
ABSTRACT
Expressed by cancer stem cells of various epithelial cell origins, CD133 is an attractive therapeutic target for cancers. Autologous chimeric antigen receptor-modified T-cell directed CD133 (CART-133) was first tested in this trial. The anti-tumor specificity and the postulated toxicities of CART-133 were first assessed. Then, we conducted a phase I clinical study in which patients with advanced and CD133-positive tumors received CART-133 cell-infusion. We enrolled 23 patients (14 with hepatocellular carcinoma [HCC], 7 with pancreatic carcinomas, and 2 with colorectal carcinomas). The 8 initially enrolled patients with HCC were treated by a CART-133 cell dose escalation scheme (0.05–2 × 106/kg). The higher CAR-copy numbers and its reverse relationship with the count of CD133+ cells in peripheral blood led to the determination of an acceptable cell dose is 0.5–2 × 106/kg and reinfusion cycle in 23 patients. The primary toxicity is a decrease in hemoglobin/platelet (≤ grade 3) that is self-recovered within 1 week. Of 23 patients, three achieved partial remission, and 14 achieved stable disease. The 3-month disease control rate was 65.2%, and the median progression-free survival was 5 months. Repeated cell infusions seemed to provide a longer period of disease stability, especially in patients who achieved tumor reduction after the first cell-infusion. 21 out of 23 patients had not developed detectable de novo lesions during this term. Analysis of biopsied tissues by immunohistochemistry showed CD133+ cells were eliminated after CART-133 infusions. This trial showed the feasibility, controllable toxicities, and effective activity of CART-133 transfer for treating patients with CD133-postive and late-stage metastasis malignancies.
Introduction
In recent years, chimeric antigen receptor-specific T (CAR-T) cells have emerged as a tool for the clinical treatment of cancers. T cells modified by CAR have a potent cellular effector mechanism, called HLA-independent recognition, to recognize and kill tumor cells expressing the corresponding antigen.Citation1 Especially in B cell acute lymphoblastic leukemia (B-ALL), the target CD19-modified CAR-T cell therapy achieved a 74% to 90% complete remission rate.Citation2–Citation6 CAR-T cells targeting CD19 or CD20 have also shown encouraging antitumor activity in relapsed or refractory B-cell lymphomas.Citation6,Citation7 In solid tumors, epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2, disialoganglioside 2, and mesothelin and interleukin-13 Ra2 are current targets of CAR-T cells. Although the strategy for the use of CAR-T cells has been shown to be safe and effective in the treatment of B-ALL it has not yet been shown same effective for solid tumors as for hematologic malignancies.Citation8–Citation11
Hepatocellular carcinoma (HCC) is a leading cause of cancer-related morbidity and mortality. Despite evaluation of many chemotherapy and targeted therapy agents, the only proven treatments for advanced disease are sorafenib, regorafenib, and lenvatinib, and overall survival benefits are modest.Citation12,Citation13 Additionally, liver metastases may escape immune surveillance due to the immunosuppressive nature of the intrahepatic space,Citation11,Citation14 and once the tumor metastasizes to the liver, a poor prognosis is suggested. China also has a relatively high incidence of liver cancer. Clinical trials being conducted in China are evaluating MG7 and EpCAM antigen-targeted CAR-T cells in liver cancer.Citation15
CD133 is a pentaspan transmembrane glycoprotein that is overexpressed in various solid tumors. CD133 was found here to be highly expressed in 50% of case of HCC, pancreatic cancer, gastric cancer, and intrahepatic cholangiocarcinomas.Citation16,Citation17 Especially for HCC, high CD133 expression in HCC cells corresponds with higher stage tumors, and indicating a poor prognosis for most patients.Citation18,Citation19 Moreover, CD133 is a marker of cancer stem cells (CSCs) and endothelial progenitor cells (EPCs) that had been verified to participate in tumor metastasis and recurrence.Citation20,Citation21 These characteristics make CD133 a reasonable target for immunotherapy for patients with advanced CD133-postive tumors.
In this study, we have developed a CD133-specific CAR-modified T cell, termed CART-133. This CAR T eliminates CD133-expressing tumors in vitro and in a xenograft tumor model. Then, we are conducting a clinical trial to assess the antimalignancy efficacy, feasibility, and toxicity of CART-133 cells in patients with advanced HCC or other malignancies with liver metastases, and no serious adverse events were observed.
Methods
Patients
This study was an open-label and single-arm phase I trial (ClinicalTrials.gov identifier: NCT02541370) that was approved by the Institutional Review Board at the Chinese PLA General Hospital. Adult patients (aged 18–70 years) were eligible for inclusion if they had a diagnosis of progressive disease (PD) of CD133-positive HCC or solid malignant tumor with multiple metastases based on Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1) after receiving at least two previous systemic therapies. Primary exclusion criteria were severe organ dysfunction, a history of or active systemic autoimmune/immunodeficiency disease, and a treatment history of immunosuppressive agents or glucocorticoids within a month of the study. All patients provided written informed consent before enrolling in the study.
Generation and transduction of CART-133 cells
Generation of lentiviral.
CAR.133 containing anti-CD133 scFv derived from HW350341.1, human CD137 and CD3z signaling domains (). The CAR construct was verified by DNA sequencing. A pseudotyped, clinical-grade lentiviral vector supernatant was produced by standard transient transfection as McGinley et al. described. According to the manufacturer's instructions, Lipofectamine 3000 transfection reagent (Invitrogen, USA), pWPT-anti-CD133 CAR plasmid, ps-PAX2 packaging plasmid, and pMD2.G envelope plasmid were transfected into 293 T cells. The lentiviral supernatants were collected and stored at −80°C.
Generation of CART-133 Cells.
Figure 1. Treatment plan and treatment protocol. (A) Schematic representation of the CAR-133-CD137ζ chimeric T-cell receptor cDNA plasmid, not to scale. (B) Consort flow diagram of the clinical trial. (C) One standard cycle of CART-133 treatment procedure. CART-133 treatment for non-HCC patients included a chemotherapy pre-regimen (red dashed line).
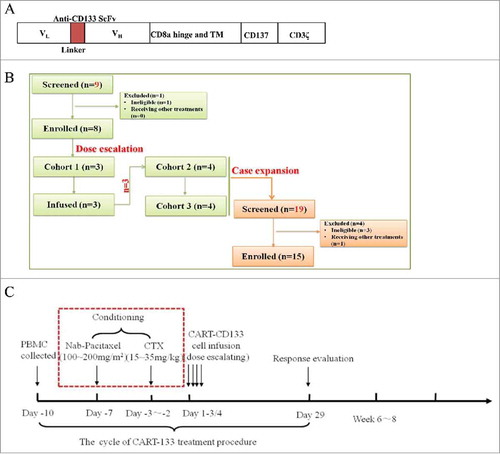
CART-133 cells were generated as previously described.Citation7,Citation22–Citation24 Briefly, PBMCs from the 80-100 ml fresh blood were directly suspended in the GT-T551 medium (Takara, Japan) with the anti-CD3 mAb (500 ng/μL) and IL-2 (interleukin-2) (300 U/mL) (Peprotech, USA). Lentivirus-mediated CAR transduction was performed on day 3 of cell culture in six-well plates precoated with a recombinant fibronectin fragment. After transduction, the cells were expanded ex vivo in the presence of IL-2 added three times weekly until the specified cell dose achieved.
Study description and clinical response criteria
All patients had imaging with computed tomography (CT), magnetic resonance imaging (MRI), and/or positron emission tomography to assess overall disease burden before CART-133 cell infusion. Peripheral blood (PB) samples were obtained before CART-133 cell infusion and at predetermined time points after infusion to evaluate the activity of CART-133 cells and toxicity. In the initial phase of the study, patients1 to 8 were enrolled in doses escalation scheme (). HCC patients were not given conditioning treatment before cell infusion due to liver dysfunction and non-HCC patients were conditioned with cyclophosphamide (30 mg/kg) and nab-paclitaxel (150 mg/m2) conditioning treatment. Protocol details are shown in the study flow chart (). Patients were eligible to receive an additional cell treatment cycle if they could tolerate the toxicity and had a clinical benefit. The second treatment cycle was performed at least 4 weeks after the first infusion. All patients were recruited to receive CART-133 cell therapy and to undergo follow-up between June 15, 2015, and June 30, 2017.
Clinical response was evaluated by using contrast-enhanced computed tomography (CT) or/and magnetic resonance imaging (MRI) and performed 1 month after the cell infusion. The clinical responses were defined according to Response Evaluation Criteria in RECIST 1.1 and immune-related response criteria.Citation25 Adverse events were documented and graded based on the Common Terminology Criteria for Adverse Events version 4.0 (CTCAE 4.0). Cytokines release syndrome (CRS) graded and managed based on a previous report from Lee et al.Citation26 Dose-limiting toxicity (DLT) was defined as any grade 3 or above toxicity was considered possibly or likely related to CART-133 cells. The following toxicities were excluded from DLT: tumor lysis syndrome and liver dysfunction resolved to below grade 2 within 2 weeks of onset; infusion-related toxicities (within 24 hours); grade 3 diarrhea, nausea, and anorexiathat that would be resolved to grade 2 within 2 weeks; grade 3 CRS that would be resolved within 1 week; transient increase of grade 3 liver enzymes (<72 hours); and grade 3 fever lasting 1 week or less.
Flow cytometry analysis and cell sorting
Evaluation of CAR expression was performed by staining with a goat anti-mouse Fab antibody (Jackson ImmunoResearch, USA). In addition, the following anti-human antibodies were also used in this study: CD133 (phycoerythrin, PE), CD3 (chlorophyll protein complex PerCP), CD4 (fluorescein isothiocyanate, FITC), CD8-PE, CD45RO (allophycocyanin, APC), CD56-APC, CD62 L-PE, and CCR7-PE-Cy7 were purchased from Becton Dickinson. Isotype-matched control mAbs were applied in all the procedures. FACS data were analyzed by a FACS Calibur flow cytometer (BD Biosciences) and FlowJo software (Version 10.0.7, FlowJo, Ashland, OR).
Quantitative real-time PCR
Real-time quantitative polymerase chain reaction (Q-PCR) was used to assess the level of the CAR fusion genes according to a previously described protocol.Citation7 A 153-bp (base pair) fragment containing portions of the CD8 a chain and adjacent CD137 chain was amplified using forward primer 5′-GGTCCTTCTCCTGTCACTGGTT-3′ and reverse primer 5′-TCTTCTTCTTCTGG AAATCGGCAG-3′to detect the CAR gene; amplification of β-actin was used as an internal control and for normalization of DNA quantities. Q-PCR was performed using SYBR Green Master Mix (Toyobo, Japan) and run on an ABI prism 7500 sequence detection system (Applied Biosystems). A 7-point standard curve that consisted of 100 to 108 copies/μL plasmid DNA containing the CAR gene was prepared.
The expression of the CD133 gene in PBMC from patients was detected by Q-PCR. Total RNA was isolated from peripheral blood using Trizol reagent (Takara, Japan) and reverse transcribed into cDNA using the cDNA Synthesis Kit (Takara, Japan) according to the manufacturer's protocol. The following primers (forward and reverse primers) were used: CD133 (5′-GGCAACAGCGATCAAGGAG-3′ and 5′-GATGGATGCACCAAGCACAG-3′) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (5′-GTGGAGTCCACTGGCGTCTT-3′, and 5′-GTGCAGGAGGCATTGCTGAT-3′). The relative fold change in expression was calculated by normalizing against GAPDH to adjust for loading variation.
Cytokine measurements
Serum IL-2, IL-6, IL-8, IL-10, IFN-γ, TNF-α levels were batch analyzed using a BD Biosciences microbead sandwich immunoassay according to the manufacturer's instructions. Briefly, analyte concentration was determined using a standard curve prepared with each assay.
Statistical analysis
The results are shown as the mean ± standard error of the mean (SEM) of triplicate determinants (wells). Data were plotted using GraphPad Prism version 6.0. Two-way analysis of variance (ANOVA) was used to determine the significance of the differences between the means in all experiments. The survival curve and progression-free survival (PFS) were determined by the Kaplan-Meier method. P value < 0.05 was considered to be statistically significant. Detailed descriptions of statistical analyses are provided in Supplement Methods.
Results
CART-133 exhibits enhanced antitumor activity against CD133+ cell line
CART-133 cells used for in vitro experiments and animal models were generated from three healthy donors. Mean transfection efficiencies of 34.22% ± 4.00% and 32.95% ± 4.76% were verified in the final CART-133 and mock T-cell populations, respectively (Supplement ). Six kinds of tumor-cell lines (SW1990, HT29, DLD1, SW480, Hep3B, and LOVO) were divided into three groups (high, medium, and negative expression of CD133). CART-133 cells showed remarkable lysis ability and produced higher cytokines than to mock and NT (non-transduced T) cells against CD133high/medium+ cells but not CD133− cells after co-culture for 8 hours (Supplement ). The subcutaneous xenotransplanted tumor model of CD133+ cells was established in BALB/c nude mice. As shown in Supplement , tumor growth was significantly inhibited and the high level of CAR-gene copy in tumor tissue was detected in the CART-133 cell group compared to other groups. (p < 0.05)
Figure 2. CART-133 cell dose escalation. (A) Dose group and CART-133 infusion cell dose pattern in all patients. (B) Hemoglobin (Hgb), reticulocyte, CD133+ cells and CAR-gene copy numbers in PB were detected before and at serial time points after CART-133 cell infusion in each patient from every cohort. (C) Tumor biomarkers in serum from each patient were detected before and at serial time points after CART-133 cell infusion. The blue dashed line on the plots is the normal range of each tumor biomarker. Red represents the increase, and green represents the decrease. N = cell infusion cycle; n = case number.
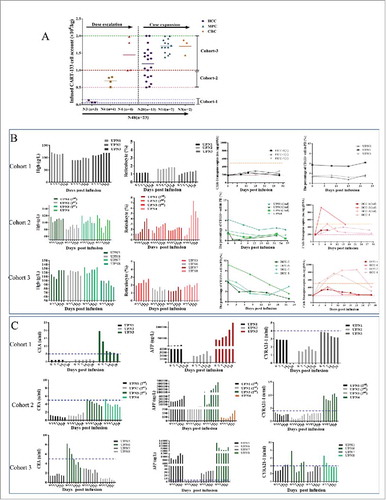
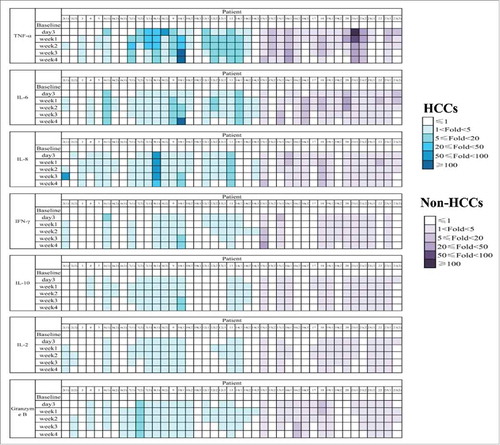
Figure 3. Safety of CART-133 cells. Cytokines from the serum of each patient's PB, which was collected before and at serial time points after cell infusion, was measured by fluorescence-activated cell sorting. The color shades represent different fold-changes with the baseline.
Patient characteristics
Twenty-three patients were enrolled in this study. The clinical and disease-specific characteristics of patients are listed in . Their median age was 56 years (range, 36–66 years). Fourteen patients had received a diagnosis of advanced HCC, 7 patents had advanced pancreatic cancer, and the other 2 patients had advanced colorectal cancer. CD133 positivity was confirmed by immunohisto- chemistry, as shown in Supplement . All patients had refractory/recurrent metastatic advanced disease and had experienced treatment failure with two or more conventional regimens. Twenty-two patients had stage IV carcinoma. Twelve patients had their primary lesion removed by surgery and presented with metastasis primarily in the lymph node, liver, and a wide range of anatomic sites. In HCC patients, 12 had sorafenib resistance, 10 had bulky disease burdens (lesion diameter > 10 cm), and 9 had portal vein tumor thrombus.
Table 1. Characteristics of patients (n = 23).
Generation and characterization of CART-133 cells
CART-133 cells were successfully generated from each patient. A mean of 95.254 ±7.286% of the infused cells were CD3+ cells principally composed of CD8+ subset (62.906±18.834%). The transduction efficiency ranged from 11.23% to 56.47%. Approximately 26.425% ±14.395% of the total cell population was Tcm (central memory cells) (CD3+/CD45RO+/CD62 L+/CCR7+; range: 6.34%–63.48%). The detailed data of infused cells for each patient are summarized in Supplement Table S2 and .
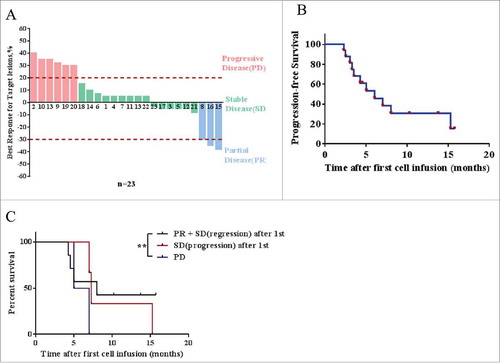
Figure 4. Response of CART-133 cells. (A) Maximum reduction in target lesion size after CART-133 cell infusion. PD: progressive disease, SD: stable disease, PR: partial remission. (B) Progression-free survival and overall survival by the Kaplan-Meier method. (C) Patients were divided into 3 groups based on response from the first cell infusion. PR + SD (lesion-regression), SD (lesion-progression), PD (progressive disease). Statistical analysis of PFS differences between the 3 groups.
CART-133 cell dose escalation scheme
The 8 initially enrolled patients were involved in dose-escalation scheme. Patients 1 to 3 in cohort 1 received CART-133 cells for a cell dose range of 0.05-0.15 × 106/kg (). Although no obvious adverse reactions occurred after CART-133 cell infusion for up to 1 month, all patients had no obvious decrease in CD133 cells and an increase in CAR-gene copy numbers in PB; the results of cytokines and tumor markers also had no distinct fluctuations (). Therefore, the dose of cohort 1 was closed because it was noneffective, and then the treatment dose of CART-133 cells was then amended in subsequent cohorts. Three patients in cohort 1 and patient 4 were eligible to receive doses of CART-133 cells in cohort 2 (0.5-1.0 × 106/kg). After cell infusion, all patients experienced mild (≤ Grade 2) hematologic toxicities and self-recovered within 1 week. In addition, increased CAR-gene copy number and reduced CD133+ cells in PB occurred in all four patients after cell infusion; two kinds of tumor markers were decreased in patients 3 and 4 (). Based on cohort 2's effective clinical immune response and tolerated/recovery toxicities, the CART-133 cell dose was increased to 1.0-2.0 × 106/kg for patients 5 to 8 in cohort 3. Similar toxicities and effective activity were all observed in cohort 3 (). No DLT was observed in cohorts 2 and 3. These results determined the acceptable CART-133 cell dose was 0.5 to 2 × 106/kg, which was subsequently used in the additional 15 patients. All further analyses started from the completed effective dose (excluding cohort-1) in this study.
Administration and safety of CART-133 cells
In this trial, 3 patients received two cell cycles, and 9 patients received three to four cycles ( and ). Patients received a mean of 1.43 × 106/kg CAR-positive T-cells (range, 0.5-2.0 × 106/kg) in total during the infusion. All toxicities except infusion-associated toxicities are listed in and Supplement Table S3. Nearly all the patients experienced reduction of hemoglobin, lymphocytes, and thrombocytes. Hematologic toxicities generally occurred 3 to 5 days after cell-infusion and self-recovered within 1 week. All the non-HCC patients experienced grade 2–4 lymphopenia whereas all HCC patients had less than grade 2. In particular, grade 3 adverse event (AE) that lasted 3 weeks with hyperbilirubinemia (direct bilirubin) was observed in three patients (4, 9, 22), who had an obstructed of the biliary tract accompanied by a high bilirubin background before cell infusion. Concentrations of plasma cytokines were determined after infusion by multiplex analysis at serial time points before and after CART-133 cell infusion (). There was a significant increase in the level of tumor necrosis factor-alpha (TNF-α), interleukin (IL)-6, and IL-8 at 1 and 2 weeks after cell infusion.
Table 2. Patients’ response and toxicity.
Overall clinical responses
As shown in and , the duration of responses in all patients currently ranges from 9 to 63 weeks. Three patients achieved PR, and 14 patients had SD for 9 weeks to 15.7 months (), and 3 patients have a continued response at the time of this writing. HCC patients’ median PFS was 7 months, and the median PFS was 5 months in all patients (range, 2.0-15.2 months) (). The 3-month disease control rate (DCR) was 65.2%, and the 6-month DCR was 30.4%. Tumor remission was observed in nine patients, and 21 patients had not developed detectable de novo metastatic lesions during the trial. In patients who achieved SD or PR after the first infusion, repeated cell infusion seems to have provided a longer period of disease stability ().
Table 3. CART-133 efficacy in all patients.
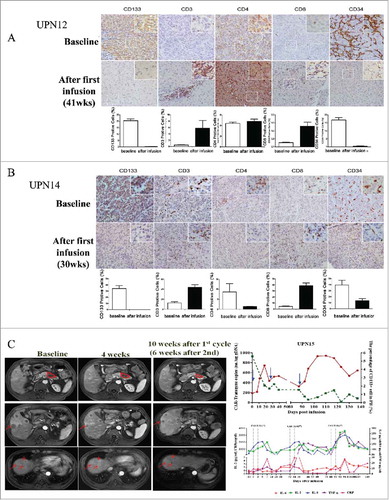
Special presentations: (i) Patients 6 and 12 were the only 2 patients who had a lower tumor burden (lesion diameter <5 cm) among HCC patients. Patient 6 obtained the longest stable disease period (15.2 months), and patient 12 attained a 13.7-month ongoing SD by having three CART-133 monotherapies. (ii) Patient 14 had stable disease for 6 months, and then his tumor rapidly progressed with a 50% increase of the primary lesion after the third cell infusion. We were surprised to observe that the liver biopsy pre- and post-treatment presented a dramatic clearance of CD133+ tumor cells and a rapid proliferation of CD133-negative tumor cells (). (iii) Patient 15 was diagnosed as having progressed stage IV pancreatic cancer accompanied by multiple metastases before cell treatment. His tumor was significantly reduced by approximately 40% after the first cell infusion, and he maintained this status for 4 months. A grade 2 CRS was first occurred after 1 week of the third cell infusion. The above data are shown in the .
Figure 5. Special presentations. (A) Immunohistochemical examination (diaminobenzidine with hematoxylin counterstaining) of a punch biopsy of liver lesion from patient 12 before and 41 weeks after the first CART-133 cell infusion. (B) Immunohistochemical examination of patient 14 before and 30 weeks after the first CART-133 cell infusion showed that tumor cells were CD133- after cell infusion. Notably, scattered CD3+ and CD8+ cells infiltrated the tumor after infusion, and CD34+ cells significantly decreased. (C) Left: Representative tumor response images for patient 15 before and after CART-133 cell infusion, contrast-enhanced MRI scans show pancreas and liver lesions reduced significantly 4 weeks after the first CART-133 cell infusion and remained reduced after the second cell infusion. Right: CD133+ cells in PB, CAR-gene copy numbers in PB and cytokines in serum were detected before and at serial time points after CART-133 cell infusion in patient 15.
Bioactivity and persistent of CART-133 cells in vivo
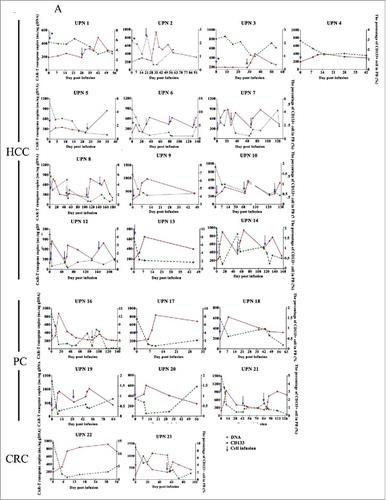
The reverse relationship between CART cells and CD133+ cells in PB was confirmed. The copy numbers of CAR-gene in PB reached their peak (646.1±257.1/μg gDNA) between 7 and 21 days in almost all patients. Meanwhile, during this period, CART-133 cells resulted in a significant decrease of the CD133+ cells and CD133 gene expression in PB ( and Supplement ). CAR-gene copies decreased after approximately 4 weeks (), but lower-level signals (364.4±117.8/μg gDNA) could be detected after more than 2 months in 7 patients. For repeated cell infusion, a peak of CAR-gene copies in PB was also observed in each cell cycle (Supplement ).
Biological evidence of CART-133 cells. (A) Quantitative real-time PCR was performed on genomic DNA harvested from each patient's PB mononuclear cells collected before and at serial time points after CART-133 cell infusion, using primers specific for the transgene. CD133+ cells count change from baseline in the blood after the infusion of CART-133 cells in 21 patients. CART-133 cell infusion in cohort 1 with * is shown. (B) CAR-gene copy numbers analysis from 22 patients (except patient 11, who refused to offer blood for detection) before and during the 1 month after cell infusion; the number of CART-133 cell infusion cycle was 41. (C) Patients were divided into 4 groups based on time of duration of PFS.
T lymphocyte cell infiltration and CD133+ cell elimination in tumor lesions. Immunohistochemical staining of biopsies from two patients at 2 weeks after the third cell infusion demonstrated an increasing augmentation of CD3+ and CD8+ T cells scattered within tumor parenchyma; on the contrary, CD133+ cells were not detected in tumor tissue ( and ).
Antitumor ability of CART-133 cells is positively correlated with the cytokine secretion ability. During the 4 weeks after cell infusion, the median of increased fold change in IFN-γ, TNF-α, and IL-6 in patients who achieved more than 8 months of PFS was significantly higher than in the other patients ().
Discussion
In this trial, our results exhibited the feasibility, safety, and efficacy of CART-133 in patients with advanced relapsed/refractory and metastatic tumor. Especially for HCC patients, previous studies in advanced HCC of first-line sorafenib have shown response rates of 2%–3%,Citation13,Citation27 and if patients whose conditions failed to first-line treatment and sorafenib, the median overall survival (OS) is no more than 4 months.Citation13 It was encouraging that HCC patients receiving CART-133 treatment have demonstrated median PFS of 7 months in our study; patients whose disease even failed to respond to first-line treatement and sorafenib might achieve relatively lengthy stable disease after CART-133 repeat infusion.
“On-target, off-tumor” effects are the most common side effects in the clinical application of CAR-T cells and result in an autoimmune response against normal tissues that express the targeted antigen in solid tumor therapies. CD133 was initially localized to CD34+ hematopoietic stem cells, and CD133 was also confirmed on the surface of CD34+ progenitor cells in adult bone marrow, umbilical cord blood, and PB.Citation28 Hematopoietic system toxicities were observed in almost all patients in this study, including the reduction of hemoglobin, lymphocytes, and thrombocytes after cell infusion for 2 to 5 days. The first reported that grade 2 to 3 bilirubinemia occurred in 3 patients with underlying illness backgrounds of either bile duct stenosis or high levels of bilirubin in this study. Bilirubinemia toxicity may in part be attributable to CD133, which was confirmed to be a marker of endothelial cells in the lesions. CART-133 cells targeted on CD133 antigen expressed on the bile duct endothelium in vivo after cell infusion, inducing an immune response that resulted in the release of inflammatory factors, increased bile duct blockage, and in turn, increased direct bilirubin secretion and caused severe toxicity. On the other hand, in this study, five HCC patients had lung metastasis, 7 pancreatic cancer patients and 2 colorectal carcinoma patients with lymph node metastasis in this study, and no obvious inflammatory response was observed in the liver, lung, pancreatic, colorectal, and multiple lymph nodes of these patients. So, this serious “on-target, off-tumor” effect of CART-133 seems to have occurred only in patients with bile duct stenosis. Given that the serious toxicity was different from that in other reports, CART-133 cell therapy must be approached with great caution in patients with a background of bile duct stenosis.
Persistence was the most common problem in the treatment of solid tumors by CART. We showed that CART-133 cells may persist for a long period in vivo by repeat infusion, and persistence of CAR-gene may achieve effective disease clearance and protection from recurrence. Some studies have reported that the second anti-CD19 CAR-T cell infusion did not achieve amplified peak as it had in the first infusion in vivo. One mechanism is that CD19 is still present but cannot be detected and recognized by CART-19 cells as its cell surface fragment containing cognate epitope is absent due to deleterious mutation and alternative splicing.Citation29 The other probable mechanism, a cellular immune response specific for murine scFv epitopes, of anti-CD19 CAR formed, resulting in the failure of the second infusion.Citation30,Citation31 In this study, the peak of amplification of the CAR-gene can be detected after CART-133 repeat infusion, and the CD133+ cells in patients’ PB was sustained at low levels. These results suggest that the homologous CD133 epitopes of the cell surface may not be prone to mutations, and patients have low probability to produce a humoral immune response to the CAR with murine scFv after receiving repeated CART-133 infusion; repeated infusions can extend the presence of CART-133 in vivo. In the previous study, CRS and tumor lysis symptoms correlated with the tumor burden at the time of the infusion of the CAR-directed T cells.Citation32,Citation33 On the other hand, patients 6 and 12 were the only patients who had a lower-load tumor among HCC patients; they both achieved longer stable disease than other HCC patients, which suggested the superiority of CART-133 cell treatment for HCC patients with lower tumor burden, as was endorsed by previous reports.Citation3,Citation7 These findings suggested that the CART cells were used in patients earlier in the course of their disease or immediately after surgery help eliminate the minimal residual disease and reduce the chance for recurrence. It is noteworthy that the 1-month DCR of non-HCC patients with a pre-treated regimen is 100%. Many mechanisms have been proposed to underline the enhanced in vivo antitumor activity of transferred T cells through prior conditioning chemotherapy. Lymphocytes and interstitial cell clearance can contribute to the expansion of CART cells in vivo and infiltrate the solid tumor.Citation6,Citation34,Citation35 Therefore, lymphocyte clearance and reduced tumor load conditioning regimens are necessary under the premise that patients can tolerate a conditioning chemotherapy regimen to enhance the CART-133 antitumor effect in vivo.
Many studies have shown that the high level of CD133 predicts a poor prognosis in human HCC patients, is associated with lower survival rate, and also positively correlates with a high level of microvessel density and CD34+ cells infiltration in the tumor bed.Citation18,Citation19 On the other hand, CD133 is currently the most mature surface marker on EPCs, and EPCs have been shown to have important correlations with tumor recurrence and distal metastasis.Citation36,Citation37 We found that patients seem can achieved long-term stability without new metastasis presented. It suggested that CART-133 cells seem to effectively affect the occurrence of new metastasis by reducing the angiogenesis in the tumor and the number of EPC in vivo. These results need to be further confirmation in future clinical trials with large samples.
Tumor antigen heterogeneity is an important factor limiting CART's efficacy in solid tumors. In this study, puncture specimens of two patients showed that CD133+ tumor cells were cleared after cell infusion, which confirmed that CART-133 can home to the tumor lesions and specifically kill CD133+ cells in vivo; on the other hand, tumor recurrences maintained by cells expressed nontargeted antigen after CART-133 cells eliminate target expression cells. Further improvement of the cure rate of solid tumor by CART treatment and the avoidance of the rapid growth of antigen-negative cells after antigen-positive cell clearance is very important. In addition, T cell exhaustion is reportedly a significant barrier that limits the antitumor responses of engineered T cells in the setting of chronic antigen exposure.Citation38 Refueling the exhausted CART cells via the PD-1/PD-L1 pathway using anti-PD-1 therapy is considered an important approach with potential benefits in solid tumors.Citation39,Citation40 One case report involved a PD-1 blocking antibody was administered to a patient with refractory diffuse large B-cell lymphoma after CART-19 cells infusion. Following PD-1 blockade, the patient had a clinically significant antitumor response, and a reexpansion of CART 19 cells.Citation41 However, massive T cells infiltrated the tumor tissue after CART-133 cell treatment ( and ); the status suggested that combined anti-PD-1 therapy may augment the efficacy of CART-133 cells in advanced solid tumors after CART cell infusion. In our other case report, one patient achieved a longer period of disease remission through treatment with CART-EGFR combined with CART-133 and anti-PD-1.Citation23 We are evaluating the combination treatment of PD-1 antibody checkpoint blockade and CAR-redirected T-cell therapy in our other ongoing clinical trial.
In conclusion, adoptive immunotherapy with anti-CD133 CAR-modified T cells is a feasible and possibly effective treatment. Patients can achieve longer stable survival time or even partial remission of disease after CART-133 cell therapy. HCC patients with lower tumor burden or who maintain an early stage of tumor may have a favorable clinical response even with repeated CART-133 monotherapy. Finally, patients with biliary obstruction should be cautious during CART-133 treatment. These data will facilitate subsequent clinical trials to further augment the expansion, function, and persistence of CART-133 cells in the future.
Disclosure of potential conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
supp_data.zip
Download Zip (610.4 KB)Additional information
Funding
References
- Ramos CA, Dotti G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin Biol Ther. 2011;11:855–73. doi:10.1517/14712598.2011.573476. PMID:21463133.
- Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi:10.1126/scitranslmed.3008226. PMID:24553386.
- Dai H, Zhang W, Li X, Han Q, Guo Y, Zhang Y, Wang Y, Wang C, Shi F, Zhang Y, et al. Tolerance and efficacy of autologous or donor-derived T cells expressing CD19 chimeric antigen receptors in adult B-ALL with extramedullary leukemia. Oncoimmunology. 2015;4:e1027469. doi:10.1080/2162402X.2015.1027469. PMID:26451310.
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi:10.1126/scitranslmed.3002842. PMID:21832238.
- Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–102. doi:10.1182/blood-2010-04-281931. PMID:20668228.
- Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–9. doi:10.1200/JCO.2014.56.2025. PMID:25154820.
- Wang Y, Zhang WY, Han QW, Liu Y, Dai HR, Guo YL, Bo J, Fan H, Zhang Y, Zhang YJ, et al. Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin Immunol. 2014;155:160–75. doi:10.1016/j.clim.2014.10.002. PMID:25444722.
- Guo Y, Wang Y, Han W. Chimeric antigen receptor-modified T cells for solid tumors: challenges and prospects. J Immunol Res. 2016;2016:3850839. doi:10.1155/2016/3850839. PMID:26998495.
- Yu S, Li A, Liu Q, Li T, Yuan X, Han X, Wu K. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J Hematol Oncol. 2017;10:78. doi:10.1186/s13045-017-0444-9. PMID:28356156.
- Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33:1688–96. doi:10.1200/JCO.2014.58.0225. PMID:25800760.
- Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA, DeMatteo RP, Ayala A, Joseph Espat N, Junghans RP, et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. 2015;64:817–29. doi:10.1007/s00262-015-1692-6. PMID:25850344.
- Colagrande S, Inghilesi AL, Aburas S, Taliani GG, Nardi C, Marra F. Challenges of advanced hepatocellular carcinoma. World J Gastroenterol: WJG. 2016;22:7645–59. doi:10.3748/wjg.v22.i34.7645..
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90. doi:10.1056/NEJMoa0708857. PMID:18650514.
- Hurwitz HI, Uppal N, Wagner SA, Bendell JC, Beck JT, Wade SM, 3rd, Nemunaitis JJ, Stella PJ, Pipas JM, Wainberg ZA, et al. Randomized, Double-Blind, Phase II study of ruxolitinib or placebo in combination with capecitabine in patients with metastatic pancreatic cancer for whom therapy with gemcitabine has failed. J Clin Oncol. 2015;33:4039–47. doi:10.1200/JCO.2015.61.4578. PMID:26351344.
- Liu B, Song Y, Liu D. Clinical trials of CAR-T cells in China. J Hematol Oncol. 2017;10:166. doi:10.1186/s13045-017-0535-7. PMID:29058636.
- Schmohl JU, Vallera DA. CD133, Selectively targeting the root of cancer. Toxins. 2016;8:e165. doi:10.3390/toxins8060165. PMID:27240402.
- Ferrandina G, Petrillo M, Bonanno G, Scambia G. Targeting CD133 antigen in cancer. Expert Opin Ther Targets. 2009;13:823–37. doi:10.1517/14728220903005616. PMID:19530986.
- Kohga K, Tatsumi T, Takehara T, Tsunematsu H, Shimizu S, Yamamoto M, Sasakawa A, Miyagi T, Hayashi N. Expression of CD133 confers malignant potential by regulating metalloproteinases in human hepatocellular carcinoma. J Hepatol. 2010;52:872–9. doi:10.1016/j.jhep.2009.12.030. PMID:20395004.
- Yang XR, Xu Y, Yu B, Zhou J, Qiu SJ, Shi GM, Zhang BH, Wu WZ, Shi YH, Wu B, et al. High expression levels of putative hepatic stem/progenitor cell biomarkers related to tumour angiogenesis and poor prognosis of hepatocellular carcinoma. Gut. 2010;59:953–62. doi:10.1136/gut.2008.176271. PMID:20442200.
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi:10.1038/35102167. PMID:11689955.
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi:10.1158/0008-5472.CAN-06-3126. PMID:16990346.
- Wang CM, Wu ZQ, Wang Y, Guo YL, Dai HR, Wang XH, Li X, Zhang YJ, Zhang WY, Chen MX, et al. Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory hodgkin lymphoma: An open-label phase I trial. Clin Cancer Res. 2017;23:1156–66. doi:10.1158/1078-0432.CCR-16-1365. PMID:27582488.
- Feng KC, Guo YL, Liu Y, Dai HR, Wang Y, Lv HY, Huang J-h, Yang Q-m, Han W-d. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol. 2017;10:4. doi:10.1186/s13045-016-0378-7. PMID:28057014.
- Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, Han W. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci. 2016;59:468–79. doi:10.1007/s11427-016-5023-8. PMID:26968708.
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47. doi:10.1016/j.ejca.2008.10.026. PMID:19097774.
- Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, Mackall CL. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–95. doi:10.1182/blood-2014-05-552729. PMID:24876563.
- ruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56–66. doi:10.1016/S0140-6736(16)32453-9. PMID:27932229.
- Weigmann A, Corbeil D, Hellwig A, Huttner WB. Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc Natl Acad Sci U S A. 1997;94:12425–30. doi:10.1073/pnas.94.23.12425. PMID:9356465.
- Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, et al. Convergence of Acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5:1282–95. doi:10.1158/2159-8290.CD-15-1020. PMID:26516065.
- Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, Smithers H, Jensen MC, Riddell SR, Maloney DG, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127:2406–10. doi:10.1182/blood-2015-08-665547. PMID:26907630.
- Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, Hawkins R, Chaney C, Cherian S, Chen X, et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8:355ra116. doi:10.1126/scitranslmed.aaf8621. PMID:27605551.
- Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20:119–22. doi:10.1097/PPO.0000000000000035. PMID:24667956.
- Xu XJ, Tang YM. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett. 2014;343:172–8. doi:10.1016/j.canlet.2013.10.004. PMID:24141191.
- Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi:10.1126/science.1076514. PMID:12242449.
- Kochenderfer JN, Somerville RP, Lu T, Shi V, Bot A, Rossi J, Xue A, Goff SL, Yang JC, Sherry RM, et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum Interleukin-15 levels. J Clin Oncol. 2017;35:1803–1813. JCO2016713024.
- Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105:71–7. doi:10.1172/JCI8071. PMID:10619863.
- Khakoo AY, Finkel T. Endothelial progenitor cells. Annu Rev Med. 2005;56:79–101. doi:10.1146/annurev.med.56.090203.104149. PMID:15660503.
- Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354:1165–9. doi:10.1126/science.aae0491. PMID:27789799.
- Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21:687–92. doi:10.1158/1078-0432.CCR-14-1860. PMID:25501578.
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Science Translational Med. 2016;8:328rv4. doi:10.1126/scitranslmed.aad7118..
- Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, June CH, Schuster SJ PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. 2017;129:1039–41. doi:10.1182/blood-2016-09-738245. PMID:28031179.