MK-2206 or 2.5 μM GDC-0068 for 48 hours (right panel). (C) FACS analysis of cell surface levels of CD20 in clones of Raji cells (top panel) and SU-DHL4 (lower panel) with sgFOXO1, sgFOXO3 and control clones (with either empty vector or sgEGFP) incubated with AKT inhibitors for 48 h (same clones as in ). (D) Impaired response of control clone (with sgEGFP), but not the clone with sgFOXO1 to low concentrations of rituximab in the presence of human serum, as estimated by CDC assay. Cells were incubated with either MK-2206 (left panel) or GDC-0068 (right panel) for 48 hours before rituximab application and estimation of cell survival.
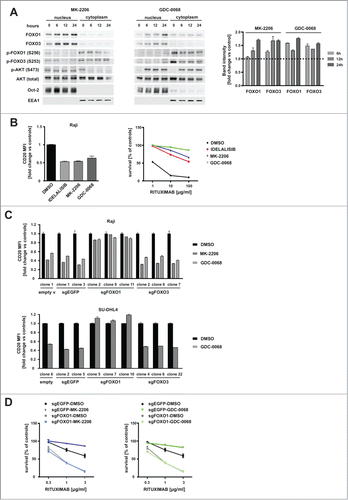
Consistently, MK-2206 or GDC-0068-dependent downregulation of CD20 translated into impaired complement-dependent rituximab efficacy in vitro (, right panel). While the survival of control cells in the presence of complement decreased by 90% at the highest tested rituximab concentration (100 µg/ml), the pretreatment with MK-2206 or GDC-0068 rendered Raji cells partially resistant to rituximab-mediated CDC by 55% and 80%, respectively. SU-DHL4 cells were more sensitive than Raji cells to rituximab-induced CDC (most likely due to higher surface levels of CD20 antigen; MFI in ). Low concentrations of rituximab (1 µg/ml) decreased SU-DHL4 survival by 80% (Suppl. , right panel), while the pretreatment with GDC-0068 impaired the cytotoxic effects of rituximab and decreased cell survival by 55% only (Suppl. , right panel).
The decline in surface CD20 upon incubation with AKT inhibitors was clearly dependent on FOXO1 levels. Only sgFOXO1 clones (but not sgFOXO3 clones) were resistant to MK-2206 or GDC-0068 effects on CD20 levels (). We also confirmed our previous observations that BCR inhibitors (including inhibitors currently tested in clinical trials) strongly downregulate CD20 expression in tumor cells.Citation23 Here, we further demonstrated that FOXO1 is the transcription factor required for the decline in CD20 upon incubation with BTK, SYK, PI3K and AKT inhibitors (Suppl. ). Consistently, the inhibitor of mTOR (rapamycin), an alternative AKT downstream target, had no influence on CD20 levels (Suppl. ).
The results of CDC assay showed that the incubation with MK-2206 (, left panel) or GDC-0068 (, right panel) impaired the cytotoxic effects of rituximab in control sgEGFP cells, while it did not change the sensitivity of sgFOXO1 cells to rituximab-mediated CDC. Besides being sensitive to rituximab sgFOXO1 cells were also slightly more sensitive than control cells to chemotherapy (components of R-CHOP) in vitro (Suppl. ).
Regulation of MS4A1 promoter activity by FOXO1
To determine whether expression of MS4A1 gene (encoding CD20) is modulated by AKT inhibitors at the transcriptional level, MS4A1 transcript was quantified in both Raji and SU-DHL4 cells using reverse transcription polymerase chain reaction (qRT-PCR). The results showed that MK-2206 and GDC-0068 downregulated the levels of MS4A1 mRNA (). The activity of MS4A1 promoter (location −394/+89bp according to NCBI Reference Sequence NM_152866.2) was then assessed using reporter constructs. In agreement with ours qRT-PCR results, the activity of MS4A1 promoter was decreased upon incubation with AKT inhibitors ().
Figure 4. Binding of FOXO1 to the promoter and transcriptional regulation of MS4A1 expression are necessary for the downregulation of CD20 levels by AKT inhibitors. (A) The mRNA levels of MS4A1 (CD20) estimated by RT-PCR in Raji and SU-DHL4 cells 18 hours upon incubation with AKT inhibitors (MK-2206 and GDC-0068). (B) Luciferase reporter assays showing reduced activity of MS4A1 promoter (−394/+89 bp) cloned into pGL4 reporter and introduced by nucleofection into Raji and SU-DHL4 cells. Assays performed 24 hours upon incubation with AKT inhibitors. (C) Increased activity of FOXO transcription factors 24 h upon expression of exogenous wild-type FOXO1, but not the FOXO1-H215R mutant, measured in luciferase reporter assays in Raji cells transfected with 3xIRS-Luc construct (left panel). Luciferase reporter assays showing increased activity of MS4A1 promoter (−394/+89 bp) upon expression of FOXO1-H215R mutant relative to FOXO1-WT (right panel). (D) ChIP-Seq data from independent experiments conducted by Dominguez-Sola et al., 2015,Citation2 using anti-FOXO1 antibody, showing approximate FOXO1 binding site at MS4A1 locus near the transcription start site (TSS). ChIP-seq reads are mapped mostly to upstream region located immediately before the TSS. (E-G) ChIP experiments showing increased binding of acetylated histone H3 (acetyl-K27) (E) and FOXO1 (F) to fragment −182/−88 bp of MS4A1 promoter as well as to the promoters of known FOXO transcriptional targets (GADD45a and IL7R) (G) upon incubation of SU-DHL4 and Raji cells with GDC-0068 for 24 hours.
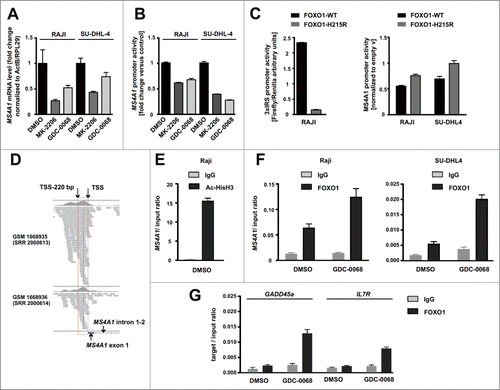
We also compared the effects of DNA binding-defective mutant of FOXO1 (H215R) and overexpressed wild-type FOXO1 on both FOXO and MS4A1 promoter's activities. FOXO activity (measured using the 3 × IRS-Luc reporter) was increased only by the overexpressed wild-type FOXO1, but not by the DNA binding-defective H215R mutant (, left panel). The mutant H215R FOXO1 had weak or no effect on the MS4A1 promoter activity in comparison to the negative effect of wild-type FOXO1 (, right panel), although the levels of expression of both FOXO1 constructs were comparable (data not shown). In summary, the results clearly indicated that DNA-binding activity of FOXO1 is critical for inhibiting the MS4A1 promoter activity.
Although we are uncertain of the exact location of FOXO consensus binding site in the MS4A1 promoter, we analyzed the published ChIP-seq data of B cells isolated from human tonsils.Citation2 We found that the MS4A1 gene was among the FOXO1 target gene repertoires with a 594 bp-wide peak. Evaluating the distribution of sequencing reads indicating FOXO1 binding sites (), we found that the highest peak corresponded to a 220 bp-long region located upstream of TSS (transcription start site) in the MS4A1 promoter, commonly in two biological replicates of ChIP-seq experiments. These results were further supported by our ChIP assays, where immunoprecipitation was performed with anti-acetylated histone H3 (K27) and anti-FOXO1 antibody followed by qRT-PCR () using primers flanking the −182/ −88 bp fragment of MS4A1 promoter. We observed abundant histone H3 acetylation within this sequence, which is commonly seen in chromatin regions permissive for gene expression ().Citation25 Moreover, within the −182/ −88 bp fragment of MS4A1 promoter we detected specific binding of FOXO1 in both Raji and SU-DHL4 cells, which was strongly increased upon incubation with GDC-0068 (). Importantly, we observed lack of FOXO1 binding to MS4A1 promoter in sgFOXO1 cells as compared with control (sgEGFP), which served as a validation of the specificity of ChIP assay (Suppl. ). As a control, we found increased binding of FOXO1 to the promoters of known FOXO1 targets, GADD45a and IL7R (). Collectively, we demonstrated that wild-type FOXO1 downregulates CD20 transcription and binds MS4A1 promoter as other promoters of known FOXO1 target genes in a GDC-0068-activated manner.
Mutants of FOXO1 downregulate CD20 expression
Since N-terminal mutations of FOXO1 in DLBCL are linked to its increased transcriptional activity,Citation8 we analyzed the expression of MS4A1 mRNA in primary FOXO1-wild type and FOXO1-mutated lymphoma samples (ICGC, MALY-DE project). We found that the levels of MS4A1 mRNA in FOXO1-mutated samples (M1V, R21C, R21H, T24A and T24I mutations) were significantly lower than in wild-type FOXO1 samples (averaged normalized expression 1699 versus 2871, respectively, ).
Figure 5. Expression of mutated FOXO1 results in decreased levels of CD20 antigen. (A) Averaged normalized expression of MS4A1 mRNA in malignant lymphoma samples collected by International Cancer Genome Consortium (ICGC, MALY-DE project), including samples with wild-type FOXO1 (n = 10) and with N-terminal mutations of FOXO1 (n = 6). Results were visualized using GraphPad Prism software. Statistical significance was determined with Mann Whitney test, *p < 0.05 (B) FOXO transcriptional activity was measured using luciferase assays with 3 × IRS-Luc reporter (IRS, insulin-responsive signal), introduced to cells together with constructs encoding either wild-type FOXO1 (FOXO1-WT) or FOXO1-AAA mutant, mixed with constructs encoding either myr-AKT1 and GFP or GFP alone (as control). (C-D) FACS analysis showing levels of surface CD20 in Raji cells (panel C) and SU-DHL4 cells (panel D) expressing exogenous FOXO1 (WT or mutant). Cells were cotransfected with construct encoding EGFP followed by the FACS analysis of CD20 surface level (mean fluorescence intensity; MFI) in EGFP-positive cells (48 h post transfection) and visualization using FlowJo software (left panel in D). (E) Luciferase reporter assays showing reduced activity of MS4A1 promoter (−394/+89 bp) introduced by nucleofection into Raji cells (left panel) and SU-DHL4 cells (right panel), together with constructs encoding either wild-type or mutant FOXO1.
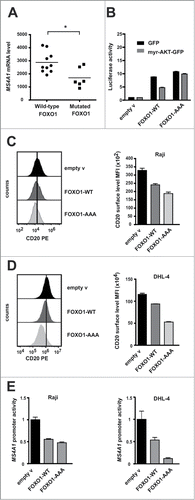
This finding prompted us to further elucidate the role of FOXO1 mutants in the regulation of CD20 expression. We employed a commonly used FOXO1-AAA mutant, located in the nucleus and resistant to the inactivation by AKT,Citation26,Citation27 and therefore mimicking the constitutive activity of recurrently mutated FOXO1 in human B-NHL tumors.Citation8 We confirmed that FOXO1 transcriptional activity (as measured by 3 × IRS-Luc reporter) is greater with mutated than wild type FOXO1. Unlike the activity of exogenous wild-type FOXO1, inhibited by AKT, the activity of mutated FOXO1 remains high and unchanged by the expression of the constitutive active myr-AKT1 ().
In both Raji and SU-DHL4 cell lines, FOXO1-AAA downregulated CD20 surface levels ( and D, respectively) and inhibited MS4A1 promoter activity in luciferase assays (). These effects were found to be even greater with mutant FOXO1-AAA than with wild-type FOXO1 overexpression.
Discussion
FOXO1 transcription factor has recently been identified as one of the top eight candidate genes implicated in the resistance of DLBCL to R-CHOP therapy.Citation7 In particular, primary cells isolated from relapsed and refractory DLBCL patients are enriched in FOXO1 mutations in comparison to untreated DLBCLs (27% vs. 8.6% of cases). Consistently, clonal expansion of cells with FOXO1 mutation has been documented by comparison of DLBCL samples from patients at diagnosis versus the relapse stage.Citation7 Moreover, upon R-CHOP treatment DLBCL patients with mutations in FOXO1 have a decreased overall survival, when compared to patients without such mutations.Citation8 In DLBCL, the majority of mutations are activating, located to the N-terminal region of FOXO1. They result in diminished phosphorylation of FOXO1 by negative regulators (such as AKT) and in nuclear retention of FOXO18.
Although the contribution of FOXO1 mutations to the therapeutic resistance of B-NHLs becomes apparent, the molecular mechanism underlying such phenomenon has not been explained so far. Herein, we report for the first time that FOXO1 is a negative transcriptional regulator of CD20 expression. Importantly, we demonstrate the negative correlation between recurrent FOXO1 mutations and the expression of MS4A1 mRNA in human primary lymphoma samples. Our results provide strong evidence that activating N-terminal hot spot mutations of FOXO1 in human B-NHL inhibit expression of CD20 and we hypothesize that it might be one of the key mechanisms responsible for the increased resistance to rituximab-based therapies. In this scenario, tumor cells with activating mutations of FOXO1 would be characterized by the reduced levels of CD20 molecule and undergo clonal expansion under the selective pressure of rituximab treatment. Therefore, FOXO1 may play active role in the resistance of B-cell lymphomas to R-CHOP therapy by affecting the expression of the rituximab target, CD20 antigen. However, we cannot exclude the contribution of other mechanisms causing R-CHOP resistance, particularly resistance to chemotherapy in tumors with FOXO1 mutations.
The model cell lines used in our study, SU-DHL4 and Raji, represent DLBCL and Burkitt lymphoma respectively, the types of human B-NHL most commonly affected by FOXO1 mutations.Citation5,Citation8 Our results demonstrate that the expression of exogenous constitutively-active mutant of FOXO1 (AAA mutant) in both cell lines diminishes cell surface levels of CD20 to a higher extent than the overexpression of wild-type FOXO1. From these experiments a new role of FOXO1 as a negative regulator of CD20 expression has emerged. Moreover, we report here that disruption of FOXO1 expression (but not FOXO3) by CRISPR/Cas9 gene editing results in upregulation of CD20 levels and improved efficacy of rituximab both in vitro and in vivo, suggesting that the actions of FOXO1 and FOXO3 on CD20 gene regulation are not redundant. Their functions are unlikely dependent on similar posttranslational modifications (phosphorylation by AKT), interacting partners, factors causing their translocation to nucleus as reported in human embryonic stem cells (hESCs).Citation28-30
Based on the knockout mice studies, both FOXO1 and FOXO3 were proposed to act as tumor suppressors.Citation31 Tumor suppressor role of FOXO1 was reported in classical Hodgkin lymphoma (cHL).Citation32 The traditional notion of FOXO3 as tumor suppressor inducing apoptosis and differentiation was reported in hindering lymphoma and leukemia development.Citation33-35 However, recent evidence suggests that FOXO1 and FOXO3 may behave as either tumor suppressors or oncogenes depending on tumor type.Citation34,Citation36 Unlike FOXO1, the hot spot mutations of FOXO3 in human lymphoma samples have not been identified so far.
Recently, the lists of genes positively and negatively regulated by FOXO1 in mouse germinal center B cells upon conditional deletion of FOXO1 have been published, together with a list of potential FOXO1 targets that were identified by ChIP-seq analysis in human tonsil-derived B cells.Citation2 The MS4A1 gene was listed among genes downregulated in the dark zone (when compared to light zone) of germinal center, where FOXO1 activity is present exclusively, and is strictly required for dark zone formation.Citation2 Our findings demonstrating that FOXO1 is a negative regulator of human CD20 are consistent with the ChIP-seq results, showing that MS4A1 promoter is among the promoters specifically bound by FOXO1. Although we did not determine the exact localization of a putative binding site for FOXOs in the promoter of CD20, our ChIP results suggest that a DNA-binding element present between bp −182 and −88 could be responsible for the indirect recruitment of FOXO1 to the MS4A1 promoter. Importantly, this region of MS4A1 promoter falls within the 220bp-long region upstream of TSS, that we mapped using the published ChIP-seq data.Citation2 Moreover, our ChIP results show that FOXO1 protein bound to the MS4A1 promoter was even more abundant than FOXO1 bound to the promoters of known target genes, such as GADD45α or IL-7R.
FOXO1 seems to bind indirectly and potentially downregulate the expression of many genes.Citation2 However, what is most studied and mechanistically elucidated, is the downregulation of cyclins D1/D2 expression, previously reported in renal carcinoma, fibroblasts, colon carcinoma, lymphoid cells and pancreatic β-cells.Citation37-40 In these cases, the negative effects of FOXOs on the expression of cyclins D1/D2 was suggested to be mediated via either Bcl-6 transcriptional repressor or cdx transcription factors. Indeed, ChIP-seq data from tonsil-derived B cells identified MS4A1 among many potential targets of Bcl-6, however the consensus binding sites for Bcl-6 were not found in the sequence of MS4A1 promoter,Citation2 suggesting again that the binding to DNA might be mediated by a complex of transcriptional regulators.
The development of therapeutic strategies to inhibit function of FOXO1 has been stimulated by research conducted on the crucial role of FOXO1 in metabolic disorders such as diabetes and obesity. Specific FOXO1 inhibitor AS1842856 has been developed and administered in vivo to diabetic or to asthmatic mice for short period of time.Citation41,Citation42 However, long-term in vivo studies using this inhibitor are lacking, and most studies were limited to in vitro applications so far. Importantly, our studies suggest that pharmacological inhibition of either PTEN or FOXO1 activity could be beneficial for patients with low CD20 levels on tumor cells, as both inhibitors upregulate surface levels of CD20 antigen.
Collectively, our results indicate that FOXO1- activating mutations are strong negative regulators of CD20 expression and add new insights into the mechanisms underlying the contribution of FOXO1 mutations to the resistance of B-NHLs to R-CHOP therapy. Results of our study have also implications for understanding CD20 regulation in DZ and LZ of human tonsillar GC B cells, as well as in GCB centrocyte (LZ-derived) subtype of DLBCL patients characterized by superior outcome after R-CHOP treatment, as compared with the GCB centroblast (DZ-derived) subtype.Citation24
In light of the current knowledge, the inhibition of FOXO1 function seems to be rational and beneficial for the therapy of B-NHL types of tumors. Future development of more specific and well-tolerated FOXO1 inhibitors may lead to novel therapeutic regimens for B-NHL patients. Emerging technologies such as proteolysis-targeting chimaeras (PROTACs) seem to offer feasible means to target proteins that were considered therapeutically undraggable, such as transcription factors or regulatory proteins.Citation43
Materials and methods
Generation of sgFOXOs and lentiviral transduction
sgFOXO1, sgFOXO3 and sgEGFP were generated with oligonucleotide pairs (Table S1) and cloned into pLenti-CRISPRv2. 293T cells were used for the production of a replication-incompetent lentivirus. Transduced Raji and SU-DHL4 cells were selected upon puromycin treatment and single clones were obtained from resistant cell pools by limiting dilution (for details see Supplementary Information).
Luciferase assays
Luciferase assays were performed as previously described.Citation44 The pGL4-MS4A1 promoter-Luc reporter constructs were previously used.Citation45 To assess FOXOs activity the 3 × IRS Luciferase reporter was used (Addgene, plasmid #13511, gift of Kunliang GuanCitation27). The construct encoding constitutively active AKT1 isoform pMIG-myrAKT1-IRES-GFP was described earlier.Citation46
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed according to the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling) protocol (for details see Supplementary Information).
Animal studies
The SCID FOX Chase female mice (8–9 weeks old) were intravenously (i.v.) inoculated with 2 × 106 Raji cells (mix of 3 clones with either empty or sgFOXO1 vector). Mice were intraperitoneally (i.p.) treated with rituximab (10 mg/kg) three times a week starting on day 5 after inoculation of tumor cells. Bioluminescence was measured once a week (starting from the day of injection). Mice were i.p. injected with D-luciferin (150 mg/kg), anaesthetized with isoflurane and visualized using IVIS Imaging System (Xenogen, Alameda, CA, USA). Images were analyzed with the Living Image 4.2 software (Caliper Life Science, Hopkinton, MA, USA). Statistical significance was determined with the Mann-Whitney Rank Sum test.
Statistics
Results were plotted with GraphPad Prism, and statistical significance was assessed by appropriate tests provided in Figure legends. The p-value were marked with the asterisks on the charts (* p < 0.05, ***p < 0.001).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
2017ONCOIMM0690R-s01.docx
Download MS Word (2.3 MB)Acknowledgments
We acknowledge Prof. Margaret A. Shipp (Dana-Farber Cancer Institute, Boston, USA) for the kind guidance of BP's scientific career in the frame of the Mentoring Programme (sponsored by the Foundation for Polish Science, FNP, Poland), Mrs. Elzbieta Gutowska (Medical University of Warsaw) for PBMC isolation and The Francis Crick Institute facilities employees for their excellent technical assistance.
Additional information
Funding
References
- Basso K, Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nat Rev Immunol. 2015;15:172–84. doi:10.1038/nri3814. PMID:25712152.
- Dominguez-Sola D, Kung J, Holmes AB, Wells VA, Mo T, Basso K, Dalla-Favera R. The FOXO1 Transcription Factor Instructs the Germinal Center Dark Zone Program. Immunity. 2015;43:1064–74. doi:10.1016/j.immuni.2015.10.015. PMID:26620759.
- Sander S, Chu VT, Yasuda T, Franklin A, Graf R, Calado DP, Li S, Imami K, Selbach M, Di Virgilio M. PI3 Kinase and FOXO1 Transcription Factor Activity Differentially Control B Cells in the Germinal Center Light and Dark Zones. Immunity. 2015;43:1075–86. doi:10.1016/j.immuni.2015.10.021. PMID:26620760.
- Szydlowski M, Jablonska E, Juszczynski P. FOXO1 transcription factor: a critical effector of the PI3K-AKT axis in B-cell development. International reviews of immunology. 2014;33:146–57. doi:10.3109/08830185.2014.885022. PMID:24552152.
- Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490:116–20. doi:10.1038/nature11378. PMID:22885699.
- Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6:130–40. doi:10.1016/j.celrep.2013.12.027. PMID:24388756.
- Morin RD, Assouline S, Alcaide M, Mohajeri A, Johnston RL, Chong L, Grewal J, Yu S, Fornika D, Bushell K, et al. Genetic Landscapes of Relapsed and Refractory Diffuse Large B-Cell Lymphomas. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:2290–300. doi:10.1158/1078-0432.CCR-15-2123. PMID:26647218.
- Trinh DL, Scott DW, Morin RD, Mendez-Lago M, An J, Jones SJ, et al. Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood. 2013;121:3666–74. doi:10.1182/blood-2013-01-479865. PMID:23460611.
- Perez-Callejo D, Gonzalez-Rincon J, Sanchez A, Provencio M, Sanchez-Beato M. Action and resistance of monoclonal CD20 antibodies therapy in B-cell Non-Hodgkin Lymphomas. Cancer treatment reviews. 2015;41:680–9. doi:10.1016/j.ctrv.2015.05.007. PMID:26045227.
- Camicia R, Winkler HC, Hassa PO. Novel drug targets for personalized precision medicine in relapsed/refractory diffuse large B-cell lymphoma: a comprehensive review. Molecular cancer. 2015;14:207. doi:10.1186/s12943-015-0474-2. PMID:26654227.
- van Meerten T, van Rijn RS, Hol S, Hagenbeek A, Ebeling SB. Complement-induced cell death by rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:4027–35. doi:10.1158/1078-0432.CCR-06-0066. PMID:16818702.
- Ushmorov A, Leithauser F, Sakk O, Weinhausel A, Popov SW, Moller P, Wirth T. Epigenetic processes play a major role in B-cell-specific gene silencing in classical Hodgkin lymphoma. Blood. 2006;107:2493–500. doi:10.1182/blood-2005-09-3765. PMID:16304050.
- Sugimoto T, Tomita A, Hiraga J, Shimada K, Kiyoi H, Kinoshita T, Naoe T. Escape mechanisms from antibody therapy to lymphoma cells: downregulation of CD20 mRNA by recruitment of the HDAC complex and not by DNA methylation. Biochem Biophys Res Commun. 2009;390:48–53. doi:10.1016/j.bbrc.2009.09.059. PMID:19769942.
- Jilani I, O'Brien S, Manshuri T, Thomas DA, Thomazy VA, Imam M, Naeem S, Verstovsek S, Kantarjian H, Giles F, et al. Transient down-modulation of CD20 by rituximab in patients with chronic lymphocytic leukemia. Blood. 2003;102:3514–20. doi:10.1182/blood-2003-01-0055. PMID:12893761.
- Beers SA, French RR, Chan HT, Lim SH, Jarrett TC, Vidal RM, Wijayaweera SS, Dixon SV, Kim H, Cox KL, et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood. 2010;115:5191–201. doi:10.1182/blood-2010-01-263533. PMID:20223920.
- Manshouri T, Do KA, Wang X, Giles FJ, O'Brien SM, Saffer H, Thomas D, Jilani I, Kantarjian HM, Keating MJ, et al. Circulating CD20 is detectable in the plasma of patients with chronic lymphocytic leukemia and is of prognostic significance. Blood. 2003;101:2507–13. doi:10.1182/blood-2002-06-1639. PMID:12446458.
- Taylor RP, Lindorfer MA. Antigenic modulation and rituximab resistance. Semin Hematol. 2010;47:124–32. doi:10.1053/j.seminhematol.2010.01.006. PMID:20350659.
- Bobrowicz M, Dwojak M, Pyrzynska B, Stachura J, Muchowicz A, Berthel E, Dalla-Venezia N, Kozikowski M, Siernicka M, Miazek N, et al. HDAC6 inhibition upregulates CD20 levels and increases the efficacy of anti-CD20 monoclonal antibodies. Blood. 2017;130:1628–38. PMID:28830887.
- Winiarska M, Bil J, Wilczek E, Wilczynski GM, Lekka M, Engelberts PJ, Mackus WJ, Gorska E, Bojarski L, Stoklosa T, et al. Statins impair antitumor effects of rituximab by inducing conformational changes of CD20. PLoS Med. 2008;5:e64. doi:10.1371/journal.pmed.0050064. PMID:18366248.
- Kennedy AD, Beum PV, Solga MD, DiLillo DJ, Lindorfer MA, Hess CE, Densmore JJ, Williams ME, Taylor RP. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J Immunol. 2004;172:3280–8. doi:10.4049/jimmunol.172.5.3280. PMID:14978136.
- Bowles JA, Weiner GJ. CD16 polymorphisms and NK activation induced by monoclonal antibody-coated target cells. J Immunol Methods. 2005;304:88–99. doi:10.1016/j.jim.2005.06.018. PMID:16109421.
- Szydlowski M, Kiliszek P, Sewastianik T, Jablonska E, Bialopiotrowicz E, Gorniak P, et al. FOXO1 activation is an effector of SYK and AKT inhibition in tonic BCR signal-dependent diffuse large B-cell lymphomas. Blood. 2016;127:739–48. doi:10.1182/blood-2015-06-654111. PMID:26585955.
- Bojarczuk K, Siernicka M, Dwojak M, Bobrowicz M, Pyrzynska B, Gaj P, et al. B-cell receptor pathway inhibitors affect CD20 levels and impair antitumor activity of anti-CD20 monoclonal antibodies. Leukemia. 2014;28:1163–7. doi:10.1038/leu.2014.12. PMID:24492323.
- Dybkaer K, Bogsted M, Falgreen S, Bodker JS, Kjeldsen MK, Schmitz A, et al. Diffuse large B-cell lymphoma classification system that associates normal B-cell subset phenotypes with prognosis. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2015;33:1379–88. doi:10.1200/JCO.2014.57.7080. PMID:25800755.
- Clayton AL, Hazzalin CA, Mahadevan LC. Enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell. 2006;23:289–96. doi:10.1016/j.molcel.2006.06.017. PMID:16885019.
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi:10.1016/S0092-8674(00)80595-4. PMID:10102273.
- Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. The Journal of biological chemistry. 1999;274:16741–6. doi:10.1074/jbc.274.24.16741. PMID:10358014.
- Zhang X, Yalcin S, Lee DF, Yeh TY, Lee SM, Su J, Mungamuri SK, Rimmelé P, Kennedy M, Sellers R, et al. FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nature cell biology. 2011;13:1092–9. doi:10.1038/ncb2293. PMID:21804543.
- Monsalve M, Olmos Y. The complex biology of FOXO. Current drug targets. 2011;12:1322–50. doi:10.2174/138945011796150307. PMID:21443460.
- van der Vos KE, Coffer PJ. The extending network of FOXO transcriptional target genes. Antioxidants & redox signaling. 2011;14:579–92. doi:10.1089/ars.2010.3419.
- Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–23. doi:10.1016/j.cell.2006.12.029. PMID:17254969.
- Xie L, Ushmorov A, Leithauser F, Guan H, Steidl C, Farbinger J, et al. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood. 2012;119:3503–11. doi:10.1182/blood-2011-09-381905. PMID:22343918.
- Birkenkamp KU, Essafi A, van der Vos KE, da Costa M, Hui RC, Holstege F, et al. FOXO3a induces differentiation of Bcr-Abl-transformed cells through transcriptional down-regulation of Id1. The Journal of biological chemistry 2007;282:2211–20. doi:10.1074/jbc.M606669200. PMID:17132628.
- Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nature reviews Cancer. 2007;7:847–59. doi:10.1038/nrc2223. PMID:17943136.
- Vandenberg CJ, Motoyama N, Cory S. FoxO3 suppresses Myc-driven lymphomagenesis. Cell death & disease. 2016;6:e2046. doi:10.1038/cddis.2015.396.
- Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276–88. doi:10.1038/onc.2008.21. PMID:18391970.
- Ramaswamy S, Nakamura N, Sansal I, Bergeron L, Sellers WR. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer cell. 2002;2:81–91. doi:10.1016/S1535-6108(02)00086-7. PMID:12150827.
- Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJ, Lam EW, et al. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Molecular and cellular biology. 2002;22:7842–52. doi:10.1128/MCB.22.22.7842-7852.2002. PMID:12391153.
- Fernandez de Mattos S, Essafi A, Soeiro I, Pietersen AM, Birkenkamp KU, Edwards CS, et al. FoxO3a and BCR-ABL regulate cyclin D2 transcription through a STAT5/BCL6-dependent mechanism. Molecular and cellular biology. 2004;24:10058–71. doi:10.1128/MCB.24.22.10058-10071.2004. PMID:15509806.
- Glauser DA, Schlegel W. The FoxO/Bcl-6/cyclin D2 pathway mediates metabolic and growth factor stimulation of proliferation in Min6 pancreatic beta-cells. Journal of receptor and signal transduction research. 2009;29:293–8. doi:10.3109/10799890903241824. PMID:19929250.
- Nagashima T, Shigematsu N, Maruki R, Urano Y, Tanaka H, Shimaya A, et al. Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: improvement of fasting glycemia in diabetic db/db mice. Molecular pharmacology. 2010;78:961–70. doi:10.1124/mol.110.065714. PMID:20736318.
- Chung S, Lee TJ, Reader BF, Kim JY, Lee YG, Park GY, Karpurapu M, Ballinger MN, Qian F, Rusu L, et al. FoxO1 regulates allergic asthmatic inflammation through regulating polarization of the macrophage inflammatory phenotype. Oncotarget. 2016;7:17532–46. doi:10.18632/oncotarget.8162. PMID:27007158.
- Deshaies RJ. Protein degradation: Prime time for PROTACs. Nat Chem Biol. 2015;11:634–5. doi:10.1038/nchembio.1887. PMID:26284668.
- Zerrouqi A, Pyrzynska B, Brat DJ, Van Meir EG. P14ARF suppresses tumor-induced thrombosis by regulating the tissue factor pathway. Cancer Res. 2014;74:1371–8. doi:10.1158/0008-5472.CAN-13-1951. PMID:24398474.
- Winiarska M, Bojarczuk K, Pyrzynska B, Bil J, Siernicka M, Dwojak M, Bobrowicz M, Miazek N, Zapala P, Zagozdzon A, et al. Inhibitors of SRC kinases impair antitumor activity of anti-CD20 monoclonal antibodies. mAbs. 2014;6:1300–13. doi:10.4161/mabs.32106. PMID:25517315.
- Kharas MG, Okabe R, Ganis JJ, Gozo M, Khandan T, Paktinat M, Gilliland DG, Gritsman K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood. 2010;115:1406–15. doi:10.1182/blood-2009-06-229443. PMID:20008787.