
ABSTRACT
Memory CD8+ T cell responses have the potential to mediate long-lasting protection against cancers. Resident memory CD8+ T (Trm) cells stably reside in non-lymphoid tissues and mediate superior innate and adaptive immunity against pathogens. Emerging evidence indicates that Trm cells develop in human solid cancers and play a key role in controlling tumor growth. However, the specific contribution of Trm cells to anti-tumor immunity is incompletely understood. Moreover, clinically applicable vaccination strategies that efficiently establish Trm cell responses remain largely unexplored and are expected to strongly protect against tumors. Here we demonstrated that a single intradermal administration of gene- or protein-based vaccines efficiently induces specific Trm cell responses against models of tumor-specific and self-antigens, which accumulated in vaccinated and distant non-vaccinated skin. Vaccination-induced Trm cells were largely resistant to in vivo intravascular staining and antibody-dependent depletion. Intradermal, but not intraperitoneal vaccination, generated memory precursors expressing skin-homing molecules in circulation and Trm cells in skin. Interestingly, vaccination-induced Trm cell responses strongly suppressed the growth of B16F10 melanoma, independently of circulating memory CD8+ T cells, and were able to infiltrate tumors. This work highlights the therapeutic potential of vaccination-induced Trm cell responses to achieve potent protection against skin malignancies.
Introduction
Immunotherapy is emerging as a new form to treat cancer by harnessing the activity of cytotoxic CD8+ T lymphocytes (CTLs) that specifically recognize tumor-associated antigens. Transfusion of autologous tumor-specific CTLsCitation1-Citation4 and blockade of T cell inhibitory receptorsCitation5-Citation7 have demonstrated to elicit durable clinical benefit in a significant proportion of patients with melanoma, leukemia, lymphoma and other cancers, who failed to respond to conventional treatments. Vaccination strategies eliciting CTL responses specific for tumor-specific and self-antigens have shown promising results in recent clinical trials.Citation8-Citation10 Long-lasting protective immunity relies on the efficient establishment of long-lived memory CD8+ T cells, which have the potential to eradicate primary and disseminated tumors.Citation11 They have been typically classified in two subsets: effector-memory (Tem) and central-memory (Tcm) CD8+ T cells.Citation12 Tcm cells express high levels of both CD62L and CCR7, which enable them to recirculate between blood and secondary lymphoid organs.Citation12 On the contrary, Tem cells lack the expression of CD62L and CCR7, but express tissue-homing receptors that enable them to recirculate through the blood and non-lymphoid tissues.Citation12 Tcm cells have been shown to mediate more potent anti-tumor immunity than Tem cells, due to their enhanced ability to proliferate upon antigen re-encounter.Citation13
In addition to circulating Tem and Tcm subsets, resident-memory CD8+ T (Trm) cells stably reside in non-lymphoid tissues, such as skin, lung, intestine, brain, female reproductive tract, and others.Citation14 Trm cells do not express CD62L or CCR7Citation15,Citation16 and constitutively up-regulate CD69 and integrin αE(CD103)β7 (commonly referred to as CD103), which are responsible for avoiding tissue egress and facilitating interaction with epithelial barriers, respectively.Citation16,Citation17 Trm cells are long-lived and represent the first line of defense against pathogen reinfection.Citation18 After antigen recognition, Trm cells undergo a rapid and potent activation, characterized by the production of high amounts of effector molecules, such as IFN-γ, TNF-α, and granzyme B, as well as proinflammatory cytokines, chemokines and antimicrobial molecules.Citation19 As a result, Trm cells trigger an innate-like alarm state that can protect against antigenically unrelated pathogens and recruit other immune cells to the site of infection, such as circulating memory T cells and B cells.Citation19,Citation20 The remarkable ability of Trm cells to mediate protective adaptive and innate immune responses has attracted the attention for developing vaccination strategies that exploit the ability of Trm cells to prevent infectious diseases.Citation21,Citation22
Although most of studies on Trm cells have been performed in the context of viral infections, studies analyzing the infiltration of CD8+ T cells in human solid tumors have shed some light on the possible role of Trm cells in anti-tumor immunity. Tumor infiltration of CD8+ T cells displaying a Trm phenotype, mainly based on CD103 expression, was shown to predict a more favorable prognosis in patients with ovarian, endometrial, breast and lung cancers.Citation23-Citation28 One of these studies showed that Trm cells infiltrating lung tumors exhibited cytotoxic activity against autologous tumor cells after blocking inhibitory receptors.Citation27 CD103 enables tissue residency and additionally, has been shown to promote anti-tumor cytolytic activity of CD8+ T cells.Citation27,Citation29,Citation30 In melanoma, metastatic lesions contain a significant proportion of tumor-infiltrating Trm cells with CD69+CD103− and CD69+CD103+ phenotypes.Citation31 Interestingly, a recent report indicated that acquisition of a distinct transcriptional program is required for T cell residency in both tumors and non-lymphoid tissues.Citation32
A causal contribution of Trm cells against melanoma has been recently depicted from mouse models. Results indicated that Trm cells generated by a model of autoimmune vitiligo inhibit the development of melanoma tumors in a CD103-dependent manner.Citation33 More recent reports evidenced a cooperative role of Trm and Tcm cells, since cutaneous infection with recombinant ovalbumin (OVA)-expressing vaccinia virus generated Trm cells and delayed growth of melanoma tumors expressing full-length OVA.Citation34 Although encouraging, this study did not rule out the potential contribution of vaccinia virus-induced OVA-specific CD4+ T cells,Citation35 which have been previously shown to acquire anti-tumor activity.Citation36 Therefore, the contribution of the vaccination-induced Trm cell compartment against melanoma has not been completely understood yet. Moreover, clinically applicable strategies that efficiently establish Trm cell responses need to be described and are expected to have a big impact in cancer immunotherapy.
Here, we demonstrated that intradermal administration of clinically relevant vaccines efficiently induces Trm cells specific for tumor-specific and self-antigens that accumulate in vaccinated and non-vaccinated skin. Interestingly, vaccination-induced Trm cells strongly suppress the growth of melanoma, independently of circulating CD8+ T cells, and were able to infiltrate melanoma tumors. Therefore, our work highlights the therapeutic potential of vaccination-induced Trm cells to achieve potent protection against skin malignancies.
Results
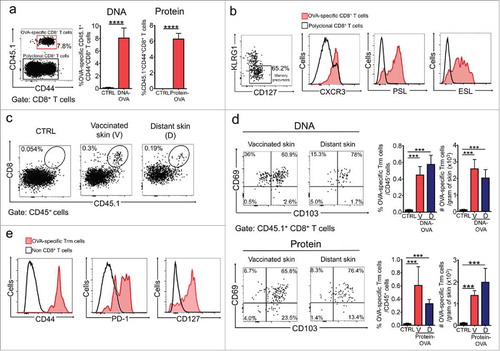
We first sought to identify clinically applicable vaccination strategies that efficiently generate Trm cell responses in skin. We tested intradermal vaccination using two types of vaccines known to induce strong CD8+ T cell responses: DNA vaccines assisted by electroporationCitation37,Citation38; and dendritic cell-targeted protein vaccines, using anti-DEC-205 antibodies.Citation39,Citation40 Initially, we vaccinated mice with a DNA vaccine encoding ovalbumin (DNA-OVA) as a model of tumor-specific non-self antigen. Prior to vaccination, mice were transferred with naïve CD8+ T cells from T cell receptor (TCR) transgenic OTI mice, which recognize the OVA(257-264) epitope. Effector and memory responses were analyzed two and four-five weeks later, respectively. We observed that DNA vaccination efficiently generated antigen-specific CD8+ T effector (Teff) cells (), which predominantly displayed a memory precursor phenotype characterized by low expression of KLRG1 and higher expression of IL-7 receptor (CD127) (, left panel). These memory precursors expressed CXCR3, as well as the skin homing molecules E- and P-selectin ligands (ESL and PSL, respectively) (), which are important for efficient establishment of Trm cells in the skin.Citation18 Of note, CD45.1+ CD8+ T cells produced IFN-γ after ex vivo OVA(257-264) peptide stimulation, while CD45.1− CD8+ T cells did not (data not shown). This indicates that only transferred OTI CD8+ T cells became expanded after vaccination, outcompeting the endogenous repertoire, as demonstrated by other authors.Citation19 At the memory phase, we detected antigen-specific Trm cells defined by the co-expression of CD69 and CD103 in vaccinated skin and, interestingly, also in distant non-vaccinated skin (-d). This could be a result of skin-wide seeding of Trm cell precursors at the effector phase of the response,Citation16,Citation32,Citation41 and subsequent dissemination through the epidermis.Citation42 Additionally, a significant proportion of CD69+CD103− OVA-specific CD8+ T cells were present in vaccinated skin (), that may correspond to inflammation-driven Trm cells, which have been described to accumulate at the site of infection.Citation43 We next tested a protein-based vaccine that specifically delivers antigen to cross-presenting dendritic cells (DCs) by fusing OVA protein to a DEC-205-specific antibody (αDEC-OVA).Citation44 Similar to the DNA vaccine, intradermal vaccination with αDEC-OVA, in combination with poly(I:C) as adjuvant (Protein-OVA), efficiently generated Teff cells (), as well as Trm cells lodged in both vaccinated and distant skin (, lower panels). In contrast to DNA vaccination, αDEC-OVA did not induce a significant accumulation of CD69+CD103− OVA-specific CD8+ T cells in the vaccinated site. As expected, vaccination-induced Trm cells displayed elevated expression of CD44, PD-1 and CD127 ().
Figure 1. DNA- and protein-based intradermal vaccination generates Trm precursors in blood and Trm cell responses in the skin. C57 BL/6 mice were intravenously transferred with OVA-specific CD45.1+ OTI CD8+ T cells and a day later, intradermally vaccinated with DNA-OVA or Protein-OVA. Control mice (CTRL) were vaccinated with empty plasmid (for DNA vaccination) or unvaccinated (for Protein vaccination). a, b Analysis of Teff responses in blood twelve days after vaccination by flow cytometry. (a) Representative dot-plot showing the expression of CD44 and CD45.1 in total CD8+ T cell population (left panel). Graphs with the percentage of CD44+ CD45.1+ OVA-specific Teff cells. (b) Representative dot-plot of KLRG1 and CD127 expression in CD45.1+ Teff cells (left panel). Representative histograms showing the expression of CXCR3, P-selectin ligand (PSL) and E-selectin ligand (ESL) in OVA-specific memory precursors (KLRG1low CD45.1+ Teff cells). c-e Analysis of memory responses in skin 4–5 weeks after vaccination by flow cytometry. (c) Representative dot-plots of total CD45+ live cells showing the presence of OVA-specific memory CD8+ T cells in vaccinated (V) and distant (D) skin. (d) Representative dot-plots and graphs showing OVA-specific Trm cells generated in vaccinated and distant skin after DNA-OVA (top) and Protein-OVA (bottom) vaccination. OVA-specific Trm cells were defined as CD3+CD8+CD45.1+CD103+CD69+ cells. (e) Representative histograms showing expression of CD44, PD-1 and CD127 analyzed in CD45.1+ OVA-specific Trm cells. (a, d) Pooled data of two independent experiments, n = 10 mice per group in a, and n = 7 mice per group in d. Bars are the mean ± SEM. ***p < 0.001; ****p < 0.0001 by Mann-Whitney unpaired t test.
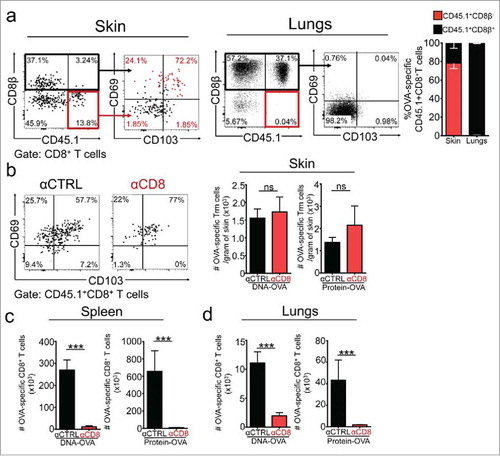
To demonstrate the residency of OVA-specific CD8+ T cells found in the skin, we carried out intravascular stainingCitation45 and showed that vaccination-induced OVA-specific CD8+ T cells were largely refractory to CD8β staining, and positive for CD69 and CD103 expression (). In contrast, antigen-specific memory CD8+ T cells found in other tissues, such as lungs, were positive for CD8β staining and lacked expression of CD69 and CD103 (), indicating that they derived from circulation. Since previous publications have reported that skin Trm cells are resistant to antibody-dependent depletion in mice and humans,Citation46,Citation47 we treated mice with an anti-CD8 (αCD8)-depleting antibody four weeks after vaccination, at the memory phase of the response, to eliminate all circulating CD8+ T cells, while sparing Trm cells. As a control, mice were treated with isotype-matched control (αCTRL) antibody. As expected, αCD8 administration efficiently depleted total CD8+ T cells in peripheral tissues such as lungs, spleen and lymph nodes (Supplementary ). As also anticipated, CD69+CD103+ OVA-specific skin Trm cells were largely resistant to antibody-dependent depletion (). In opposition, OVA-specific circulating memory CD8+ T cells were efficiently depleted from spleen and lungs (). It is worth mentioning that DNA vaccine-induced CD69+CD103− OVA-specific CD8+ T cells present in vaccinated skin resulted positive for CD8β staining and were depleted after αCD8 administration (data not shown), indicating that they are in equilibrium with the circulation. Overall, these results demonstrate that vaccination-induced skin Trm cells share distinctive characteristics broadly attributed to tissue residency.
Figure 2. Vaccination-induced skin Trm cells are largely refractory to intravascular staining and antibody-dependent depletion. C57 BL/6 mice were intravenously transferred with OVA-specific CD45.1+ OTI CD8+ T cells, the day after were intradermally vaccinated with DNA-OVA or Protein-OVA. Trm cell and circulating memory responses were analyzed 4–5 weeks later by flow cytometry. a Intravascular staining was performed by injecting mice with anti CD8β antibody intraperitoneally before the analysis. Representative dot-plots and quantification of CD8β− and CD8β+ OVA-specific CD45.1+ OTI CD8+ T cells in skin and lungs after intravascular staining. b-d Mice were treated with isotype-matched control (αCTRL) or anti-CD8 (αCD8) antibodies to eliminate circulating CD8+ T cells. Vaccination-induced memory responses were analyzed one week after antibody-dependent depletion. Representative dot-plot and graphs showing CD45.1+ OTI memory CD8+ T cells cells in skin (b), spleen (c) and lungs (d). Pooled data of two independent experiments, n = 7 mice per group. Bars are the mean ± SEM. ***p < 0.001, ****p < 0.0001, ns = non-significant by Mann-Whitney unpaired t test.
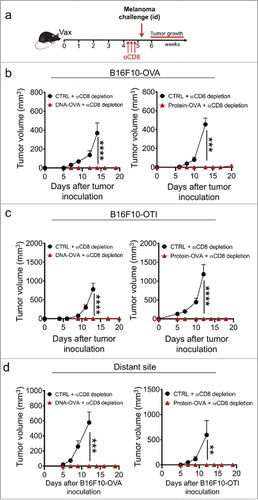
We next sought to dissect the contribution of the Trm cell compartment to anti-melanoma immunity in mice bearing vaccination-induced skin Trm cell responses, but depleted of circulating CD8+ T cells. Although the S1PR1 antagonist FTY720 has been broadly employed for restraining circulating CD8+ T cells in lymphoid organs and inhibiting their migration to peripheral tissues,Citation18,Citation34,Citation48 we observed that CD8+ T cell depletion is far more efficient than FTY720 at reducing circulating CD8+ T cell numbers in peripheral blood (Supplementary ). Moreover, evidence indicates that FTY720 may affect tumor growth by directly suppressing tumor cell viability, impairing T cell function or interfering with the ability of T cells to infiltrate tumors.Citation49-Citation54 As schematized in , vaccinated mice were treated with αCD8 antibody four weeks after vaccination and one week later, intradermally challenged with B16F10 melanoma cells expressing full-length OVA (B16F10-OVA).Citation55 DNA and protein-based vaccination completely abrogated the development of otherwise fast-growing melanoma cells injected in the flank close to the vaccinated skin of αCD8-treated mice (). To discard the contribution of other arms of the adaptive immunity that are induced with vaccines comprising full-length OVA, vaccinated mice were challenged with B16F10 cells engineered to express H-2Kb-restricted OVA(257-264) peptide (B16F10-OTI). These cells exhibited similar tumor growth kinetics compared to B16F10-OVA and parental B16F10 cells in non-treated mice (Supplementary ). In this setting, we also observed full protection in OVA-vaccinated mice (), indicating that depletion-resistant Trm cell responses are sufficient to provide strong anti-tumor immunity, independently of circulating CD8+ T cells. Anti-tumor protection was antigen-specific since mice bearing OVA-specific Trm cells were not protected against control B16F10 cells (Supplementary ). Remarkably, OVA-vaccinated mice were also fully protected against B16F10-OVA or B16F10-OTI cells injected in skin distant from the vaccination site (), suggesting that the vaccination-induced Trm cell compartment mediates strong and skin-wide protection against melanoma.
Figure 3. Vaccination-induced skin Trm cells mediate strong protection against cutaneous melanoma independently of circulating CD8+ T cells. C57 BL/6 mice were intravenously transferred with OVA-specific CD45.1+ OTI CD8+ T cells and the day later, were intradermally vaccinated with DNA-OVA or Protein-OVA. Control mice (CTRL) were vaccinated with empty plasmid (for DNA vaccination) or left unvaccinated (for protein vaccination). After 4–5 weeks, mice were depleted of circulating CD8+ T cells by administering anti-CD8 antibody and one week later, were intradermally challenged with melanoma cells. a-c Experimental scheme (a) and tumor growth curves of mice challenged with B16F10-OVA (b) and B16F10-OTI (c) close to the vaccinated site. (d) Tumor growth curves of mice challenged at distant skin sites with B16F10-OVA (left) and B16F10-OTI (right). Data is representative of two independent experiments, n = 5 and n = 4 for DNA and protein vaccines, respectively. Bars are the mean ± SEM. **p < 0.01, ***p < 0.001, ****p < 0.0001 by Two-way ANOVA and Bonferroni post-hoc test.
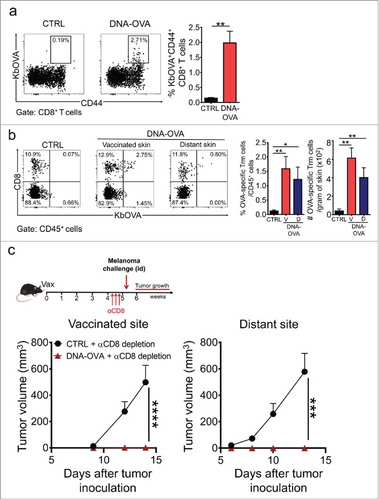
To establish the potential of intradermal vaccination to elicit protective Trm cell responses from the endogenous T cell repertoire, which is closer to a clinical setting, DNA vaccination was carried out in mice that did not receive adoptive transfer of TCR-transgenic OTI CD8+ T cells. Two weeks after vaccination, OVA-specific Teff cells were detected in blood (), although at lower magnitude as compared with the previous setting using OTI CD8+ T cell transfer. At the memory phase, comparable OVA-specific Trm cell responses were generated in vaccinated and distant skin (). More importantly, mice bearing OVA-specific Trm cells raised from the endogenous repertoire suppressed the formation of melanoma tumors in skin close to the vaccination site and also at distant sites (). These results indicate that Trm cell responses induced from the endogenous repertoire can also mediate potent skin-wide protection against melanoma.
Figure 4. Skin Trm cells generated from the endogenous T cell repertoire protect against melanoma. C57 BL/6 mice were intradermally vaccinated with DNA-OVA. Control mice were vaccinated with empty pVAX plasmid (CTRL). a. Effector responses were analyzed in blood twelve days after vaccination by H-2Kb/OVA(257–264) multimer and CD44 stainings followed by flow cytometry. Representative dot-plots and graphs showing OVA-specific Teff cells in total CD8+ T cell population. b. The generation of OVA-specific skin Trm cell responses was evaluated 4–5 weeks after vaccination by H-2Kb/OVA(257–264) (KbOVA) multimer staining followed by flow cytometry. Representative dot-plots and quantification of OVA-specific (KbOVA+) memory CD8+ T cells in total CD45+ live cells present in vaccinated (V) and distant (D) skin. OVA-specific Trm cells were defined as CD3+CD8+KbOVA+CD103+CD69+ cells. c. Mice were depleted of circulating CD8+ T cells 4–5 weeks after vaccination and one week later, were intradermally challenged with B16F10-OVA cells near the vaccinated site (bottom left) or at a distant site (bottom right). Data is representative of two independent experiments, bars are the mean ± SEM, n = 5 mice per group. *p < 0.05, **p < 0.01 for (a, b) by Mann-Whitney unpaired t test; ***p < 0.001 and ****p < 0.0001 for (c) by Two-way ANOVA and Bonferroni post hoc test.
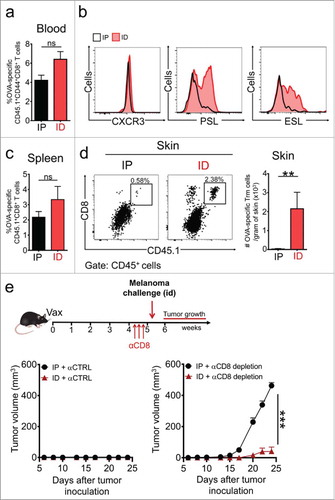
To confirm that αCD8 depletion abrogates protective circulating memory, we then delivered the αDEC-OVA vaccine intraperitoneally, attempting to avoid the generation of resident memory, while maintaining circulating memory responses. Both intradermal and intraperitoneal immunizations yielded similar levels of OVA-specific Teff cell responses (). However, intradermal, but not intraperitoneal, vaccination led to the generation of specific memory precursors displaying the skin-homing molecules PSL and ESL (). As anticipated, intradermal and intraperitoneal vaccinations generated similar levels of circulating memory CD8+ T cells (), but only intradermal vaccination efficiently generated specific Trm cells in skin (). Interestingly, both vaccination routes led to protection against intradermal challenge with B16F10-OVA in non-depleted αCTRL-treated mice (, left panel). As expected, tumor protection was severely impaired in intraperitoneally vaccinated mice devoid of circulating CD8+ T cells, and all these mice succumbed to melanoma challenge (, right panel). In contrast, intradermally vaccinated mice carrying OVA-specific Trm cell responses were completely protected. These results demonstrate that vaccination-induced skin resident and circulating memory compartments are sufficient to protect against melanoma tumors expressing a tumor-specific model antigen.
Figure 5. Route of vaccine administration determines the generation of Trm precursors and protective Trm cell responses against melanoma. C57 BL/6 mice were intravenously transferred with OVA-specific CD45.1+ OTI CD8+ T cells and a day later, were intradermally (ID) or intraperitoneally (IP) vaccinated with a Protein-OVA (αDEC-OVA plus polyI:C). a, b Analysis of Teff responses in blood by flow cytometry, twelve days after vaccination. (a) Graph showing quantification of OVA-specific CD45.1+ OTI CD8+ T cells. (b) Representative histograms showing expression of CXCR3, E-selectin ligand (ESL) and P-selectin ligand (PSL) in CD45.1+ OTI CD8+ T cells. c, d Analysis of memory responses in spleen (c) and skin (d) by flow cytometry, 4–5 weeks after vaccination. (c) Quantification of CD45.1+ OTI CD8+ T cells in spleen. (d) Representative dot-plot of CD8 and CD45.1 expression in CD45+ total live cells and quantification of OVA-specific Trm cells generated in skin after ID or IP vaccination. e Experimental scheme (top) and tumor growth curves (bottom) of mice treated with isotype-matched control (αCTRL, left) or anti-CD8 (αCD8, right) antibodies, and challenged with B16F10-OVA melanoma cells. (a, c, d) Pooled data of two independent experiments, n = 7 mice per group. (e) Data is representative of two independent experiments, n = 4 mice per group. Bars are the mean ± SEM. **p < 0.01, ns = non-significant for (a, c, d) by Mann-Whitney unpaired t test. ***p < 0.001 for (e) by Two-way ANOVA and Bonferroni post hoc test.
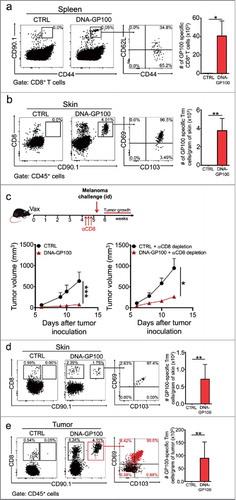
We next evaluated the anti-tumor role of vaccination-induced Trm cells in the context of a melanoma-associated self-antigen that involves CD8+ T cells bearing TCRs with lower affinity,Citation56 which have been reported to inefficiently induce Trm cell responses in a viral infection model.Citation57 Hence, mice were intradermally vaccinated with a plasmid encoding the melanocyte/melanoma antigen GP100, which is subject of peripheral tolerance.Citation58 Interestingly, specific circulating and resident responses were efficiently elicited in DNA-GP100 vaccinated mice (,b). Importantly, Trm cells suppressed the growth of B16F10 melanoma tumors in mice devoid of circulating CD8+ T cells (). Tumor growth suppression was apparently at a greater extent in non-depleted mice, although this difference was not statistically significant and all mice developed tumors (), further emphasizing the anti-tumor efficacy of Trm cells. The lack of fully protective responses in GP100-vaccinated mice may reflect the influence of mechanisms of central and peripheral tolerance, such as low TCR affinity and Treg-mediated regulation, respectively.Citation59 We then sought to analyze the infiltration of GP100-specific Trm cells into melanoma tumors. Interestingly, CD90.1+ CD8+ T cells exhibiting distinctive CD69+ CD103+ phenotype were present in skin surrounding tumors and within tumors of GP100-vaccinated αCD8-depleted mice (,e). In contrast, other CD8+ T cells infiltrating tumors did not display a Trm phenotype. Collectively, our results allow us to conclude that vaccination-induced Trm cells recognizing tumor-specific and self-antigens efficiently suppress melanoma growth and are highly desirable to achieve potent anti-tumor protection.
Figure 6. Vaccination with a melanoma-associated self-antigen generates specific Trm cell responses and suppresses tumor growth. C57 BL/6 mice were intravenously transferred with GP100-specific CD90.1+ pMEL-1 CD8+ T cells and the day afterwards, intradermally vaccinated with a DNA vaccine encoding the melanoma-associated antigen GP100 (DNA-GP100). Control mice were vaccinated with empty pcDNA plasmid (CTRL). a-b GP100-specific memory responses were analyzed 4–5 weeks after vaccination in spleen (a) and skin (b) by flow cytometry. Representative dot-plots of CD90.1+ GP100-specific memory CD8+ T cells in total CD8+ T cell (a) and CD45+ live (b) populations. Memory phenotype and total number quantifications are also shown for each case. c-e Mice were either non-depleted or depleted of circulating CD8+ T cells by injecting anti-CD8 antibody (αCD8) 4–5 weeks after vaccination. One week later, mice were intradermally challenged with B16F10 melanoma cells. (c) Experimental scheme and tumor growth curves are shown for non-depleted (bottom left) and CD8+ T cell-depleted (bottom right) mice. (d-e) CTRL and DNA-GP100 vaccinated mice depleted of circulating CD8+ T cells were sacrificed twelve-days after tumor challenge, and skin (d) and tumors (e) were analyzed by flow cytometry. Representative dot-plots of CD8 and CD90.1 expression in CD45+ live cells (left panels), and CD103 and CD69 expression in GP100-specific CD90.1+ CD8+ T cells (middle panel). In the case of tumor analysis, CD90.1+ (red dots) and CD90.1− (black dots) CD8+ T cells were overlaid. Percentages of CD90.1+ CD8+ T cells for each quadrant are indicated. Quantification of total GP100-specific Trm cells, defined as CD3+CD8+CD90.1+CD103+CD69+ T cells are shown for each case (right panels). (a, b, d, e) Pooled data of two independent experiments, bars are the mean ± SEM, n = 6 mice per group. (c) Data is representative of two independent experiments, n = 3 mice per group. Bars are the mean ± SEM. *p<0.05, **p<0.01 for (a, b, d, e) by Mann-Whitney unpaired t test. *p<0.05 and ***p<0.001 by Two-way ANOVA and Bonferroni post hoc test.
Discussion
During a long time, the existence of Trm cells was surprisingly ignored, considering that accumulating evidence supports that they provide superior protection and can be more abundant than their circulating counterpart.Citation19 Their remarkable ability to mediate protective adaptive and innate immune responses has prompted the development of vaccination strategies that harness Trm cells to fight tumors. Here, we report that clinically applicable vaccination strategies efficiently establish protective Trm cell responses, unveiling a major contribution of the tissue-resident compartment to vaccination-induced immunity against melanoma. We demonstrated that a single intradermal administration of gene- or protein-based vaccines efficiently induces specific Trm cell responses against models of tumor-specific and self-antigens that protect against highly aggressive B16F10 melanoma. Tumor-specific Trm cells sufficiently and completely prevented the growth of melanoma, independently of circulating CD8+ T cells or other arms of the adaptive immunity elicited by intradermal vaccination, such as CD4+ T cells.Citation37 To this end, we specifically dissected the contribution of vaccination-induced Trm cell responses by eliminating circulating CD8+ T cells through the administration of an αCD8 antibody at the memory phase of the response. This strategy enabled us to avoid the use of FTY720, which has been reported to interfere with the ability of T cells to infiltrate tumors, impair T cell function and even directly suppress tumor growth.Citation49-Citation54 Total anti-tumor protection was observed in B16F10 models expressing either full length OVA or the immunodominant MHC class I-restricted OVA peptide; in the latter case, discarding the potential contribution of OVA-specific CD4+ T cells and antibody responses. We also confirmed the involvement of Trm cells by delivering protein vaccines intraperitoneally, which bypasses the generation of skin-resident memory, while eliciting comparable levels of circulating memory CD8+ T cells, compared to intradermal vaccination. Consistently, both vaccination routes prevented the growth of melanoma in non-depleted mice, but only intradermal vaccination effectively suppressed tumor growth in mice depleted of circulating CD8+ T cells.
Interestingly, protective Trm cell responses were present in skin distant from the vaccinated site, which probably results from skin-wide seeding of Trm cell precursors expressing skin-homing molecules during the effector phase of the response, as has been observed for infection models.Citation16,Citation32,Citation41 Additionally, skin Trm cells exhibit a rather persistent crawling behavior that allows them to continuously patrol the skin at a speed of ∼2 mm/day.Citation42 This persistent migration is essential for Trm cells to encounter their target cells and can further contribute to their dissemination through the skin. Importantly, we show that vaccination can efficiently rise skin-wide Trm cell responses from the endogenous T cell repertoire, which is pertinent in case these results are considered to be translated into a clinical setting.
We further validated the anti-tumor role of vaccination-induced Trm cell responses against a model of melanocyte-melanoma self-antigen, GP100. To our knowledge, this is the first study to report that Trm cell responses against a self-antigen can be efficiently generated by vaccination. Interestingly, GP100 vaccination triggered the development of vitiligo, which has been shown to be necessary for the establishment of Trm cell responses.Citation33 However, in our experimental setting, Trm cell responses can be efficiently elicited in the absence of vitiligo, at least with the OVA model. If vitiligo eventually promotes Trm cell formation induced by GP100 vaccination remains to be addressed. GP100-specific Trm cell responses drastically reduced the growth of B16F10 melanoma tumors in mice devoid of circulating CD8+ T cells, but in contrast to the OVA model, all vaccinated mice developed tumors. Moreover, anti-tumor protection was not significantly improved in non-depleted mice carrying both resident and circulating memory responses against GP100, emphasizing the protective nature of Trm cells. Incomplete tumor protection observed in this setting is probably a consequence of central and peripheral tolerance mechanisms that constrain responses against self-antigens.Citation59 This is consistent with the weaker ability of pMEL-1 TCR-bearing CD8+ T cells to recognize the mouse GP100(25-33) peptide, as evidenced by reduced IFN-γ production in response to TCR stimulation.Citation60 Interestingly, this model enabled us to identify tumor-infiltration of GP100-specific Trm cells displaying a distinctive CD69+CD103+ phenotype, which was not observed in the rest of tumor-infiltrating CD8+ T cells. Considering that our setting involved the depletion of the circulating memory, these results strongly suggest that vaccination-induced skin Trm cells can get positioned within tumors to fight melanoma. Collectively, our data demonstrate that vaccination-induced Trm cells mediate strong and skin-wide protection against cutaneous melanoma.
Vaccination is gaining great interest from the cancer immunotherapy field. Despite the limited success observed in initial trials, more effective vaccination platforms and the identification of antigens that drive tumor rejection have fostered the efficacy of cancer vaccines.Citation8,Citation61 In this context, gene- and protein-based vaccination used in this study has demonstrated safety and immunogenicity in different clinical trials.Citation39,Citation62 Both vaccination platforms can be used for either personalized vaccines targeting tumor-specific antigens resulting from missense mutations,Citation63 or vaccines targeting highly prevalent tumor-associated self-antigens, which are suitable for broader use, including cancers with a low mutational load.Citation64 Emerging evidence indicates that effective anti-tumor protection requires coordinated immunity across different tissues.Citation65,Citation66 Furthermore, the ability of CD8+ T cells to acquire a characteristic gene expression signature that enables them to become established in non-lymphoid tissues, is essential to defeat tumors.Citation32 These recent findings are backed up by studies describing that tumor infiltration of CD8+ T cells displaying a Trm phenotype predicts better clinical outcomes in patients with different types of cancer.Citation23-Citation28,Citation31 Hence, we conclude that cancer vaccines should evoke tissue resident memory responses to effectively protect against tumors.
Methodology
Animals. C57 BL/6/J wild-type (CD45.2), B6.Cg-Thy1 a/Cy Tg(TcraTcrb)8Rest/J (pMEL-1), C57 BL/6-Tg(TcraTcrb)1100Mjb/J (OTI) and CBy.SJL(B6)-Ptprca/J (CD45.1) mice were purchased from Jackson Laboratories, kept at the animal facility of Fundacion Ciencia & Vida and maintained according to the “Guide to Care and Use of Experimental Animals, Canadian Council on Animal Care”. This study was carried out in accordance with the recommendations of the “Guidelines for the welfare and use of animals in cancer research, Committee of the National Cancer Research Institute”. The protocol was approved by the “Committee of Bioethics and Biosafety” from Fundacion Ciencia & Vida. Blinding or randomization strategy were done whenever it was possible, no animals were excluded from the analysis and male and female mice were used indistinctly. Mice were allocated randomly in the different experimental procedures.
Immunizations. Mice were intradermally immunized in the lower back skin with 40 µg of the following vectors: pVAX-OVA (DNA-OVA), encoding the membrane bound form of chicken OVA; pcDNA-GP100 (DNA-GP100), encoding the melanoma antigen GP100; and empty pVAX and pcDNA plasmids were used as controls. Immediately, DNA electroporation was performed by placing a parallel needle array electrode (two rows of four 2 mm pins, 1.5 × 4 mm gaps) over the injected blebs to deliver the electric pulses (two 1125 V/cm, 0.05 ms pulses followed by eight 275 V/cm, 10 ms pulses) using the Derma Vax™ DNA Vaccine Skin Delivery System (Cyto Pulse Sciences, Inc.). Plasmids were purified using the NucleoBond® PC10000 EF (Macherey-Nagel, ref 740548) or the NucleoBond XtraMidi EF (Macherey-Nagel, ref 740420.50). In the case of protein vaccination (Protein-OVA), mice were intradermally or intraperitoneally immunized with 0.5 µg of an antibody targeting the C-type lectin receptor DEC-205 fused to the full-length OVA protein (αDEC-OVA)Citation44 in combination with 25 µg of polyI:C (InvivoGen, ref 31852-Citation29-6).
Cells: Mouse melanoma cell line B16F10 (ATCC CLR-6475) was obtained from American Type Culture Collection. B16F10-OVA cells were previously generated.Citation45 B16F10-OTIx5-ZsGreen cells (B16F10-OTI) were generated by lentiviral transduction of B16F10 cell line with the pLVX-OT1 × 5-ZsGreen vector encoding the OVA(257-264) (OTI) epitope minigene fused to ZsGreen. All melanoma cell lines were cultured in complete RPMI 1640 medium (ThermoFisher Scientific, ref 61870-036), supplemented with penicillin, streptomycin (ThermoFisher Scientific, ref 15140122), non-essential amino acids (ThermoFisher Scientific, ref 11140050), sodium pyruvate (ThermoFisher Scientific, ref 11360070) and heat-inactivated fetal bovine serum (ThermoFisher Scientific, ref 10437010) in a humidified incubator at 37°C with 5% CO2. All cell lines were routinely tested for mycoplasma contamination.
Intravenous transfer of CD8+ T cells: Naïve CD45.1+CD45.2+ OTI and CD90.1+ pMEL-1 TCR-transgenic CD8+ T cells were purified from secondary lymphoid organs of transgenic mice using the EasySep™ Mouse CD8+ T Cell Enrichment Kit (StemCell Technologies, ref 19853). Mice were intravenously injected with 2 × 105-3 × 105 cells in 100 µL of sterile PBS (ThermoFisher Scienfic, ref 10010023).
Flow cytometry staining: Monoclonal antibodies specific for mouse molecules were purchased from Biolegend: CD3-FITC (clone 17A2), CD3-APC (clone 17A2), CD3-PerCp/Cy5.5 (clone 17A2), CD8-Brillant Violet 421 (clone 53–6.7), CD45-PE (clone 30-F11), CD45-PerCP(clone 30-F11), CD45.1-PE/Cy7 (clone A20), CD45.1-FITC (clone A20), CD103-APC (clone 2E7), CD103-PerCP (clone 2E7), CD69-APC/Cy7 (clone H1.2F3), CD69-APC (clone H1.2F3), CD44-PerCP (clone IM7), CD62 L-PE (clone MEL-14), CD8β-FITC (clone YTS156.7.7), KLRG1-APC/Cy7 (clone 2F1/KLRG1), KLRG1-Brillant Violet 421 (clone 2F1/KLRG1), CXCR3-PerCP/Cy5.5 (clone CXCR3–173), CD127-FITC (clone A7R34), PD-1-APC (clone 29 F.1A12), TruStain fcX (clone 93) and viability dye Zombie Aqua (ref 423101). E- and P-selectin ligands were detected by using hFc-E-selectin and hFc-P-selectin fusion proteins (R&D Systems, ref 575-ES, 737-PS), followed by staining with anti hIgG-APC antibody (clone HP6017). In the case of MHC-I multimer staining, PE-labelled H-2Kb/OVA(257-264) dextramer (Immudex, ref JD2163) was used according to the manufacturer's protocol. Samples were analyzed in a FACSCanto II cytometer (BD Bioscience) and data analyzed using FlowJo version X.0.7 (Tree Star, Inc.).
Intravascular staining: Mice were injected intravenously with 3 µg of CD8β-FITC antibody (clone YTS156.7 Biolegend) in 300 µL of sterile PBS (ThermoFisher Scienfic, ref 10010023), as previously described.Citation45 Mice were euthanized 3 min after injection and blood, skin and lungs were analyzed by flow cytometry.
CD8+ T cell depletion: Mice were intraperitoneally injected with three 20 µg doses of rat monoclonal anti-CD8 antibody (BioXCell, clone YTS169.4, ref BE0117) in three consecutive days, 4–5 weeks after vaccination. Control mice were injected with the same amounts of rat IgG2b isotype-matched control anti-KLH (BioXCell, clone LTF2, ref BE0090). Alternatively, mice were intraperitoneally injected with seven 25 µg doses of FTY720 (Sigma-Aldrich, ref SML0700) during consecutive days before CD8+ T cell analysis in blood.
Preparation of tissue cell suspensions: Spleens from vaccinated mice were mechanically disaggregated using microscope slides with ground edges (Sail Brand, ref 7105), and single-cell suspension was obtained using a 70 μm cell strainer (BD Falcon, ref 352350). For skin preparations, vaccinated and non-vaccinated skin was excised, cut in small fragments and digested in 1 mL non-supplemented RPMI 1640 medium (ThermoFisher Scientific, ref 61870–036) containing 5 mg/mL of collagenase type IV (Gibco, ref 17104019) and 5 µg/mL of DNAse I (AppliChem, ref A3778,0010) for 30 min at 37°C. Skin pieces were then disaggregated using microscope slides with ground edges (Sail Brand, ref 7105), and single-cell suspension was obtained using a 70 μm cell strainer (BD Falcon®, ref 352350) followed by a second digestion with 5 mL of supplemented RPMI 1640 medium containing 25 µg/mL of DNAse I (AppliChem, ref A3778,0010) during 5 min on ice, to obtain cell suspensions. For lung preparations, lungs were excised, cut in small fragments and digested in 1 mL of non-supplemented RPMI 1640 medium (ThermoFisher Scientific, ref 61870-036) containing 5 mg/mL of collagenase type IV (Gibco, ref 17104019) and 5 µg/mL of DNAse I (AppliChem, ref A3778,0010) for 60 min at 37°C, with shaking at 230 RPM. Blood was obtained by tail bleeding in 1.5 mL tubes containing heparin (Sanderson Laboratories), and red blood cells were lysed with RBC lysis buffer (Biolegend, ref 420301).
Tumor challenge: Three days after the last dose of depleting antibody, 50 μL of PBS containing 1 × 106 of B16F10-OVA or B16F10-OTI melanoma cells were injected intradermally in the lower back near the vaccination site or at a distant site in the upper back. Tumor growth was monitored by measuring perpendicular tumor diameters with calipers. Tumor volume was calculated using the following formula: V = (D x d2)/2 where V is the volume (mm3), D is the larger diameter (mm) and d is the smaller diameter (mm). Mice were sacrificed when moribund or when the mean tumor diameter was ≥ 15 mm, according to the approved ethical protocol.
Statistical analysis: Statistical analysis was performed using Graphpad Prism software (Graphpad Software Inc.). Unpaired t tests were performed pairwise between relevant groups. In the case of tumor growth, Two-way ANOVA was performed between relevant groups. Error bars in Figs. indicate the mean plus SEM. P value < 0.05 was considered statistically significant; *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Disclosures of potential conflicts of interest
There are no conflict of interest or financial disclosures between authors.
supp_data.zip
Download Zip (1.3 MB)Acknowledgments
We would like to thank Dr. Ana María Avalos for proofreading the manunscript.
Additional information
Funding
References
- Prickett TD, Crystal JS, Cohen CJ, Pasetto A, Parkhurst MR, Gartner JJ, Yao X, Wang R, Gros A, Li YF, et al. Durable complete response from metastatic melanoma after transfer of autologous t cells recognizing 10 mutated tumor antigens. Cancer Immunol Res. 2016;4(8):669–78. doi:10.1158/2326-6066.CIR-15-0215. PMID:27312342.
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–33. doi:10.1056/NEJMoa1103849. PMID:21830940.
- Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, Zheng Z, Vogl DT, Cohen AD, Weiss BM, et al. Chimeric antigen receptor t cells against cd19 for multiple myeloma. N Engl J Med. 2015;373(11):1040–7. doi:10.1056/NEJMoa1504542. PMID:26352815.
- Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, et al. chemotherapy-refractory diffuse large b-cell lymphoma and indolent b-cell malignancies can be effectively treated with autologous t cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–9. doi:10.1200/JCO.2014.56.2025. PMID:25154820.
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–28. doi:10.1056/NEJMoa1501824. PMID:25891174.
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44. doi:10.1056/NEJMoa1305133. PMID:23724846.
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. doi:10.1056/NEJMoa1003466. PMID:20525992.
- Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–6. doi:10.1038/nature23003. PMID:28678784.
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–21. doi:10.1038/nature22991. PMID:28678778.
- Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, Gailani F, Riley L, Conlon K, Pockaj B, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–27. doi:10.1056/NEJMoa1012863. PMID:21631324.
- Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–24. doi:10.1111/j.0105-2896.2006.00391.x. PMID:16824130.
- Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708–12. doi:10.1038/44385. PMID:10537110.
- Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102(27):9571–6. doi:10.1073/pnas.0503726102. PMID:15980149.
- Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. 2015;21(7):688–97. doi:10.1038/nm.3883. PMID:26121195.
- Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10(5):524–30. doi:10.1038/ni.1718. PMID:19305395.
- Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. 2013;14(12):1294–301. doi:10.1038/ni.2744. PMID:24162776.
- Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. 2013;14(12):1285–93. doi:10.1038/ni.2745. PMID:24162775.
- Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature. 2012;483(7388):227–31. doi:10.1038/nature10851. PMID:22388819.
- Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, Jacobs H, Haanen JB, Schumacher TN. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science. 2014;346(6205):101–5. doi:10.1126/science.1254803. PMID:25278612.
- Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol. 2013;14(5):509–13. doi:10.1038/ni.2568. PMID:23542740.
- Zens KD, Chen JK, Farber DL. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight. 2016;1(10) pii:e85832. doi:10.1172/jci.insight.85832. PMID:27468427.
- Davies B, Prier JE, Jones CM, Gebhardt T, Carbone FR, Mackay LK. Cutting edge: tissue-resident memory t cells generated by multiple immunizations or localized deposition provide enhanced immunity. J Immunol. 2017;198(6):2233–7. doi:10.4049/jimmunol.1601367. PMID:28159905.
- Webb JR, Milne K, Watson P, Deleeuw RJ, Nelson BH. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res. 2014;20(2):434–44. doi:10.1158/1078-0432.CCR-13-1877. PMID:24190978.
- Webb JR, Wick DA, Nielsen JS, Tran E, Milne K, McMurtrie E, Nelson BH. Profound elevation of CD8+ T cells expressing the intraepithelial lymphocyte marker CD103 (alphaE/beta7 Integrin) in high-grade serous ovarian cancer. Gynecol Oncol. 2010;118(3):228–36. doi:10.1016/j.ygyno.2010.05.016. PMID:20541243.
- Webb JR, Milne K, Nelson BH. PD-1 and CD103 Are Widely Coexpressed on Prognostically Favorable Intraepithelial CD8 T Cells in Human Ovarian Cancer. Cancer Immunol Res. 2015;3(8):926–35. doi:10.1158/2326-6066.CIR-14-0239. PMID:25957117.
- Wang ZQ, Milne K, Derocher H, Webb JR, Nelson BH, Watson PH. CD103 and Intratumoral Immune Response in Breast Cancer. Clin Cancer Res. 2016;22(24):6290–7. doi:10.1158/1078-0432.CCR-16-0732. PMID:27267849.
- Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpréville V, Validire P, Besse B, Mami-Chouaib F. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol. 2015;194(7):3475–86. doi:10.4049/jimmunol.1402711. PMID:25725111.
- Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, Samaniego-Castruita D, Singh D, Seumois G, Alzetani A, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol. 2017;18(8):940–50. doi:10.1038/ni.3775. PMID:28628092.
- Franciszkiewicz K, Le Floc'h A, Jalil A, Vigant F, Robert T, Vergnon I, Mackiewicz A, Benihoud K, Validire P, Chouaib S, et al. Intratumoral induction of CD103 triggers tumor-specific CTL function and CCR5-dependent T-cell retention. Cancer Res. 2009;69(15):6249–55. doi:10.1158/0008-5472.CAN-08-3571. PMID:19638592.
- Boutet M, Gauthier L, Leclerc M, Gros G, de Montpreville V, Théret N, Donnadieu E, Mami-Chouaib F. TGFbeta Signaling intersects with CD103 integrin signaling to promote t-lymphocyte accumulation and antitumor activity in the lung tumor microenvironment. Cancer Res. 2016;76(7):1757–69. doi:10.1158/0008-5472.CAN-15-1545. PMID:26921343.
- Boddupalli CS, Bar N, Kadaveru K, Krauthammer M, Pornputtapong N, Mai Z, Ariyan S, Narayan D, Kluger H, Deng Y, et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight. 2016;1(21):e88955. doi:10.1172/jci.insight.88955. PMID:28018970.
- Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature. 2017;552(7684):253–7. PMID:29211713
- Malik BT, Byrne KT, Vella JL, Zhang P, Shabaneh TB, Steinberg SM, Molodtsov AK, Bowers JS, Angeles CV, Paulos CM, et al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci Immunol. 2017;2(10) pii:eaam6346. doi:10.1126/sciimmunol.aam6346. PMID:28738020.
- Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martínez-Cano S, Mejías-Pérez E, Esteban M, Melero I, Hidalgo A, et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat Commun. 2017;8:16073. doi:10.1038/ncomms16073. PMID:28714465.
- Iborra S, Izquierdo HM, Martínez-López M, Blanco-Menéndez N, Reis e Sousa C, Sancho D. The DC receptor DNGR-1 mediates cross-priming of CTLs during vaccinia virus infection in mice. J Clin Invest. 2012;122(5):1628–43. doi:10.1172/JCI60660. PMID:22505455.
- Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PAet al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207(3):637–50. doi:10.1084/jem.20091918. PMID:20156971.
- Lladser A, Mougiakakos D, Tufvesson H, Ligtenberg MA, Quest AF, Kiessling R, Ljungberg K. DAI (DLM-1/ZBP1) as a genetic adjuvant for DNA vaccines that promotes effective antitumor CTL immunity. Mol Ther. 2011;19(3):594–601. doi:10.1038/mt.2010.268. PMID:21157438.
- Ligtenberg MA, Witt K, Galvez-Cancino F, Sette A, Lundqvist A, Lladser A, Kiessling R. Cripto-1 vaccination elicits protective immunity against metastatic melanoma. Oncoimmunology. 2016;5(5):e1128613. doi:10.1080/2162402X.2015.1128613. PMID:27467944.
- Dhodapkar MV, Sznol M, Zhao B, Wang D, Carvajal RD, Keohan ML, Chuang E, Sanborn RE, Lutzky J, Powderly J, et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci Transl Med. 2014;6(232):232ra51. doi:10.1126/scitranslmed.3008068. PMID:24739759.
- Idoyaga J, Lubkin A, Fiorese C, Lahoud MH, Caminschi I, Huang Y, Rodriguez A, Clausen BE, Park CG, Trumpfheller C, et al. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9 A. Proc Natl Acad Sci U S A. 2011;108(6):2384–9. doi:10.1073/pnas.1019547108. PMID:21262813.
- Kadoki M, Patil A, Thaiss CC, Brooks DJ, Pandey S, Deep D, Alvarez D, von Andrian UH, Wagers AJ, Nakai K, et al. Organism-Level Analysis of Vaccination Reveals Networks of Protection across Tissues. Cell. 2017;171(2):398–413 e21. doi:10.1016/j.cell.2017.08.024. PMID:28942919.
- Ariotti S, Beltman JB, Chodaczek G, Hoekstra ME, van Beek AE, Gomez-Eerland R, Ritsma L, van Rheenen J, Marée AF, Zal T, et al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc Natl Acad Sci U S A. 2012;109(48):19739–44. doi:10.1073/pnas.1208927109. PMID:23150545.
- Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat Immunol. 2015;16(4):406–14. doi:10.1038/ni.3108. PMID:25706747.
- Idoyaga J, Cheong C, Suda K, Suda N, Kim JY, Lee H, Park CG, Steinman RM. Cutting edge: langerin/CD207 receptor on dendritic cells mediates efficient antigen presentation on MHC I and II products in vivo. J Immunol. 2008;180(6):3647–50. doi:10.4049/jimmunol.180.6.3647. PMID:18322168.
- Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc. 2014;9(1):209–22. doi:10.1038/nprot.2014.005. PMID:24385150.
- Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, Carbone FR, Mueller SN. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature. 2011;477(7363):216–9. doi:10.1038/nature10339. PMID:21841802.
- Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, Dorosario AA, Chaney KS, Cutler CS, Leboeuf NR, et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med. 2012;4(117):117ra7. doi:10.1126/scitranslmed.3003008. PMID:22261031.
- Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–60. doi:10.1038/nature02284. PMID:14737169.
- Li MH, Hla T, Ferrer F. FTY720 inhibits tumor growth and enhances the tumor-suppressive effect of topotecan in neuroblastoma by interfering with the sphingolipid signaling pathway. Pediatr Blood Cancer. 2013;60(9):1418–23. doi:10.1002/pbc.24564. PMID:23704073.
- Tsai HC, Huang Y, Garris CS, Moreno MA, Griffin CW, Han MH. Effects of sphingosine-1-phosphate receptor 1 phosphorylation in response to FTY720 during neuroinflammation. JCI Insight. 2016;1(9):e86462. doi:10.1172/jci.insight.86462. PMID:27699272.
- Ntranos A, Hall O, Robinson DP, Grishkan IV, Schott JT, Tosi DM, Klein SL, Calabresi PA, Gocke AR. FTY720 impairs CD8 T-cell function independently of the sphingosine-1-phosphate pathway. J Neuroimmunol. 2014;270(1-2):13–21. doi:10.1016/j.jneuroim.2014.03.007. PMID:24680062.
- Azuma H, Takahara S, Ichimaru N, Wang JD, Itoh Y, Otsuki Y, Morimoto J, Fukui R, Hoshiga M, Ishihara T, et al. Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Cancer Res. 2002;62(5):1410–9. PMID:11888913
- Ng KT, Man K, Ho JW, Sun CK, Lee TK, Zhao Y, Lo CM, Poon RT, Fan ST. Marked suppression of tumor growth by FTY720 in a rat liver tumor model: the significance of down-regulation of cell survival Akt pathway. Int J Oncol. 2007;30(2):375–80. PMID:17203219.
- Thompson ED, Enriquez HL, Fu YX, Engelhard VH. Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J Exp Med. 2010;207(8):1791–804. doi:10.1084/jem.20092454. PMID:20660615.
- Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105(27):9331–6. doi:10.1073/pnas.0710441105. PMID:18599457.
- Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz de Miera E, Darvishian F, McGary K, Huang K, Boyer J, Corse E, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci U S A. 2013;110(17):6973–8. doi:10.1073/pnas.1221609110. PMID:23576742.
- Frost EL, Kersh AE, Evavold BD, Lukacher AE. Cutting Edge: Resident Memory CD8 T Cells Express High-Affinity TCRs. J Immunol. 2015;195(8):3520–4. doi:10.4049/jimmunol.1501521. PMID:26371252.
- Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198(4):569–80. doi:10.1084/jem.20030590. PMID:12925674.
- Stone JD, Harris DT, Kranz DM. TCR affinity for p/MHC formed by tumor antigens that are self-proteins: impact on efficacy and toxicity. Curr Opin Immunol. 2015;33:16–22. doi:10.1016/j.coi.2015.01.003. PMID:25618219.
- Badou A, Savignac M, Moreau M, Leclerc C, Foucras G, Cassar G, Paulet P, Lagrange D, Druet P, Guéry JC, et al. Weak TCR stimulation induces a calcium signal that triggers IL-4 synthesis, stronger TCR stimulation induces MAP kinases that control IFN-gamma production. Eur J Immunol. 2001;31(8):2487–96. doi:10.1002/1521-4141(200108)31:8%3c2487::AID-IMMU2487%3e3.0.CO;2-L. PMID:11500833.
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi:10.1126/science.aaa4971. PMID:25838375.
- Kim TJ, Jin HT, Hur SY, Yang HG, Seo YB, Hong SR, Lee CW, Kim S, Woo JW, Park KS, et al. Clearance of persistent HPV infection and cervical lesion by therapeutic DNA vaccine in CIN3 patients. Nat Commun. 2014;5:5317. doi:10.1038/ncomms6317. PMID:25354725.
- Walters JN, Ferraro B, Duperret EK, Kraynyak KA, Chu J, Saint-Fleur A, Yan J, Levitsky H, Khan AS, Sardesai NY, et al. A Novel DNA Vaccine Platform Enhances Neo-antigen-like T Cell Responses against WT1 to Break Tolerance and Induce Anti-tumor Immunity. Mol Ther. 2017;25(4):976–88. doi:10.1016/j.ymthe.2017.01.022. PMID:28237837.
- Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res. 2013;119:421–75. doi:10.1016/B978-0-12-407190-2.00007-1. PMID:23870514.
- Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell. 2017;168(3):487–502 e15. doi:10.1016/j.cell.2016.12.022. PMID:28111070.
- Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martínez-Cano S, Mejías-Pérez E, Esteban M, Melero I, Hidalgo A, et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat Commun. 2017;8:16073. doi:10.1038/ncomms16073. PMID:28714465.