
ABSTRACT
CD96 is a transmembrane glycoprotein Ig superfamily receptor, expressed on various T cell subsets and NK cells, that interacts with nectin and nectin-like proteins, including CD155/polio virus receptor (PVR). Here, we have compared three rat anti-mouse CD96 mAbs, including two that block CD96-CD155 (3.3 and 6A6) and one that does not block CD96-CD155 (8B10). Using flow cytometry, we demonstrated that both mAbs 3.3 and 6A6 bind to the first Ig domain of mouse CD96 and compete with CD155 binding, while mAb 8B10 binds to the second Ig domain and does not block CD155. While Fc isotype was irrelevant concerning the anti-metastatic activity of 3.3 mAb, in four different experimental metastases models and one spontaneous metastasis model, the relative order of anti-metastatic potency was 6A6 > 3.3 > 8B10. The metastatic burden control of all of the anti-CD96 clones was highly dependent on NK cells and IFN-γ. Consistent with its inability to block CD96-CD155 interactions, 8B10 retained anti-metastatic activity in CD155-deficient mice, whereas 3.3 and 6A6 lost potency in CD155-deficient mice. Furthermore, 8B10 retained most of its anti-metastatic activity in IL-12p35-deficient mice whereas the activity of 3.3 and 6A6 were partially lost. All three mAbs were inactive in CD226-deficient mice. Altogether, these data demonstrate anti-CD96 need not block CD96-CD155 interactions (ie. immune checkpoint blockade) to promote NK cell anti-metastatic activity.
KEYWORDS:
Introduction
Tumor growth and progression to a metastasis is a multistage process in which the primary cancer cells conditions the pre-metastatic niche in distant organs and proceeds to spread and colonize those sites.Citation1,Citation2 NK cells are important host protective cells in such a process.Citation3 Recently we described in a series of mouse metastasis models the ability of the 3.3 anti-mouse CD96 monoclonal antibody (mAb) to protect the host from spontaneous and experimental metastasis via a NK cell-dependent mechanism.Citation4 In these settings, anti-CD96 was superior as a monotherapy to either anti-PD-1 or anti-CTLA-4 mAb, highlighting a particular niche role for this pathway in regulating NK cell anti-metastatic activity.Citation4 With the success of immune checkpoint blockade anti-PD-1/PD-L1 and anti-CTLA-4 alone and in combination in a proportion of cancer patients with advanced disease and in some cases in the adjuvant setting, there is now great interest in additional receptor/ligand pathways that regulate T cell and NK cell anti-tumor activity. One of these includes CD96, CD226/DNAM-1Citation5, and TIGITCitation6 which belong to the Ig superfamily of receptors that interact with nectin and nectin-like proteins, including CD155 (NECL5:PVR)Citation4,Citation7,Citation8 and CD112 (NECTIN2:PVRL2).Citation9 CD226/DNAM-1 activates NK cell-mediated functions and plays an essential role for in vivo immune surveillance,Citation10,Citation11 whereas CD96 and TIGIT reportedly counterbalance DNAM-112.
Human CD96, first described as TACTILE, is a transmembrane glycoprotein which possesses a complex extracellular domain composed of three Ig-like domains and a short 45 amino acid cytoplasmic domainCitation8,Citation13 and is expressed by a variety of lymphocyte subsets, but not myeloid or B cells.Citation14-16 CD96 triggers T cell and NK cell adhesion and function,Citation14,Citation17 by interacting with its high affinity ligand CD155/poliovirus receptor (PVR), and has been shown to play a critical negative role as regulating NK cell-mediated immune surveillance.Citation7,Citation18 Both human and mouse CD96 contain a cytoplasmic ITIM-like domain. Past studies using chimeric human/murine CD96 receptor constructs have revealed that CD96 mediates its interaction with CD155 via the first Ig-like domain, with some modulation by the second domain of CD96.Citation19 Recently, we showed that targeting CD96 with a CD96-CD155 blocking mAb (mAb) clone 3.3 efficiently decreased spontaneous and experimental metastases in mice.Citation4 However, the role of CD155 in the mechanism of action of mAbs targeting CD96 and the comparative anti-tumor efficacies of different anti-CD96 mAbs targeting different epitopes of CD96 are poorly understood. Here we examine the mechanism of action and anti-metastatic efficacy of three different anti-mouse CD96 mAbs that stimulate and promote NK cell control of metastases.
Results
Anti-mCD96 mAbs bind different Ig domains of the CD96 molecule
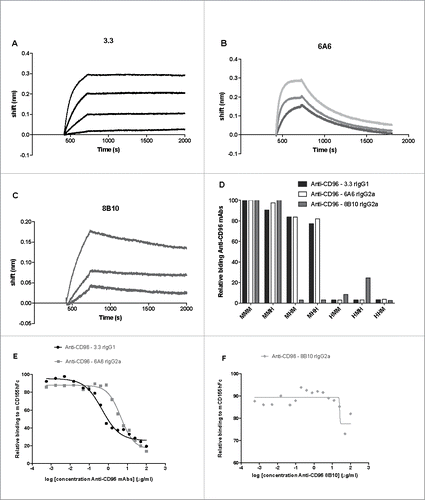
Initially we tested the binding activity of three different parental anti-mouse CD96 clones [3.3, rat IgG14; 6A6, rat IgG2a and 8B10, rat IgG2aCitation14] to mouse CD96 by Octet analysis (). The 3.3 had a KD of 1.43 × 10−10 M with a slow off rate (Kd = 3.58 × 10−5), while 6A6 had a KD of 8.3 × 10−9 M with a fast off rate (Kd = 2.1 × 10−3). The KD of 8B10 was slightly weaker 1.38 × 10−8 M (Kd = 2.09 × 10−4). Human and mouse CD96 both have three extracellular Ig-like domains/loops and mouse/human chimeric CD96 constructs generated by domain exchange are powerful tools for an initial screen of the binding mechanism of different anti-CD96 mAbs.Citation19 We investigated the binding patterns of the parental anti-mouse CD96 clones using flow cytometry of HEK-293 cells transiently transfected with various chimeric CD96 constructs ()(Supplementary Figure 1). Two clear patterns of mAb binding were obtained, as the first mouse CD96 Ig domain was sufficient for binding of 3.3 and 6A6 anti-CD96 clones, whereas 8B10 bound to the second Ig domain of CD96. These observations were in concert with the previous description of both 3.3 and 6A6 mAbs as CD96-CD155 blocking, whereas 8B10 binds CD96, but does not block the CD96-CD155 interaction.Citation14,Citation18 In order to determine the degree of competition of these anti-CD96 mAbs for CD155 binding, we next used HEK-293 cells transiently transfected with a fully mouse CD96 and incubated with recombinant mouse CD155hFc (). The presence of saturating amounts of 6A6 or 3.3 decreased binding of CD155 to CD96 and blocking was concentration dependent (). Interestingly, mAb 3.3 blocked CD155 binding approximately 10-fold more efficiently than mAb 6A6 (), with calculated IC50 of 0.47 μg/ml and 3.21 μg/ml for the two antibodies, respectively. By contrast 8B10 clone displays no concentration dependent blocking of CD155 ().
Figure 1. Anti- mCD96 mAbs bind different Ig domains of the CD96 molecule. Octet binding profiles of (A) 3.3, (B) 6A6 and (C) 8B10 anti-CD96 mAbs to recombinant CD96. (D) Relative binding of anti-mCD96 mAbs (3.3, 6A6 and 8B10) to mouse/human CD96 three domain chimeric molecules (M = mouse and H = human) expressed in HEK-293 cells as measured by flow cytometry where binding to mouse CD96 is standardized as 100%. Relative binding curves of mCD155-Fc (3 μg/ml) binding to HEK-293 cells transfected with mCD96 in the presence of various dilutions (log concentration shown starting at 100 μg/ml) (E) 3.3 and 6A6 mAbs and (F) 8B10 mAb as indicated.
Anti-CD96 mAbs differentially suppress experimental and spontaneous metastasis
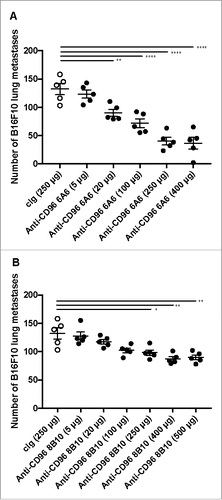
We have previously demonstrated the heightened anti-metastatic activity of CD96-deficient miceCitation18 and the metastases control of the 3.3 mAb.Citation4 Here, we wished to directly compare the anti-metastatic activity of 3.3, 6A6 and 8B10 clones in vivo, with a view to determining whether all anti-CD96 mAbs might be similar in their potency and whether CD96-CD155 interactions were critical. Using the B16F10 experimental metastases model we had previously determined a regimen of 250 μg ip of anti-CD96 mAb 3.3 on days 0 and 3 after tumor inoculation as being optimal.Citation4 We thus proceeded to test in a dose response the activities of 6A6 and 8B10 using the same model (, ). As little as 20 μg/dose of 6A6 and 250 μg 8B10 began to reduce lung metastases, and 6A6 reached an optimal anti-metastatic activity at 250 μg/dose (like previously reported for 3.34), while 8B10 appeared slightly more effective at 400–500 μg doses. Still, even at these doses, treatment was less effective when compared to clone 6A6.
Figure 2. Dose response of anti-CD96 mAbs, against B16F10 experimental lung metastases. WT mice were injected i.v. with B16F10 melanoma (1 × 105 cells). On day 0 and 3 after tumor inoculation, mice were treated with i.p. injections of cIg (250 μg) or various doses of anti-CD96 mAb (A) 6A6 (rat IgG2a) and (B) 8B10 (rat IgG2a) as indicated. The cIg group shown is common to parts A and B. The metastatic burden was quantified in the lungs after 14 days by counting colonies on the lung surface. Individual mice are represented by each symbol and means ± SEM of 5 mice per group are shown. Significant differences between groups as indicated by crossbars were determined by a one-way ANOVA with post Tukey test (*: p < 0.05, **:p < 0.01, ****: p < 0.0001).
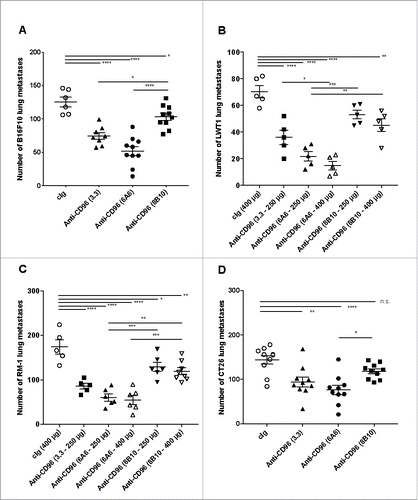
With the schedule of each anti-CD96 mAb to achieve maximal efficacy optimized, we next compared the three mAb clones in four different experimental lung metastases models (B16F10 melanoma, LWT1 BRAFV600E mutant melanoma, RM-1 prostate carcinoma and CT26 colon adenocarcinoma). Initially, we tested all clones at 400 μg/dose against B16F10 at two different cell doses ( and Supplementary Figure 2). All three clones had significant anti-metastatic activity, but 6A6 and 3.3 were more effective than 8B10 (). The same trend was observed at 250 μg or 400 μg/dose schedules in the LWT1 melanoma (), RM-1 prostate carcinoma () and CT26 colon adenocarcinoma () experimental metastasis models.
Figure 3. Anti-CD96 mAbs differentially suppress experimental lung metastasis. Comparative activities of various anti-CD96 mAbs against various experimental lung metastases. Groups of 5–10 C57BL/6 or BALB/c WT were injected i.v. with (A) B16F10 melanoma (1 × 105cells), (B) LWT1 melanoma (5 × 105 cells), (C) RM-1 prostate carcinoma (5 × 104 cells), or (D) CT26 colon adenocarcinoma (5 × 105 cells). On days 0 and 3 after tumor inoculation, mice were treated with i.p. injections of cIg (400 μg), anti-CD96 mAb 3.3 (250 μg, rat IgG1), 6A6 (250 or 400 μg, rat IgG2a), or 8B10 (400 μg, rat IgG2a), as indicated. The metastatic burden was quantified in the lungs after 14 days by counting colonies on the lung surface. Individual mice are represented by each symbol and means ± SEM of 5–10 mice per group are shown. Significant differences between groups as indicated by crossbars were determined by a one-way ANOVA with post Tukey test (*: p < 0.05, **: p < 0.01, ***: p < 0.001; ****: p < 0.0001). Part of panel A has already been published in Cancer Discovery articleCitation4.
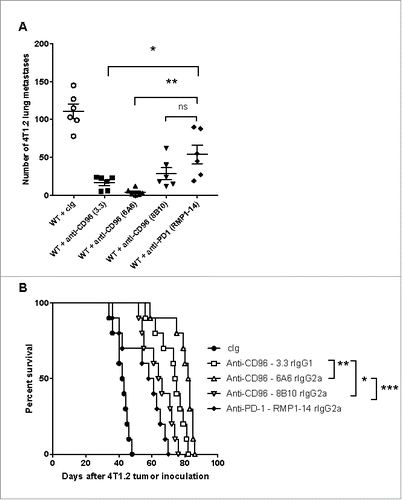
It was important to determine whether 6A6 and 3.3 were superior to 8B10 in a spontaneous tumor metastasis model. Here we employed the orthotopic 4T1.2 mammary carcinoma with primary surgery and distant lethal metastases to demonstrate neoadjuvant efficacy of these anti-CD96 mAbs. We have previously demonstrated the superior benefit of neoadjuvant immunotherapy, including anti-PD-1 alone, compared to adjuvant immunotherapy in the context of surgical resection,Citation20 although targeting CD96 has never been assessed in this manner. Once again, all three anti-CD96 mAbs displayed significant anti-metastatic activity compared to cIg () and 6A6 > 3.3 > 8B10 with respect to impact on survival ().
Figure 4. Anti-CD96 mAbs differentially suppress spontaneous metastasis. Groups of 5–10 BALB/c WT mice were injected with 5 × 104 4T1.2 mammary carcinoma cells into the mammary fat pad. Some groups of mice were treated i.p. on days 12 and 14 after tumor inoculation with neoadjuvant cIg (400 μg), anti-CD96 mAb 3.3 (400 μg, rat IgG1), 6A6 (400 μg, rat IgG2a), 8B10 (400 μg, rat IgG2a), or anti-PD-1 (RMP1-14, 250 μg) as indicated. On days 16 all primary tumors were resected. (A) Some cohorts were sacrificed on day 34 and the metastatic burden was quantified in the lungs by counting colonies on the lung surface. Individual mice are represented by each symbol and means ± SEM of 5 mice per group are shown. Significant differences between groups as indicated by crossbars were determined by a one-way ANOVA with post Tukey test (*: p < 0.05, **: p < 0.01, ***: p < 0.001). (B) Other mice were monitored for survival. The Kaplan-Meier curves for overall survival of each group are shown. Significant differences between indicated groups were determined by log-rank sum test with exact p values shown.
Anti-CD96 mAb suppression of metastases is not dependent on Fc function
Focusing on the B16F10 experimental lung metastases melanoma model, we next evaluated whether any major differences in mechanism of action existed with respect to anti-CD96 mAb Fc. We first examined whether 3.3 (), 6A6 () and 8B10 () required activating FcR, by testing these mAbs in WT versus FcR-deficient mice. Regardless of whether mice deficient in FcγRIII, FcγRIV or Fcϵγ were employed, 3.3, 6A6 and 8B10 all retained full anti-metastatic activity in these mice compared with WT mice (). Furthermore, the 3.3 mAb was produced as recombinant mAbs using four different Fc isotypes, including a mouse IgG1 D265A version containing a mutation in which all FcR binding was eliminated. Similar reduction in B16F10 experimental lung metastases with different 3.3 isotypes (rIgG1, mIgG1 D265A, and mIgG2a) compared to control Ig was observed in WT mice ( and Supplementary Fig. 3).
Figure 5. Anti-CD96 mAb suppression of metastases is not dependent on Fc function. Groups of 4–8 C57BL/6 WT or various FcR-deficient mice (FcγRIII, FcγRIV or Fcϵγ) were injected i.v. with B16F10 melanoma (1 × 105cells). On days 0 and 3 after tumor inoculation, mice were treated with i.p. injections of (A-C) cIg (250 or 400 μg), (A) anti-CD96 mAb 3.3 (250 μg, rat IgG1), (B) 6A6 (250 μg, rat IgG2a), or (C) 8B10 (400 μg, rat IgG2a) as indicated. (D) Groups of 5 C57BL/6 WT mice were injected i.v. with B16F10 melanoma (2 × 105cells). On days 0 and 3 after tumor inoculation, mice were treated with 250 μg i.p. injections of cIg (rat G1), 3.3 (rat IgG1), 3.3 (mouse IgG1), 3.3 (mouse IgG1 D265A), or 3.3 (mouse IgG2a). The metastatic burden was quantified in the lungs after 14 days by counting colonies on the lung surface. Individual mice are represented by each symbol and means ± SEM of 4–8 mice per group are shown. Significant differences between groups as indicated by crossbars were determined by a one-way ANOVA with post Tukey test (*: p < 0.05, **: p < 0.01, ***: p < 0.001).
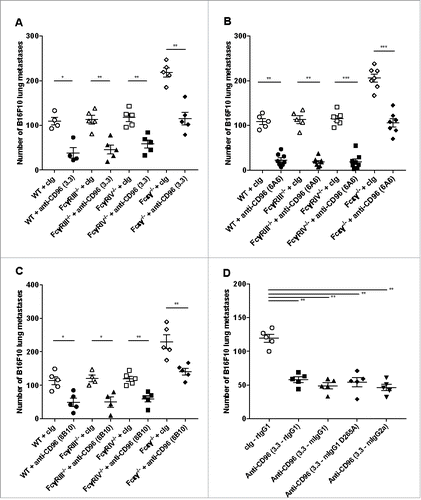
Anti-CD96 suppression of experimental lung metastasis depends upon NK cells, CD96 and IFN-γ
We next wished to examine whether the anti-metastatic mechanism of action of the 6A6 and 8B10 anti-CD96 mAbs was NK cell and IFN-γ-dependent, as previously described for 3.3 mAb.Citation4 Using the B16F10 experimental lung metastases model, the activity of the anti-CD96 6A6 and 8B10 clones was lost in WT mice in which their NK cells were depleted or IFN-γ neutralized (, ). The anti-metastatic effects of 6A6 and 8B10 were also abrogated in CD96-deficient (Cd96-/-) mice. Thus, ultimately, all three anti-mCD96 mAbs, 3.3, 6A6 and 8B10 were able to mediate metastases suppression via NK cells and IFN-γ.
Figure 6. Anti-CD96 suppression of experimental lung metastasis depends upon NK cells, CD96 and IFN-γ. C57BL/6 WT and CD96-deficient mice (Cd96−/−) mice as indicated were injected i.v with B16F10 melanoma (2 × 105cells). On days 0 and 3 after tumor inoculation, mice were treated with i.p. injections of (A, B) cIg (250 μg) or anti-CD96 mAbs (A) 6A6 (250 μg) or (B) 8B10 (250 μg). Some groups of mice were treated i.p. on days −1, 0 and 7 after tumor inoculation with cIg (100 μg), anti-asGM1 (50 μg) or anti-IFN-γ (250 μg). The metastatic burden was quantified in the lungs after 14 days by counting colonies on the lung surface. Mean ± SEM of 5 mice per group are shown. A and B were the same experiment and some of the cIg and anti-asGM1 treated control groups are replicated in each panel for visual display. Significant differences between groups as indicated by crossbars were determined by a one-way ANOVA with post Tukey test (*: p < 0.05, **: p < 0.01, ****: p < 0.0001).
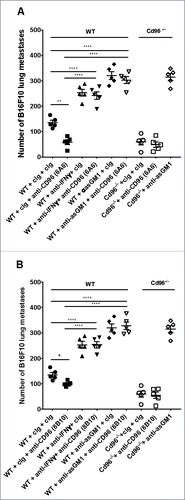
Anti-CD96 8B10 mAbs protects from metastases in the absence of CD155 and IL-12p35
CD155 has been identified as main ligand for the Ig-like receptors CD96, CD226 and TIGIT on NK cells and T cellsCitation5,Citation6 and is expressed in mice and humans on DC and other antigen presenting cells (APC).Citation21-23 Therefore, a key question was whether any of the anti-CD96 mAbs were dependent upon host CD155 expression, particularly 8B10 mAb, which does not block CD96-CD155 interactions. WT or CD155-deficient (Cd155−/−) mice were intravenously injected with B16F10 melanoma cells and mice treated with cIg or anti-CD96 mAbs on day 0 and 3 after tumor inoculation. Cd155−/− mice displayed an appreciable decrease in the number of lung metastases in comparison with WT mice (), and this decrease is postulated to be due to enhanced DNAM-1 expression and function in these mice (24; and our unpublished data). Notably, 3.3 and 6A6 anti-CD96 mAbs lost a considerable proportion of their anti-metastatic function in CD155−/− mice compared with WT mice, consistent with the importance of host CD155 (possibly on APC) and the ability of these mAbs to block CD96-CD155 interactions. By contrast, 8B10 retained its activity in CD155−/− mice, suggesting that its activity was independent of host CD155. In a similar manner, 8B10 largely retained its anti-metastatic activity in IL-12p35-deficient mice, whereas 3.3 and 6A6 activity was partially lost in IL-12p35−/− mice compared to WT mice (). Interestingly, all three mAbs lost their anti-metastatic activity in CD226-deficient mice (), suggesting a common final mechanism of control by NK cells despite the distinct role of CD155 and IL-12p35 for the blocking and non-blocking anti-CD96 mAbs.
Figure 7. Anti-CD96 8B10 mAbs protects from metastases in the absence of CD155 and IL-12p35. (A) Groups of C57BL/6 WT or CD155-deficient (Cd155−/−) mice were injected i.v. with B16F10 melanoma (1 × 105 cells). On days 0 and 3 after tumor inoculation, mice were treated with i.p. injections of anti-CD96 3.3 mAb (rat IgG1, 250 μg), 6A6 mAb (rat IgG2a, 250 μg), and 8B10 mAb (rat IgG2a, 250 μg) or the equivalent amount of cIg (250 μg). (B) Groups of C57BL/6 WT or IL-12p35-deficient (IL-12p35−/−) mice were injected i.v. with B16F10 melanoma (2 × 105 cells). On days 0 and 3 after tumor inoculation, mice were treated with i.p. injections of anti-CD96 3.3 mAb (rat IgG1, 250 μg), 6A6 mAb (rat IgG2a, 250 μg), and 8B10 mAb (rat IgG2a, 250 μg) or the equivalent amount of cIg (250 μg). (C) Groups of C57BL/6 WT or Cd226-deficient (Cd226−/−) mice were injected i.v. with B16F10 melanoma (5 × 105 cells). On days 0 and 3 after tumor inoculation, mice were treated with i.p. injections of anti-CD96 3.3 mAb (rat IgG1, 250 μg), 6A6 mAb (rat IgG2a, 250 μg), and 8B10 mAb (rat IgG2a, 250 μg) or the equivalent amount of cIg (250 μg). The metastatic burden was quantified in the lungs after 14 days by counting colonies on the lung surface. Mean ± SEM of 5–6 mice per group are shown. Significant differences between groups as indicated by crossbars were determined by a one-way ANOVA with post Tukey test (*: p < 0.05, **: p < 0.01, ***: p < 0.001; ****: p < 0.0001).
Discussion
Here, we have compared three anti-mouse CD96 mAbs including two that block CD96-CD155 (3.3 and 6A6) and one that does not block CD96-CD155 (8B10). We demonstrated that both 3.3 and 6A6 bind to the first Ig domain of mouse CD96, while 8B10 binds to epitopes mostly in the second domain of CD96 and does not compete with CD155 interactions. In four different experimental metastases models and one spontaneous metastasis model, the relative order of anti-metastatic potency was 6A6 > 3.3 > 8B10. Nevertheless, the anti-metastatic activity of 8B10 was significant and surprising and indicated that immune checkpoint blockade of CD96-CD155 interactions was not critical for anti-tumor activity. The metastatic burden control of all of the anti-CD96 clones was highly dependent on NK cells and IFN-γ, and independent of Fc function. Consistent with its inability to block CD96-CD155 interactions, 8B10 retained anti-metastatic activity in CD155-deficient and IL-12p35-deficient mice, whereas 3.3 and 6A6 lost potency in these strains of mice. Altogether, these data demonstrate that there may be several different types of mAb that can target CD96 and NK cell anti-metastatic activity with therapeutic benefit.
The distinct mechanism to completely explain the relative efficacy profiles of the 6A6 and 3.3 mAbs remains under investigation. It does not appear that mAb Fc isotype or KD for CD96 or the relative ability to block CD155 explains why 6A6 is superior. The 3.3 mAb has a comparatively very slow off rate and perhaps prolonged binding to CD96 is not a favourable property in vivo. Further investigation of mAb pharmacokinetics and epitope mapping may define characteristics that favour the anti-metastatic activity of 6A6 over 3.3, and why 8B10 displays activity despite not blocking CD96-CD155 interactions.
8B10 mAb clearly had a lower affinity for CD96 than 3.3 and 6A6, but its inability to block CD96-CD155 interactions most likely limited its anti-metastatic activity. The fact that 8B10 lost activity in mice depleted of NK cells or neutralized for IFN-γ, but fully retained activity in mice deficient in CD155 or IL-12p35 suggested that 8B10 ligation of CD96 could stimulate the anti-metastatic activity of NK cells completely independently of their interactions with APC. Perhaps here, direct NK cell:tumor interactions were more critical for the activity of 8B10. The signaling potential of mouse CD96 has not been well explored despite the presence of an ITIM motif in its cytoplasmic tail.Citation25 It remains to be determined whether some types of anti-CD96 mAbs such as 8B10 can directly block negative signaling or somehow agonize signaling through the CD96 counter receptor, CD226. It was interesting that all three anti-CD96 mAbs were inactive in CD226-deficient mice, perhaps reflecting a critical need for CD226 in any NK cell:tumor or NK cell:APC interactions, regardless of the means by which the NK cells had become activated by anti-CD96 mAbs.
Altogether, given the extensive expression of PVR/CD155 ligand on certain types of cancers and its link with enhanced tumor proliferation and migration,Citation26 targeting CD96 will be an attractive and a critical approach in the clinic for NK cell-mediated recognition of tumors and particularly in supressing tumor metastases. The activity of 8B10, independent of CD96-CD155 interaction, encourages us to believe that tumors down regulating CD155 (either on host antigen presenting cells in the tumor or tumor cells themselves) or lacking CD155 expression, might still be potential targets for such types of anti-CD96 mAbs.
Methods
Mice
C57BL/6 and BALB/c wild-type (WT) mice were obtained from Walter and Eliza Hall Institute for Medical Research and bred in house. C57BL/6 Cd96−/−, Cd226−/− and IL-12p35−/− mice were maintained as previously described.Citation4 C57BL/6 Cd155−/− mice (originally generated by Dr Yoshimi Takai, Kobe University, Kobe, Japan) were kindly provided by Dr. Stephen Gasser from the Department of Microbiology at the National University of Singapore. These mice were generated and maintained as described.Citation27 C57BL/6 FcγRIII−/− and FcγRIV−/− were kindly provided by Dr. Jeffrey Ravetch and have been already described.Citation28 C57BL/6 Fcϵγ−/− mice as previously described,Citation29 were kindly provided by Professor Mark Hogarth. All mice were bred and maintained at the QIMR Berghofer Medical Research Institute and used at the ages of 6 to 14 weeks. Groups of 5 to 10 mice per experiment were used for experimental lung metastases for confirmation of detection of biological differences. No mice were excluded on the basis on pre-established criteria in this study, and no active randomization was applied to experimental groups. Researchers were not “blinded” to the group allocation during the experiment and/or when assessing the outcome. All experiments were approved by the QIMR Berghofer Medical Research Institute Animal Ethics Committee.
Experimental lung metastases
All the tumor experiments were performed once unless specifically indicated. Single-cell suspensions of B16F10 melanoma cells (1–5 × 105), LWT1 melanoma cells (5 × 105), RM-1 prostate carcinoma cells (5 × 104), CT26 colon carcinoma (5 × 105) were injected intravenously into the lateral tail vein of the indicated strains of mice. Lungs were harvested on day 14, and metastatic tumor nodules were counted under a dissection microscope.
Spontaneous tumor metastasis
For spontaneous metastasis and post-surgery survival experiments, 5 × 104 4T1.2 tumor cells were inoculated into the fourth mammary fat pad of BALB/c mice, respectively. Neoadjuvant treatment with anti-CD96 mAbs (250 μg i.p.) was provided on day 12 and 14. On day 16 post injection, mice were anesthetized, the primary tumor surgically was removed and the wound closed with surgical clips. Mice were then either sacrificed at day 34 and lung metastases counted or cohorts were maintained and survival of the mice was monitored. All the tumors used in this study were CD155+.
Treatments and doses
Mice were treated with cIg (2A3 or 1–117), anti-CD96 (3.3, rat IgG1, mouse IgG1, mouse IgG1 D265A, mouse IgG2a),Citation18 anti-CD96 (6A6, rat IgG2a),Citation14 anti-CD96 (8B10, rat IgG2a) or anti-PD1 (CD279) (RMP1-14, rat IgG2a) using schedules and doses as indicated. Some mice additionally received either anti-asialoGM1 NK cell depleting antibody or anti-IFN-γ (H22) neutralizing antibody as previously described.Citation18,Citation30
Cell culture
B16F10 melanoma (ATCC), LWT1 melanoma, CT26 colon adenocarcinoma, 4T1.2 mammary carcinoma, and RM-1 prostate carcinoma cell lines, were maintained, injected, and monitored as previously described.Citation4,Citation31,Citation32 Human embryonic kidney 293(HEK) cell line was grown at 37°C in 5% CO2, 10% FCS, L-glutamine (Gibco) and penicillin-streptomycin (Gibco) supplemented RPMI medium (Gibco) and was kindly provided by Dr. Nathan Subramanian at QIMR Berghofer Medical Research Institute, Queensland. These cells were maintained, monitored and transfected as previously described.Citation19 All cell lines were routinely tested negative for mycoplasma, but cell line authentication was not routinely performed.
Biotinylation of antibody for immobilization onto Streptavidin Biosensors
EZ-Link Sulfo-NHS-LC-LC Biotin from Thermo Fisher Scientific (Waltham, MA, USA) was added to each CD96 mAb in a molar coupling ratio (MCR) of 1:1 and incubated 30 minutes at room temperature protected from light. The reaction was stopped by removing excess of biotin reagent using the Zeba ™ desalt spin columns (Thermo Fisher Scientific) following manufacturer's instructions for the desalting procedure.
Affinity measurements
The binding kinetics of anti-mouse CD96 mAb to recombinant CD96 (CSIRO) was measured using a Forte Bio Octet Red system (Forte Bio, Inc. USA). The assay was performed in 96-well microtiter plates at 30°C in PBS buffer with 0.5 mg/ml BSA and 0.02% Tween 20. Sensor tips were first pre-wet for 20 mins in buffer, and then the microplates were filled with 200 μl per well of diluted samples (rCD96) or buffer. The biotinylated CD96 antibody was immobilised onto Streptavidin biosensors (Forte Bio, Inc.) at 25 µg/ml. The association and dissociation of recombinant CD96 (CSIRO) was measured using descending two-fold dilutions. The association and dissociation rates were measured for 5 mins and 30 mins, respectively. The Kinetics parameters (ka and kd) and affinities (KD) were calculated by global fitting to a 1:1 interaction model using the Forte Bio Data Analysis Software V7.1 (ForteBio, Inc.). Data was exported as a Microsoft Excel file for analysis and presentation in other software packages. Multiple independent measurements were performed.
In vitro transient transfection and binding of mAbs to chimeric receptors
Different CD96 chimeric plasmids were constructed as previously described inCitation19 and were kindly provided by Dr. Günter Bernhardt at Institute of Immunology, Hannover Medical School, Germany. Following the standard FuGENE® 6 (Promega) transfection procedures, 1 μg of cDNA encoding the different human/mouse CD96 versions were transiently transfected into HEK-293 parental cells. The transfected cells were detached 48 hrs later post transfection and incubated with the different mouse anti-CD96 mAbs clones 3.3, 6A6 or 8B10 for binding assays, followed by a final incubation with a goat anti-rat AF647 secondary antibody (Thermo Fisher Scientifics) for detection.
Flow cytometry
Single-cell suspensions of either HEK-293 parental cells or transiently transfected with the diverse anti-CD96 chimeric constructs were surface stained in a two-step incubation procedure after 48 hrs post transfection.Citation19 Samples were firstly surface stained with anti-CD96 clone 3.3 (Bioxcell), 6A6 or 8B10 for 30 minutes at 4°C. Secondly, incubation with a goat anti-rat secondary antibody (Alexa Fluor 647 from Thermo Fisher Scientific) was used for detection of antibody binding. For the mouse CD155 hFc binding assay, single-cell suspensions of parental HEK −293 cells were transiently transfected as above with the full mouse construct (MMM). Fourty-eight hours post transfection cells were pre-incubated for 30 min at room temperature with serial dilutions of different anti-mouse CD96 mAbs or isotype controls. This was followed by a second 30 min incubation on ice of the transfected cells with 3 μg/ml of mouse CD155-hFc. After two washes with FACS buffer (PBS + 10% FCS) a final 30 min on ice incubation was performed with a goat anti-human secondary antibody (as above). All this data was collected on Fortessa 4B (BD) or FACSCanto II (BD) flow cytometers and analyzed with FlowJo v10 software (Tree Star, Inc.).
Statistical analysis
Statistical analysis was achieved using Graphpad Prism Software. Data was considered to be statistically significant where the p value was equal to or less than 0.05. Metastases were compared using a one-way ANOVA multiple comparisons test with post Tukey correction. Differences in survival were evaluated using a Log rank test.
Conflicts of interest
M. J. Smyth has been supported by a scientific research agreement with Bristol Myers Squibb, Corvus Pharmaceuticals, Aduro Biotech and Tizona Therapuetics. M. J. S. is on the scientific advisory board at Tizona Therapeutics. The other authors have no conflicts of interest to declare.
Author contributions
Conception and design: M.J. Smyth.
Development of methodology: A. Roman Aguilera, M. J. Smyth, V. P. Lutzky, D. Mittal, W.C. Dougall.
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): A. Roman Aguilera, V. P. Lutzky, D. Mittal, X.Y. Li, G. Bernhardt, W. C. Dougall, M. J. Smyth
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): A. Roman Aguilera, V.P. Lutzky, D. Mittal, W. C. Dougall, M. J. Smyth
Writing, review, and/or revision of the manuscript: A. Roman Aguilera, V.P. Lutzky, W. C. Dougall, M. J. Smyth
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): A. Roman Aguilera, W. C. Dougall, K. Takeda
Study supervision: M.J. Smyth & W. C. Dougall.
supp_data.zip
Download Zip (1,019.2 KB)Acknowledgments
The authors wish to thank Liam Town and Kate Elder for breeding, genotyping and maintenance and care of the mice used in this study. We thank Jeffrey Ravetch for providing the original C57 BL/6 FcγR III and FcγR IV gene-targeted breeding pairs. We thank Mark Hogarth for providing the original C57 BL/6 Fcϵγ gene-targeted breeding pairs.
Additional information
Funding
References
- Christofori G. New signals from the invasive front. Nature. 2006;441:444–50. doi:10.1038/nature04872.
- Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–95. doi:10.1016/j.cell.2006.11.001.
- Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. 2017;32:135–54. doi:10.1016/j.ccell.2017.06.009.
- Blake SJ, Stannard K, Liu J, Allen S, Yong MC, Mittal D, Aguilera AR, Miles JJ, Lutzky VP, de Andrade LF, et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 2016;6:446–59. doi:10.1158/2159-8290.CD-15-0944.
- Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, Kitamura T, Nicholl J, Sutherland GR, Lanier LL, et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity. 1996;4:573–81. doi:10.1016/S1074-7613(00)70060-4.
- Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, Tom I, Ivelja S, Refino CJ, Clark H, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10:48–57. doi:10.1038/ni.1674.
- Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol. 2015;15:243–54. doi:10.1038/nri3799.
- Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. 2017;276:112–20. doi:10.1111/imr.12518.
- Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, Cantoni C, Grassi J, Marcenaro S, Reymond N, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med. 2003;198:557–67. doi:10.1084/jem.20030788.
- Lakshmikanth T, Burke S, Ali TH, Kimpfler S, Ursini F, Ruggeri L, Capanni M, Umansky V, Paschen A, Sucker A, et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J Clin Invest. 2009;119:1251–63. doi:10.1172/JCI36022.
- Gilfillan S, Chan CJ, Cella M, Haynes NM, Rapaport AS, Boles KS, Andrews DM, Smyth MJ, Colonna M. DNAM-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J Exp Med. 2008;205:2965–73. doi:10.1084/jem.20081752.
- Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol. 2012;188:3869–75. doi:10.4049/jimmunol.1103627.
- Wang PL, O'Farrell S, Clayberger C, Krensky AM. Identification and molecular cloning of tactile. A novel human T cell activation antigen that is a member of the Ig gene superfamily. J Immunol 1992;148:2600–8
- Seth S, Maier MK, Qiu Q, Ravens I, Kremmer E, Forster R, Bernhardt G. The murine pan T cell marker CD96 is an adhesion receptor for CD155 and nectin-1. Biochem Biophys Res Commun. 2007;364:959–65. doi:10.1016/j.bbrc.2007.10.102.
- Lenac Rovis T, Kucan Brlic P, Kaynan N, Juranic Lisnic V, Brizic I, Jordan S, Tomic A, Kvestak D, Babic M, Tsukerman P, et al. Inflammatory monocytes and NK cells play a crucial role in DNAM-1-dependent control of cytomegalovirus infection. J Exp Med. 2016;213:1835–50. doi:10.1084/jem.20151899.
- Blake SJ, Dougall WC, Miles JJ, Teng MW, Smyth MJ. Molecular Pathways: Targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22:5183–8. doi:10.1158/1078-0432.CCR-16-0933.
- Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J Immunol. 2004;172:3994–8. doi:10.4049/jimmunol.172.7.3994.
- Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, Ritchie DS, Colonna M, Andrews DM, Smyth MJ. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. 2014;15:431–8. doi:10.1038/ni.2850.
- Meyer D, Seth S, Albrecht J, Maier MK, du Pasquier L, Ravens I, Dreyer L, Burger R, Gramatzki M, Schwinzer R, et al. CD96 interaction with CD155 via its first Ig-like domain is modulated by alternative splicing or mutations in distal Ig-like domains. J Biol Chem. 2009;284:2235–44. doi:10.1074/jbc.M807698200.
- Liu J, Blake SJ, Yong MC, Harjunpaa H, Ngiow SF, Takeda K, Young A, O'Donnell JS, Allen S, Smyth MJ, et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 2016;6:1382–99. doi:10.1158/2159-8290.CD-16-0577.
- Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56:855–65. doi:10.1016/0092-8674(89)90690-9.
- Lim YP, Fowler LC, Hixson DC, Wehbe T, Thompson NL. TuAg.1 is the liver isoform of the rat colon tumor-associated antigen pE4 and a member of the immunoglobulin-like supergene family. Cancer Res. 1996;56:3934–40
- Maier MK, Seth S, Czeloth N, Qiu Q, Ravens I, Kremmer E, Ebel M, Müller W, Pabst O, Förster R, et al. The adhesion receptor CD155 determines the magnitude of humoral immune responses against orally ingested antigens. Eur J Immunol. 2007;37:2214–25. doi:10.1002/eji.200737072.
- Nagumo Y, Iguchi-Manaka A, Yamashita-Kanemaru Y, Abe F, Bernhardt G, Shibuya A, Shibuya K. Increased CD112 expression in methylcholanthrene-induced tumors in CD155-deficient mice. PLoS One. 2014;9:e112415. doi:10.1371/journal.pone.0112415.
- Fuchs A, Colonna M. The role of NK cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Semin Cancer Biol. 2006;16:359–66. doi:10.1016/j.semcancer.2006.07.002.
- Sloan KE, Eustace BK, Stewart JK, Zehetmeier C, Torella C, Simeone M, Roy JE, Unger C, Louis DN, Ilag LL, et al. CD155/PVR plays a key role in cell motility during tumor cell invasion and migration. BMC Cancer. 2004;4:73. doi:10.1186/1471-2407-4-73.
- Abe A, Fukui H, Fujii S, Kono T, Mukawa K, Yoshitake N, Sekikawa A, Ichikawa K, Tomita S, Yamagishi H, et al. Role of Necl-5 in the pathophysiology of colorectal lesions induced by dimethylhydrazine and/or dextran sodium sulphate. J Pathol. 2009;217:42–53. doi:10.1002/path.2431.
- Giorgini A, Brown HJ, Lock HR, Nimmerjahn F, Ravetch JV, Verbeek JS, Sacks SH, Robson MG. Fc gamma RIII and Fc gamma RIV are indispensable for acute glomerular inflammation induced by switch variant monoclonal antibodies. J Immunol. 2008;181:8745–52. doi:10.4049/jimmunol.181.12.8745.
- Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–29. doi:10.1016/0092-8674(94)90115-5.
- Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res. 2013;19:5626–35. doi:10.1158/1078-0432.CCR-13-0545.
- de Andrade LF, Ngiow SF, Martinet L, Smyth MJ. Natural Killer cell control of BRAFV600E mutant melanoma during targeted therapy. Oncoimmunology. 2015;4:e998119. doi:10.1080/2162402X.2014.998119.
- Terabe M, Swann J, Ambrosino E, Sinha P, Takaku S, Hayakawa Y, Godfrey DI, Ostrand-Rosenberg S, Smyth MJ, et al. A nonclassical non-Valpha14Jalpha18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med. 2005;202:1627–33. doi:10.1084/jem.20051381.