
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Despite melanoma immunogenicity and remarkable therapeutic effects of negative immune checkpoint inhibitors, a significant fraction of patients does not respond to current treatments. This could be due to limitations in tumor immunogenicity and profound immunosuppression in the melanoma microenvironment. Moreover, insufficient tumor antigen processing and presentation by dendritic cells (DC) may hamper the development of tumor-specific T cells. Using two genetically engineered mouse melanoma models (RET and BRAFV600E transgenic mice), in which checkpoint inhibitor therapy alone is not efficacious, we performed proof-of-concept studies with an improved, multivalent DC vaccination strategy based on our recently developed genetic mRNA cancer vaccines. The in vivo expression of multiple chimeric MHC class I receptors allows a simultaneous presentation of several melanoma-associated shared antigens tyrosinase related protein (TRP)-1, tyrosinase, human glycoprotein 100 and TRP-2. The DC vaccine induced a significantly improved survival in both transgenic mouse models. Vaccinated melanoma-bearing mice displayed an increased CD8 T cell reactivity indicated by a higher IFN-γ production and an upregulation of activation marker expression along with an attenuated immunosuppressive pattern of myeloid-derived suppressor cells (MDSC) and regulatory T cells (Treg). The combination of DC vaccination with ultra-low doses of paclitaxel or anti-PD-1 antibodies resulted in further prolongation of mouse survival associated with a stronger reduction of MDSC and Treg immunosuppressive phenotype. Our data suggest that an improved multivalent DC vaccine based on shared tumor antigens induces potent anti-tumor effects and could be combined with checkpoint inhibitors or targeting immunosuppressive cells to further improve their therapeutic efficiency.
Introduction
During the past decade, enormous improvements have been achieved in the treatment of metastatic melanoma. In particular, the immune checkpoint inhibitors, such as antibodies against cytotoxic T-lymphocyte-associated protein (CTLA)-4 and programmed cell death protein (PD)-1, have significantly improved the treatment efficacy in a subset of melanoma patients harboring wild type BRAF.Citation1,Citation2 In addition, BRAF and MEK-kinase inhibitors achieved profound clinical responses in melanoma patients with BRAFV600 mutations.Citation3
Even with these advances, only a fraction of melanoma patients responds durably to immunotherapy.Citation4 The therapy resistance was reported to be due to chronic inflammation and immunosuppression, tumor heterogeneity, as well as to lower numbers of somatic mutations encoding neo-antigens.Citation5-Citation8 Therefore, the attention of tumor immunologists has been shifted from shared tumor-associated antigens, to mutanome-encoded, patient specific neo-antigens.Citation7 Nevertheless, tumor-associated antigens should not be forgotten since limitations in the neo-antigen expression could be overcome by boosting immune responses via targeting tumor-associated shared antigens.
We attempted to improve the presentation of shared melanoma-associated antigens (MAA) by dendritic cell (DC)-based immunotherapy since the clinical impact of such immunotherapy has been limited so far.Citation9 Efficient major histocompatibility complex (MHC)-peptide expression on DC and their activation determines the degree and quality of the T cell response. DC-based immunotherapies require improvements regarding (i) the origin and polarization of DC, (ii) the maturation stimuli by using better adjuvants, and (iii) the type and form of antigens to be loaded on DC.Citation6 To overcome these limitations, we have developed earlier a novel genetic platform for the induction of CD8 T cell responses specific for MAA, human glycoprotein (hgp)100 and tyrosinase related protein (TRP)-2 by DC vaccination.Citation10 We showed that an efficient peptide presentation through human beta 2 microglobulin (hβ2 m) can be coupled with constitutive toll-like receptor 4 (TLR4) signaling through the polypeptide product of a single gene by mRNA electroporation into bone marrow-derived DC. This modality was highly efficient in breaking immune tolerance by stimulating the activation of DC and antigen-specific CD8 T cell responses, which inhibited tumor growth and improved the overall survival in melanoma-bearing mice.Citation10,Citation11
In this study, we broadened the repertoire of the hβ2 m-platform for CD8 T cell induction by including two additional MAA, TRP-1 and tyrosinase (TYR). Moreover, we utilized this chimeric mRNA construct system to examine whether multivalent DC immunization is more effective to inhibit melanoma progression than current vaccination approaches with long peptides or peptide-pulsed DC. Importantly, we test our mRNA-based DC vaccine in two different genetically engineered mouse models (GEMM) that develop de novo tumors in a natural immune‐proficient microenvironment.Citation12 Advanced tumors in RET-transgenic (RET-tg) and BRAFV600E mutated (BRAF) melanoma models closely mimic the histopathology and clinical development of their human counterparts as they are able to progress spontaneously toward metastatic disease.Citation13,Citation14 Since they are unresponsive to sole checkpoint inhibition they represent good preclinical model systems to test our vaccination strategy based on the presentation of shared MAA by DC.Citation13-Citation15 Finally, to target the immunosuppressive melanoma microenvironment we applied either anti-PD-1 antibodies or paclitaxel (Pac) at ultra-low doses, which was reported previously by us to reduce the production of chronic inflammatory mediators and to inhibit myeloid-derived suppressor cells (MDSC) in the RET-tg model.Citation16
We found that the immunization with our improved DC vaccine induced multi-antigen-specific effector T cells and long-lasting memory T cells, leading to a significant prolongation of the survival of tumor-bearing RET-tg and BRAF mice. Moreover, both combined therapies with ultra-low dose paclitaxel or checkpoint inhibitor further improved the survival, induced stronger CD8 T cell activation and significantly attenuated an immunosuppressive pattern of MDSC and regulatory T cells (Treg). Our data suggest that mRNA-based DC vaccination with shared MAA showed a strong therapeutic effect in two melanoma GEMM and could be combined with other immunotherapeutic approaches to improve the efficacy of human melanoma treatment as an alternative to individualized neo–antigen vaccination.
Results
Chimeric β2-microglobulin molecule assembly
We have previously generated chimeric receptor constructs with MAA specific to human gp10025–33 and murine TRP-2180–188 (both H-2Db binder) and described their anti-tumor activity in melanoma-bearing mice.Citation10,Citation11 To broaden the clinical potential of the constructs we included additional MAA such as TRP-1455–463 (H-2Db binder) that was reported to confer anti-tumor immune responsesCitation17 and TYR360–368, which was predicted by SYFPEITHI prediction software as an H-2Db binder.Citation18 Both peptides were assembled into the chimeric hβ2 m-platform with the TLR4 and Kb anchors (Supplementary Fig. 1 A, B) as previously described.Citation19 The designation of new constructs is summarized in Supplementary Fig. S1 C.
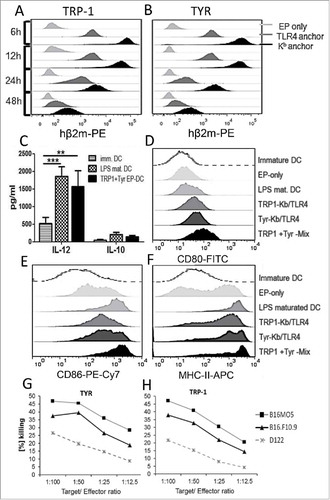
Figure 1. Chimeric MHC class I constructs induce DC maturation and CTL responses in vitro. Bone marrow-derived DC were electroporated with hβ2 m constructs: TRP-1-Kb and TRP-1-TRL4 (A) or with TYR-Kb and TYR-TLR4 (B) encoding mRNA respectively. Expression kinetics of chimeric-hβ2m constructs on DC was assessed by flow cytometry. Empty electroporated DC (EP only) served as a control. (C) Secretion of IL-12 and IL-10 by immature, LPS-maturated, and mRNA-electroporated DC (TRP-1+TYR EP-DC) was measured by ELISA and expressed as pg/mL (n = 3). (D-F) Expression of DC maturation markers (CD80, CD86, MHC-class II) was measured by flow cytometry 6 h after electroporation. Representative histograms are shown. G and H, CTL were induced by immunization of C57 BL/6 mice with DC electroporated with respective TRP1-Kb/TRL4 or TYR-Kb/TLR4 encoding mRNA (n = 3) and assessed by CTL in vitro killing assay. Results are presented as the percentage of killed syngeneic B16F10.9 and B16MO5 melanoma target cells following co-incubation with CTL induced by TYR-Kb/TLR4 (G) and TRP-1-Kb/TLR4 constructs (H). D122 Lewis lung carcinoma mouse cells that do not express TRP1 and TYR were used as a negative control. Data are representative of two independent experiments.
DC present the MHC-I constructs on the cell surface and induce cytotoxic T cells
The kinetics of MHC-I construct expression on the cell surface of bone marrow-derived DC was monitored by flow cytometry with anti-hβ2m antibodies. All constructs were found to be expressed on the DC surface for at least 48 h, although TRP-1-Kb and TYR-Kb constructs were expressed at higher levels than TRP-1-TLR4 and TYR-TLR4 ones (, ), which is consistent with previous observations.Citation10 We electroporated DC with mRNA-constructs containing Kb and TLR4 anchors for the same antigen at 1:1 ratio to ensure prolonged presentation via the Kb construct and DC maturation due to the constitutive intracellular TLR4 signaling. Upon electroporation, we observed a profound stimulation of IL-12 production by DC (similar to that induced by LPS) and no significant induction of IL-10 (). Moreover, electroporated DC displayed an upregulation of CD80, CD86 and MHC class II expression comparable to the stimulation with LPS (). This marker profile and strong IL-12 secretion supports the DC maturation, which is in line with our previous data.Citation10 In addition, the immunization with mRNA electroporated bone marrow-derived DC led to the induction of antigen-specific cytotoxic T lymphocytes (CTL), which specifically killed melanoma target cells in vitro (, ). Thus, the new TRP-1/TYR-hβ2m-TLR4/Kb chimeric constructs electroporated into DC initiated a maturated phenotype and induced antigen-specific CTL.
mRNA-electroporated DC induce stronger CTL responses than peptide-loaded DC
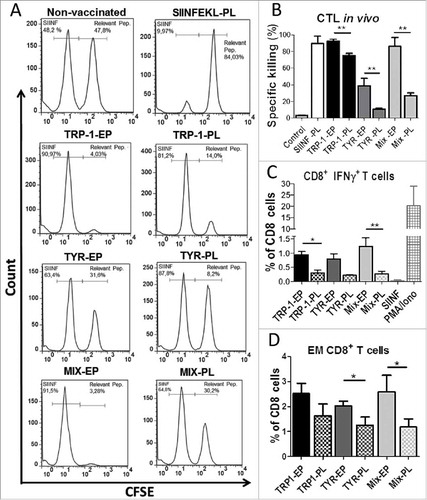
To evaluate if TRP-1/TYR-hβ2m-TLR4/Kb chimeric constructs are superior to conventional, peptide-loaded DC in inducing specific CTL killing in vivo, we transfected DC with mRNA of TRP-1-hβ2m-TLR4/Kb (TRP-1-EP), TYR-hβ2m-TLR4/Kb (TYR-EP) or with the mixture of them (Mix-EP; TRP-1/TYR- hβ2m-TLR4/Kb) at 1:1 ratio. In parallel, LPS matured DCs were loaded with TRP-1455–463 (TRP-1-PL), TYR360–368 (TYR-PL) peptide or peptide mixture (Mix-PL). Electroporated unloaded DC (control) and OVA257–264 peptide-loaded mature DC (SIINF-PL) served as controls. The induction of CTL responses was studied upon the vaccination of mice with respective DC preparations using CTL in vivo killing assay. As seen in , , all three groups of mRNA-electroporated DC (EP) were able to elicit significantly stronger CTL responses than cells loaded with the respective peptides (PL). Moreover, mice vaccinated with mRNA-electroporated DC displayed significantly higher frequency of IFN-γ secreting and effector memory CD8 T cells in spleens upon in vitro restimulation than mice receiving peptide-loaded DC vaccines (, ).
Figure 2. Comparative analysis of in vivo CTL responses induced by mRNA-electroporated and peptide-loaded DC. C57 BL/6 mice were vaccinated intraperitoneally (i.p.) three times with DC electroporated (EP) with TRP1-β2 m-Kb/TLR4, TYR-β2 m-Kb/TLR4, or their combination (Mix-EP) as well as with peptide-loaded (PL) DC with TRP-1455–463, TYR360–368 or their combination (Mix-PL) (n = 3). Mice vaccinated with SIINFEKL-loaded DC were used as a control group. (A) CTL in vivo killing assay was performed ten days after the last vaccination. Representative flow cytometry histograms showing CFSE-labeled, peptide-loaded target cells, which were injected intravenously into immunized mice. (B) CTL activity is presented as the percentage of specific killing of peptide-loaded target cells. (C) Intracellular IFN-γ production in CD8 T cells was measured by flow cytometry following in vitro restimulation of splenocytes with relevant peptides. SIINFEKL peptide served as a control. Results are shown as the percentage of IFN-γ producing CD8 T cells among total CD8 T cells. (D) Frequency of CD8+CD62 L−CD44+ effector memory (EM) T cells in spleens is shown as the percentage within total CD8 T cells. Data are representative of two independent experiments. *P < 0.05, **P < 0.01.
mRNA-transfected DC are superior to peptide-loaded DC in conferring anti-tumor effects
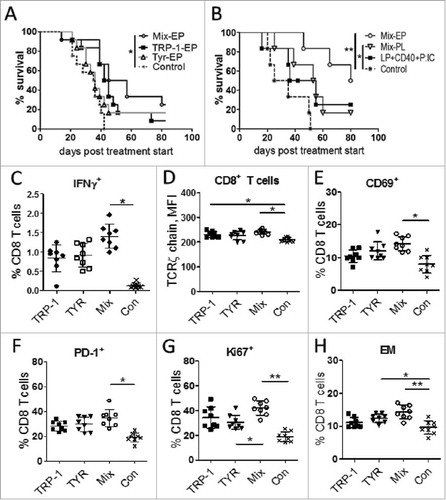
We next investigated the impact of the TRP-1/TYR DC vaccine on melanoma progression in RET-tg mice, which closely resembles human melanoma regarding histopathology and clinical development.Citation14 As seen in , the immunization with DC electroporated by the mixture of TRP-1/TYR-hβ2m-TLR4/Kb mRNA (Mix-EP) resulted in a significantly improved survival as compared to mice treated with empty DC (control group). However, the survival of mice treated with DC containing a single antigen showed only a trend for the survival prolongation, which was not significant (). Since several clinical studies described promising effects with the long-peptide vaccination combined with adjuvants,Citation20,Citation21 we applied the vaccination with peptide loaded DC (Mix-PL) as well as with long peptide mix (LP, TRP-1448–472, TYR353–377) combined with anti-CD40 mAb and poly:IC for effective T helper cell-mediated costimulationCitation22 similar to our hβ2m-TLR4 constructs. Survival monitoring displayed a non-significant tendency for improved survival for the long peptide mix (LP+CD40+P:IC) vaccinated group (). In contrast, vaccination with the mRNA-electroporated DC vaccine (Mix-EP, P < 0.01) showed a more profound and significantly higher survival prolongation than peptide-loaded DC (Mix-PL, P < 0.05; ). Consequently, the mRNA-electroporated DC vaccine (Mix-EP) showed an increased therapeutic efficiency as compared to all other vaccinated groups.
Figure 3. Vaccination with mRNA-electroporated DC induces anti-tumor responses in melanoma-bearing RET transgenic mice. Mice with palpable tumors were vaccinated i.p using mRNA-electroporated or peptide loaded DC. DC electroporation was performed by mRNA encoding TRP-1-hβ2 m-Kb/TLR4 (TRP-1-EP), TYR- hβ2 m-Kb/TLR4 (TYR-EP), or their combination (Mix-EP). LPS-maturated DCs were loaded with short peptide mix (TRP-1455–463, TYR360–368, combination; Mix-PL). Another group was vaccinated subcutaneously with a combination of long peptide mix (LP, TRP-1448–472 + TYR353–377), anti-CD40 mAb and poly:IC. Mice vaccinated with empty DC were used as a control (Con). (A, B) Survival of mice is shown as a Kaplan-Meier curve (n = 6–12/group). (C-H) Ten days after last vaccination with mRNA-electroporated DC, tumor infiltrating CD8 T cells were analyzed by flow cytometry (n = 8). Results are presented as the percentage of IFN-γ producing CD8 T cells among total CD8 T cells (C), mean fluorescence intensity (MFI) of the TCR ζ-chain in CD8 T cells (D), the percentage of CD69 (E), PD-1 (F), Ki-67 (G), and CD62 L−CD44+ EM (H) CD8 T cells among total CD8 T cells. Data are representative of two independent experiments. *P < 0.05, **P < 0.01.
Improved DC vaccine stimulates T cell-mediated immune responses
Analyzing the activation status of tumor-infiltrating T lymphocytes upon mRNA-electroporated DC vaccination, we demonstrated an increased frequency of IFN-γ producing CD8 T cells in the group immunized with TRP-1/TYR–Mix group (Mix-EP) as compared to the control group (). Furthermore, the expression of T cell activation markers such as TCR ζ-chain (), CD69 (), PD-1 () and the proliferation marker Ki67 () was significantly higher in the Mix-EP group. Moreover, these mice displayed an elevated frequency of CD44+CD62 L− effector memory (EM) CD8 T cells compared to the control group ().
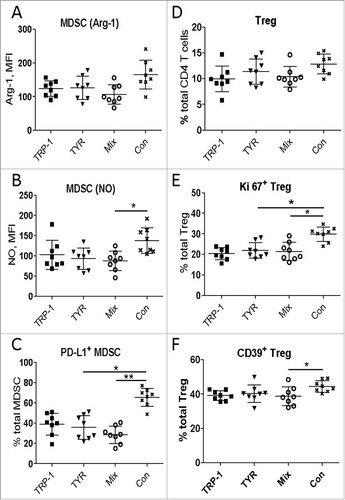
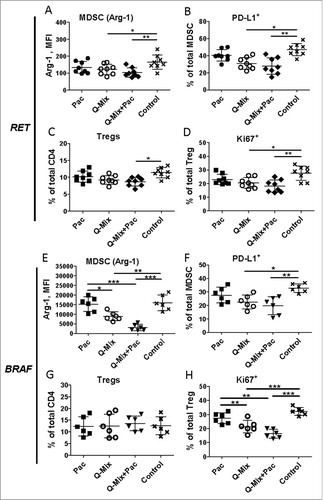
Next, we characterized immunosuppressive cells MDSC () and Treg () infiltrating skin tumors of treated mice. Although we observed no statistically significant differences in the MDSC frequency (data not shown), the level of nitric oxide (NO) production measured by the mean fluorescence intensity (MFI) showed a clear reduction in MDSC from the Mix-EP vaccinated group (). In addition, the frequency of PD-L1+ MDSC was significantly decreased (). Similar to MDSC, the frequency of Treg was not changed in the Mix-EP vaccinated group (). However, we demonstrated a reduced expression of Ki67 and CD39 on these cells (, ). This suggests that mRNA-based DC vaccination could stimulate powerful CD8 T cell responses without concomitant activation of immunosuppressive cell populations. We even detected a decrease in the immunosuppressive pattern of tumor-infiltrating MDSC and Treg.
Figure 4. Vaccination with mRNA-electroporated DC impedes immunosuppressive pattern of MDSC and Treg in melanoma-bearing RET transgenic mice. Mice with palpable tumors were vaccinated i.p. using DC electroporated with TRP1- hβ2 m Kb/TLR4 (TRP-1), TYR- hβ2 m Kb/TLR4 (TYR) encoding mRNA, or the combination (Mix). Mice vaccinated with empty DC were used as a control (Con). Tumor-infiltrating MDSC and Treg were analyzed by flow cytometry ten days after last vaccination (n = 8). Arginase (Arg)-1 expression (A) and nitric oxide (NO) production (B) by CD11b+Gr1+ MDSC is presented as MFI. C, PD-L1 expression is shown as the percentage of positive cells among total MDSC. (D) CD4+CD25+Foxp3+ Treg are presented as the percentage within total CD4 T cells. Ki67 (E) and CD39 (F) expression on Treg is shown as the percentage of respective subset within total Treg. Data are representative of two independent experiments. *P < 0.05, **P < 0.01.
Combination of multivalent DC vaccination with ultra-low dose Pac stimulates anti-tumor responses in melanoma-bearing mice
Our previous studies showed that treatment of melanoma-bearing RET-tg mice with Pac at non-cytotoxic, ultra-low doses reduced chronic inflammatory factors and immunosuppressive MDSC functions.Citation16 In addition, it was suggested that the vaccination with multiple antigens leads to improved clinical outcomes.Citation20,Citation21 Therefore, we electroporated DC with a quadruple-antigen mRNA Mix (Q-Mix), containing TRP-1/TYR-hβ2m-TLR4/Kb constructs and previously described hgp100/TRP-2-hβ2m-TLR4/Kb constructsCitation10,Citation11 and treated melanoma-bearing mice from RET-tg and BRAF models in combination with ultra-low dose Pac (Supplementary Fig. 1 C, ). Multivalent DC vaccination (Q-Mix) significantly prolonged mouse survival in both models (, ). As expected, therapeutic effects of vaccination were more profound when combined with ultra-low dose Pac (Q-Mix+Pac).
Figure 5. Combination of DC vaccination with ultra-low dose paclitaxel stimulates anti-tumor responses in melanoma-bearing mice. Mice from RET-tg (RET) or BRAF melanoma models with palpable tumors were vaccinated i.p. three times using DC electroporated with combination of hgp100 + TRP-2 + TRP-1 + TYR-hβ2 m-Kb/TLR4 (Q-Mix) mRNA. Some vaccinated mice additionally received i.p. ultra-low dose Pac (1 mg/kg) three times in weekly intervals. Other mice were treated with Pac only or with empty DC (Con). Survival of RET (A) or BRAF (B) mice is shown as a Kaplan-Meier curve (n = 9–12/group). Tumor-infiltrating T cells from RET (C-E, n = 8) or BRAF (F-H, n = 6) mice were analyzed by flow cytometry 10 days after last vaccination. Results are presented as the percentage of IFN-γ producing CD8+ T cells among total CD8 (C, F), MFI of the TCR ζ-chain in CD8 T cells (D, G) and the percentage of CD62 L−CD44+ EM (B, H) CD8 T cells among total CD8 T cells. Data are representative of two independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
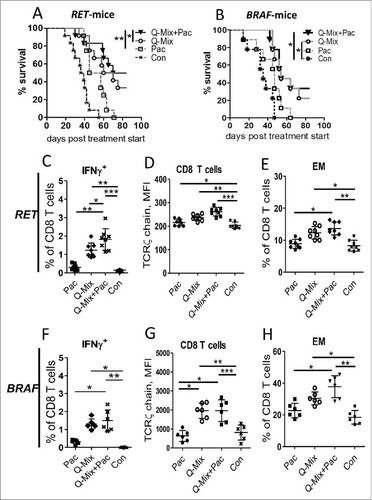
Analyzing immune cells in melanoma lesions, we demonstrated a significant increase in the frequency of IFN-γ+ CD8 T cells upon Q-Mix DC vaccination alone or in combination with Pac ( and ). In addition, an elevated frequency of antigen-specific tumor-infiltrating CD8 T cells in treated RET-tg mice was detected by tetramer staining (Supplementary Fig. 2 A, B). Furthermore, Q-Mix DC vaccination alone or combined with Pac significantly increased the frequency of tumor-infiltrating CD8 T cells (Supplementary Fig. 3 A, G) as well as the expression of TCR ζ-chain (, ), CD69 (Supplementary Fig. 3B, H) and PD-1 (Supplementary Fig. 3 C, I) on CD8 T cells. Interestingly, the frequency of EM (, ) in both models and central memory (CM) T cells (Supplementary Fig. 3 J) in the BRAF model was predominantly increased upon Q-Mix+Pac treatment. No profound effects on the frequency of tumor-infiltrating MDSC in both models were observed (data not shown). However, we detected a remarkable decrease in the expression of Arg-1 (, ), PD-L1 (, ) and NO (Supplementary Fig. 3E, K) in these cells. For tumor infiltrating Treg, we observed their decreased frequency in the RET-tg model (, ) as well as a reduced expression of Ki67 (, ) and CD39 (Supplementary Fig. 3 F, L). Interestingly, similar changes for effector T cells, MDSC and Treg were detected in spleens from RET-tg (Supplementary Fig. 4) and BRAF mice (Supplementary Fig. 5) and metastatic lymph nodes from these mice (data not shown), indicating a systemic effect of this combined treatment. Importantly, in tumor lysates of treated RET-tg mice, we detected also a significantly reduced concentration of cytokines and growth factors IL-10, TNF-α, IL-6 and VEGF (Supplementary Fig. 6) that are known to stimulate MDSC and Treg functions and expansion.Citation23
Figure 6. Combined immunotherapy with vaccine and ultra-low dose paclitaxel downregulates MDSC and Treg activity markers. Tumor-infiltrating MDSC and Treg from RET (A-D, n = 8) and BRAF mice (E-H, n = 8) were analyzed by flow cytometry 10 days upon last vaccination. Arg-1 expression (A, E) by MDSC is presented as MFI. (B, F) PD-L1 expression is shown as the percentage of positive cells within total MDSC. (C, G) CD4+CD25+Foxp3+ Treg are presented as the percentage within total CD4 T cells. (D, H) Ki67 expression on Treg is shown as the percentage of respective subset among total Treg. Data are representative of two independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Analysis of memory T cells in tumor and spleen samples from surviving BRAF and RET-tg mice at day 82 post treatment start, showed an accumulation of EM and CM T cells upon the therapy with Q-Mix vaccine and Pac (Supplementary Fig. 7), indicating a systemic, long-lasting memory T cell induction upon combinatorial treatment.
Figure 7. Enhanced anti-tumor effect of DC vaccine and anti-PD-1 antibodies in melanoma-bearing mice. RET-tg mice with established tumors were vaccinated i.p. three times in weekly intervals using DC electroporated with combination of mRNA encoding for hgp100 + TRP-2 + TRP-1 + TYR- hβ2 m-Kb/TLR4 (Q-Mix). Some vaccinated mice additionally received 4 times i.p. injections of 250 µg anti-PD-1 antibody (aPD-1) or isotype control (Control) every 3 days. (A) Survival of mice is shown as a Kaplan-Meier curve (n = 6–12/group). Ten days after last vaccination, tumor infiltrating CD8 T cells were analyzed by flow cytometry (n = 8). Results are presented as the percentage of tumor-infiltrating CD8 T cells among total leukocytes (B), MFI of the TCR ζ-chain in CD8 T cells (C), the percentage of Ki67+ (D), CD69+ (E), CD62 L−CD44+ EM (F) and IFN-γ producing CD8 T cells (G) among total CD8 T cells. Data are representative of two independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
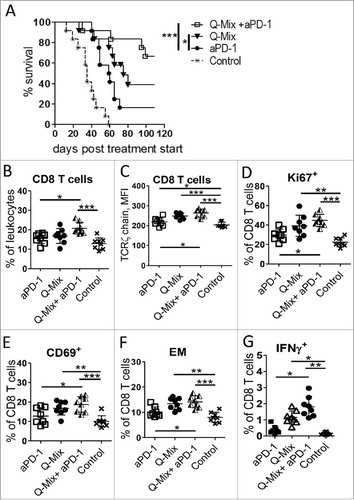
Combinatorial treatment with anti-PD-1 antibodies further increases anti-tumor efficiency
Since anti-PD-1 antibodies are approved for the therapy of melanoma patients, we addressed the question whether the combination of checkpoint inhibition with our improved DC vaccine could enhance the anti-tumor effects. Indeed, the combinatorial treatment significantly prolonged the survival of tumor-bearing RET-tg mice as compared to Q-Mix vaccination or anti-PD-1 antibodies only () This therapeutic effect was associated with a significant accumulation of CD8 T cells within the tumor () as well as with an increased expression of the TCR ζ-chain (), Ki67 () and CD69 () in these cells. In addition, tumor-infiltrating CD8 T cells displayed an elevated frequency of EM T cells () and antigen-specific IFN-γ+ cells () upon combinatorial treatment as compared to single treatments.
Interestingly, similar to the combination with ultra-low dose Pac, DC vaccination combined with anti-PD-1 antibodies had profound inhibitory effects on tumor-infiltrating MDSC (Supplementary Fig. 8 A-D). In addition, a similar impact was detected in spleens and metastatic lymph nodes (data not shown). Moreover, combination of the DC vaccine with anti-PD-1 antibodies led to a significant reduction of the frequency of intratumoral Treg (Supplementary Fig. 8E) associated with decreased frequencies of CD39+ and Ki67+ Tregs (Supplementary Fig. 8 F, G). In addition, the amount of activated conventional CD4 T cells was significantly increased (Supplementary Fig. 8 H). These results indicate that the combination of DC vaccination with chemo-modulating agents (such as ultra-low dose paclitaxel) or with the checkpoint inhibition could provide a significantly improved anti-tumor effect in preclinical melanoma GEMM.
Discussion
Limited mutation load and, therefore, reduced efficacy of neo-epitope based personalized immunotherapy along with profound immunosuppression in the tumor microenvironment may be major reasons of the resistance of a significant fraction of patients to current treatments with negative immune checkpoint inhibitors.Citation1,Citation4,Citation24 Moreover, insufficient tumor antigen processing and presentation by DC may hamper the development of tumor-specific T cells.Citation25 Many therapies aiming to boost T cell responses are now in useCitation26,Citation27 and CD8 T cells are the key players in anti-tumor immune responses since increased intra-tumoral infiltration of these cells was reported to correlate with a better patient outcome.Citation28 Vaccinations using neo-antigens are now in the focus since they could be promising in breaking immune tolerance.Citation21 However, approaches using the immunization with multiple shared tumor-associated antigens could be very efficient since the multivalent T cell response reduces the possibility of tumor escape in a larger cohort of patients.Citation20 In addition, this vaccination strategy represents a promising approach for the treatment of tumors with low mutational load.Citation7 Usually, immunotherapies include single agents that target individual steps in the anti-tumor immune response. However, such monotherapies are unlikely to overcome complex mechanisms that impede anti-tumor immunity in patients.Citation8 Therefore new multivalent and combinatorial therapies are tested now in some ongoing clinical trials that include the targeting of several immune cell populations.Citation5,Citation8
Here we applied immunization with shared MAA based on our recently developed genetic mRNA cancer vaccines,Citation10,Citation19 which was combined with chemo-modulation or checkpoint inhibition to tackle the immunosuppressive tumor microenvironment. We opted for mRNA-based DC vaccination since DC can stimulate both adaptive and innate anti-tumor immune responses and were already intensively used for cancer immunotherapy.Citation27,Citation29 Moreover, tumor-associated antigens could be delivered to DC by loading as peptides or recombinant proteins or by mRNA transfection.Citation30 Importantly, mRNA cannot integrate permanently into the genome, and therefore, has no oncogenic potential.Citation31 In addition, mRNA may act as an adjuvant by providing costimulatory signals, for example, via TLR,Citation32 yet only few studies coupled constitutively active TLR4 for this purpose.Citation33,Citation34 We have previously shown that a universal MHC-I chimeric mRNA construct DC vaccine, which couples the presentation of MAA hgp10025–33 and TRP-2180–188 and the stimulation of DC via constitutively active TLR4 domain, inhibited melanoma growth and improved mouse survival.Citation10,Citation11
In this study, we designed two additional MAA MHC-I constructs to develop a multivalent (quadruple) DC vaccine for melanoma immunotherapy. We have characterized the newly designed constructs encoding for TRP1455–463 and TYR360–368 as new MHC-I restricted MAA for this hβ2m-platform. We showed that the immunization with DC expressing the mix of TRP1/TYR-hβ2m-receptors resulted in stronger anti-tumor effects in the RET-tg melanoma mouse model as compared to TRP1/TYR long-peptide-mix vaccination with agonistic CD40 antibodies and the poly:IC adjuvant. Interestingly, the application of these adjuvants with mutated peptides was reported to induce a significant delay in the growth of MC-38 mouse colon adenocarcinoma.Citation35 Furthermore, local intratumoral administration of adjuvants might be more effective, limiting systemic inflammation yet activating APCs locally.Citation36 However, a direct comparison of different vaccination strategies is difficult, especially with regards to the adjuvant composition, the dose, and the application route (local versus systemic).Citation37
A better efficiency of our mRNA-based DC vaccination compared to long peptide vaccination could be due to the fact that peptides need protection from degradation in vivo to reach antigen presenting cells (APC) as recently demonstrated.Citation38 Moreover, longer antigen presentation by DC in our system could influence the expansion of T cells and their differentiation to memory cells,Citation39 enhancing thereby the vaccination efficiency.
To address the question whether the quadruple antigen-mix vaccination could further enhance the therapeutic efficiency, we vaccinated mice from two melanoma GEMM (RET-tg and BRAF) with electroporated DC containing TRP-1-, TYR-, hgp100- and TRP-2-hβ2m-receptors. Indeed, we found a further increase in mouse survival associated with an enhanced accumulation of antigen-specific tumor-infiltrating CD8 T cells and a stronger induction of memory T cells in both GEMM. Since MAA were described to be overexpressed in BRAF-mutated melanoma-bearing miceCitation13 and melanoma patients carrying the BRAFV600E mutation,Citation40 this DC vaccination strategy using shared MAA could open additional therapeutic options for these melanoma patients in addition to the treatment with BRAF/MEK inhibitors.Citation41
Although some Treg and MDSC markers were reduced already upon vaccination with the quadruple-mix DC vaccine, we decided to combine this treatment with the targeting of immunosuppressive tumor microenvironment. The impact of Pac on immunosuppressive myeloid cells was already described.Citation16,Citation42 Other studies reported a selective impairment of Treg by Pac in tumor-bearing mice.Citation42,Citation43 We demonstrated previously that melanoma progression in RET-tg mice was associated with the activation of MDSC and accumulation of chronic inflammatory factors,Citation15 and Pac at ultra-low doses inhibited MDSC activity and reduced production of chronic inflammatory factors, leading to the prolongation of mouse survival.Citation16 Moreover, the application of ultra-low dose Pac increased T cell responses upon peptide vaccination.Citation44 As expected, we showed here, that the administration of the quadruple DC vaccine with ultra-low dose Pac resulted in an improved survival of melanoma-bearing RET-tg and BRAF mice compared to any single therapy. Importantly, we observed also a significant decrease in intratumoral concentrations of IL-10, TNF-α, IL-6 and VEGF that are known to be involved in the expansion and stimulation of MDSC.Citation23 Chronic inflammatory factors were already described as prognostic markers in cancer patientsCitation45,Citation46 and their neutralization could lead to the restoration of anti-tumor immune responses.Citation15,Citation16 Here, upon the combined treatment, mice displayed a stronger activation of antigen-specific CD8 T cells and an induction of long-lasting memory CD8 T cells, which is considered to be a hallmark of efficient cancer vaccination.Citation47,Citation48
Next, we investigated therapeutic effects of the DC vaccination combined with anti-PD-1 antibodies that are currently applied for the treatment of advanced melanoma patients. Previous preclinical studies showed a therapeutic efficiency of vaccination together with negative immune checkpoint inhibitors, whereas either single therapy was ineffective.Citation49,Citation50 In line with these studies, we showed that the multivalent DC vaccine together with anti-PD-1 antibodies induced a more powerful immune response and prolonged survival of melanoma bearing RET-tg than DC vaccination or anti-PD-1 antibodies alone. Importantly, this combinatorial immunotherapy caused a reduction of frequency and activation markers of tumor-infiltrating Treg. This observation is in agreement with the study on melanoma patients, showing that the PD-1 blockade resulted in the down-regulation of Foxp3 expression in Treg and induced an impairment of their immunosuppressive functions.Citation51 Furthermore, we found that treated mice displayed decreased Arg-1 and PD-L1 expression as well as NO production. Similar reduction of Arg-1 expression and suppressive activity of MDSC was shown by others upon the blockade of PD-1 or PD-L1 in mice with ovarian carcinoma.Citation52
Taken together, we demonstrated in two melanoma GEMM, which closely resemble the situation in melanoma patients, a high anti-tumor efficiency of the improved, multivalent hβ2m-mRNA-based DC vaccine platform using shared MAA that induce potent tumor-infiltrating antigen-specific effector and memory CD8 T cells, without stimulating immunosuppressive cells like MDSC and Treg. The hβ2m-mRNA-based system is safe, modular and can be applied on multiple tumor antigens in different cancers. In addition, when combined with approaches targeting the immunosuppressive tumor microenvironment (such as ultra-low dose Pac or anti-PD-1 antibodies) this strategy resulted in stronger anti-tumor effects associated with further CD8 T cell stimulation and reduction of immunosuppressive cells. Based on our preclinical data, we suggest that such combinatorial strategy could significantly increase the efficiency of existing immunotherapies of patients with melanoma and other tumors.
Materials and methods
Mice
C57 BL/6 mice expressing the human RET oncogene in melanocytes (RET-tg mouse model)Citation14 were provided by I. Nakashima (Chubu University, Aichi, Japan). The C57BL/6 BRAFCA, TYR::CreER and PTENlox4-5 (BRAF) mice were kindly provided to R. Offringa by M. Bosenberg.Citation 13 C57BL/6 (H-2b) and B6.SJL (CD45.1/H-2b) were provided by the animal facility of German Cancer Research Center (DKFZ, Heidelberg, Germany). Mice were kept under pathogen-free conditions in the animal facility of DKFZ. Animal experiments were performed in accordance with government and institute guidelines and regulations.
Cell lines
B16-F10.9 is a mouse melanoma cell line (C57 BL/6 J background), which expresses relevant MAA. MO5-B16 is a mouse melanoma cell line transfected with ovalbumin (C57 BL/6 J background). 3LL-D122 Lewis lung carcinoma mouse cell line (C57BL/6J background) expressing no MAA was served as a negative control. All cells were cultured in DMEM (Invitrogen) supplemented with 10% heat-inactivated fetal calf serum (FCS, GibroBRL), 2 mmol/L glutamine, 1 mmol/L sodium pyruvate, 0.1 mmol/L nonessential amino acids, 100 U/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific). All cells were cultured at 37°C, 5%CO2.
Peptides and antibodies
The short MHC class I restricted peptides OVA257–264 (SIINFEKL), modified TRP-1455–463 (AAPDNLGYM), TYR360–368 (SSMHNALHI), hgp10025–33 (KVPRNQDWL), TRP-2180–188 (SVYDFFVWL), and the long (27mer) peptides TRP-1447–473 (VTNTEMFVTAPDNLGYA-YEVQWPGQEF) and TYR355–381 (ADPSQSSMHNALHIFMNGTMSQVQGSA) were synthesized at DKFZ. The anti-mouse CD40 Ab 3/23 in muIgG1 format was kindly provided by M. Glennie (Southampton, UK) to R. Offringa and D. Baumann in the context of the EU project IACT (Immunostimulatory Agonist Antibodies for Cancer Treatment). Anti-mouse directly conjugated monoclonal antibodies (mAbs) Gr1-PE-Cy7, CD11b-APC-Cy7, CD45.2-PerCP-Cy5.5, CD3-PerCP-Cy5.5, TCR-ζ chain (CD247)-Alexa Fluor® 647, CD4-PE-Cy7, CD8-APC-Cy7, CD25-APC-Cy7, CD69-APC, CD8-BV510, CD8-APC, CD4-BV421, TNF-α-APC, IFNγ-FITC, Ki67-PE were provided by BD Biosciences. Foxp3-FITC and Foxp3 fixation/permeabilization kit were from eBioscience. Anti-mouse PD-1-FITC, PD-L1 –APC, CD39PE-Cy7, CD44-PE-Cy7, CD62 L-APC-Cy7, CD80-FITC, CD86-PE-Cy7, MHCII (IA/IE)-APC and β2-microglobulin-PE mAbs were provided by BioLegend. Streptavidin-PE-conjugated H-2-Kb mouse MHC-class I tetramers loaded with peptides derived from TRP-1455–463 (AAPDNLGYM), TYR360–368 (SSMHNALHI) were obtained from the NIH Tetramer Core Facility (Atlanta, USA).
Cloning of plasmids and expression vectors
pGEM-4Z 5′UT-eGFP-3′UT-A64 (pGEM-4Z) vector was kindly provided by Dr. Eli Gilboa (Miami, USA). This plasmid contains a 741-bp eGFP fragment from peGFP-N1 (Clontech), flanked by the 5 ′ and 3 ′ UTRs of Xenopus laevis β-globin and 64 A-T bp. pGEM-4Z-hβ2mKb and pGEM-4Z-hβ2mTLR4 were generated previously by us and used as backbone vectors to exchange antigenic TYR360–368 and TRP1455–463 sequences by Restriction Free (RF) cloning method. Primers for RF-cloning were synthesized and PAGE purified (Sigma Aldrich). The sequences of the primers and the resulting vectors are summarized in the following. TRP1455–463-pGEM-4Z-hβ2-mKb and -mTLR4 (forward primer 5′ TGTCTCACTGACCGGCTTGTATGCTGCCGCCCCCGACAACCTGGGCTACATGGGAGGTGGCGGATCCGGAGGTGGTT and reverse primer 3′AACCACCTCCGGATC-CGCCACCTCCCATG-TAGCCCAGGTTGTCGGGGGCGGCAGCATACAAGCCGGTC-AGTGAGACA); TYR360–368-pGEM-4Z-hβ2-mKb and -mTLR4 (forward primer 5′ TGTCTCACTGACCGGCTTGTATGCTAGCAGCATGCACAACGCCCTGCACATCGGAGGTGGCGGATCCGGAGGTGGTT and reverse primer 3′ AACCACCTC-CGGATCCGCCACCTCCGATGTGCAGGGCGTTGTGCATGCTGCTAGCATACAAGCCGGTCAGTGAGACA).
In vitro mRNA transcription
Template DNA cloned in the pGEM4Z-A64 vector was prepared using NucleoBond Xtra Maxi Plus DNA purification system (Macherey-Nagel) and linearized via the SpeI restriction site positioned at the 3′ end of the poly (A) tract of the vector. One µg of linear plasmid was used for in vitro mRNA transcription with T7mScript Standard mRNA Production System (CELLSCRIPT). The concentration and quality of the mRNA was assessed by spectrophotometry.
Generation of BMDC from murine bone marrow cells
Murine bone marrow-derived DC were generated as describedCitation 53 with minor modifications. Briefly, bone marrow cells from femurs and tibiae of 4–5 weeks old C57 BL/6 female mice were cultured in BMDC growth medium, consisting of RPMI (Invitrogen) supplemented with 10% heat-inactivated FCS, 2 mmol/L glutamine, 1 mmol/L sodium pyruvate, 0.1 mmol/L nonessential amino acids, 100 U/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific) and 200 U/ml rmGM-CSF (Prospec). On day 8, non-adherent cells were harvested and cultured in fresh BMDC growth medium containing 100U/ml recombinant mGM-CSF for 24 h. BMDC were kept immature prior to β2 m-mRNA electroporation or were maturated by addition of 1 µg/ml of lipopolysaccharide (LPS, Sigma-Aldrich) for another 24 h prior to peptide loading.
mRNA electroporation of bone marrow-derived DC
2.5× 106 DC were washed twice and resuspended in OptiMEM medium containing 10–20 µg transcribed mRNA (5 μg mRNA/ construct) and electroporated in a 2 mm cuvette using Gene Pulser Xcell™ Electroporation Systems (Bio-Rad) at 400 V, 0.9 ms, one pulse. Cells were resuspended in 5 mL BMDC growth medium with 100U/ml recombinant mGM-CSF and incubated for 6 h at 37°C, 5% CO2 for β2m-mRNA-construct expression.
Therapy experiments
RET-tg mice that spontaneously develop skin melanoma were selected for the treatment at the age of 5–6 weeks when the mice displayed palpable skin tumors. BRAF mice were injected intradermally with 32 mM hydroxytamoxifen (4-HT). 21–28 days later, we initiated the therapy of animals, which developed highly pigmented, palpable melanoma skin lesions with the size of 5–10 mm3. All melanoma-bearing mice were vaccinated with 0.5 × 106 cells/in 200 μL cell suspension intraperitoneally (i.p) three times in weekly intervals using DC either electroporated with respective β2m-mRNA (5 μg/construct) or loaded with short peptides (30 μg/peptide). In some experiments, tumor-bearing mice were treated i.p. with ultra-low dose (1 mg/kg) Pac (Hexal). In other set of experiments, melanoma-bearing mice were injected i.p. four times every 3 days with 250 μg/mouse anti-PD-1 antibodies (clone 2A3) or isotype control antibodies (clone 2A3, both from BioXcell). Other tumor-bearing mice were treated subcutaneously 3 times in weekly intervals with mixture of 50 μg long peptide each (LP, TRP-1448–472, TYR353–377), 30 μg anti-CD40 mIgG1 antibodies, and 100 μg poly:IC (Invivogen) as adjuvants.
CTL in vitro killing assay
Ten days after the last vaccination mice were sacrificed and splenocytes were excised and sensitized with 50 µg/ml of respective peptide for 5 days. Afterwards lymphocytes were separated by Lympholyte-M gradient (Cedarlane) and seeded at concentrations of 5× 105- 6× 104 cells/well. 5× 103 target cells (B16F10.9, B16MO5 and 3LL-D122 cell line expressing no MAA serving as a negative control) were labeled with 35S methionine (Perkin Elmer) and co-cultured with sensitized lymphocytes in different target/effector cell ratios for 5 h at 37°C. Released 35S-methionine in the cell culture supernatant was measured as counts per minute using Packard Matrix 96 direct β-counter and the percentage of specific killing was calculated as:
CTL in vivo killing assay
Ten days following the last treatment, vaccinated mice were injected i.v. with 20× 106 splenocytes from B6.SJL (CD45.1+) mice labeled with CFSE (BioLegend) at low (0.3 µM) and high (3 µM) concentrations and loaded with respective peptides at 1:1 ratio. After 18 h, spleens of vaccinated mice were excised, stained with anti CD45.1 mAbs and analyzed by flow cytometry. The specific killing was calculated as:
Bio-Plex assay
Snap frozen skin tumor samples were mechanically disrupted and treated with lysis solution (Bio-Rad). Protein amount in lysates was determined using Pierce BCA protein assay kit (Thermo Fisher Scientific). Inflammatory factors were measured by multiplex technology (Bio-Rad) according to the manufacturer´s instruction. Acquisition and data analysis was performed by bio-plex Manager™.
Preparation of single cell suspensions
Spleen, lymph nodes and tumor lesions were mechanically disintegrated by a plunger and filtered through a 40 µm cell strainer. After the depletion of the erythrocytes by ammonium chloride lysis, cells were washed with FACS buffer (PBS+0.5% BSA+0.1% Na-Azide) and resuspended in an appropriated buffer for further analysis.
Flow cytometry
After 10 min treatment with FcγII/III block (eBioscience), cells were incubated for 30 min at 4°C with directly conjugated mAbs. To measure intracellular IFN-γ production, CD8 T cells were restimulated in vitro with 30 μg/mL specific peptide or 5 μg/mL irrelevant peptide (SIINFEKL). Stimulation with 50 ng/ml phorbol myristate acetate (PMA) and 750 ng/ml Ionomycin (Sigma Aldrich) were used as positive control. After 2 h of restimulation, 2 µM Brefeldin A and 3 μg/ml Monensin (both from eBioscience) were added. For fixation and permeabilization, the Foxp3 fixation/permeabilization kit was used followed by the treatment with FcγII/III block (both from eBioscience) for 10 min. Then, cells were incubated with antibodies for flow cytometry for 30 min at 4°C. In some experiments, cells were stained for 2 h with streptavidin-PE-conjugated H-2-Kb mouse MHC-class I tetramers followed by anti-CD8-APC mAbs. To exclude dead cells, 7-AAD (BD Bioscience, San Jose, USA) was added. Acquisition was performed by FACSCanto II with FACSDiva 6.0 software (BD Bioscience). FlowJo software (Tree Star) was used to analyze at least 500,000 events.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software. Data are presented as the mean ± SD. Statistical significance was assessed using the nonparametric Mann-Whitney test or Kruskal–Wallis according to data requirements. Bonferroni and Dunn's posttests were followed according to their appropriate use. Survival curves were generated using the product limit method of Kaplan and Meier, and comparisons were made using the log-rank test. P < 0.05 was considered as statistically significant.
Financial Support
This work was supported by the German Research Council RTG2099 (V. Umansky, J. Utikal), the German-Israeli Helmholtz Research School in Cancer Biology (M. Grees, A. Sharbi-Yunger, L. Eisenbach, V. Umansky), the K.H. Bauer Foundation (R. Offringa), the European Union FP7 program, Project no. 602262 (R. Offringa) and the DKFZ PhD program (D. Baumann)
Disclosure of Potential Conflicts of Interests
The authors disclose no conflict of interest.
2018ONCOIMM0030R-s01.docx
Download MS Word (1.1 MB)Acknowledgments
The authors thank the NIH Tetramer Core Facility for providing H-2-Kb mouse MHC-class I tetramers. We thank L. Umansky and M. Platten for the assistance with cytokine measurement.
Additional information
Funding
References
- Callahan MK. Immune Checkpoint Therapy in Melanoma. Cancer J. 2016;22(2):73–80. doi:10.1097/PPO.0000000000000183. PMID:27111901.
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(1):23–34. doi:10.1056/NEJMoa1504030. PMID:26027431.
- Long GV, Weber JS, Infante JR, Kim KB, Daud A, Gonzalez R, Sosman JA, Hamid O, Schuchter L, Cebon J, et al. Overall Survival and Durable Responses in Patients With BRAF V600-Mutant Metastatic Melanoma Receiving Dabrafenib Combined With Trametinib. J Clin Oncol. 2016;34(8):871–8. doi:10.1200/JCO.2015.62.9345. PMID:26811525.
- O'Donnell JS, Long GV, Scolyer RA, Teng MWL, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71–81. doi:10.1016/j.ctrv.2016.11.007. PMID:27951441.
- Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, Mueller B, Quaratino S, Sabatos-Peyton C, Petruzzelli L, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer. 2017;17(5):286–301. doi:10.1038/nrc.2017.17. PMID:28338065.
- Veglia F, Gabrilovich DI. Dendritic cells in cancer: the role revisited. Curr Opin Immunol. 2017;45:43–51. doi:10.1016/j.coi.2017.01.002.
- Desrichard A, Snyder A, Chan TA. Cancer neoantigens and applications for immunotherapy. Clin Cancer Res. 2016;22(4):807–12. doi:10.1158/1078-0432.CCR-14-3175. PMID:26515495.
- Smyth MJ, Ngiow SF, Ribas A, Teng MWL. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2016;13(3):143–58. doi:10.1038/nrclinonc.2015.209. PMID:26598942.
- de Rosa F, Ridolfi L, Fiammenghi L, Petrini M, Granato AM, Ancarani V, Pancisi E, Soldati V, Cassan S, Bulgarelli J, et al. Dendritic cell vaccination for metastatic melanoma. Melanoma Res. 2017;27(4):351–7. doi:10.1097/CMR.0000000000000356. PMID:28654547.
- Cafri G, Sharbi-Yunger A, Tzehoval E, Alteber Z, Gross T, Vadai E, Margalit A, Gross G, Eisenbach L. mRNA-transfected Dendritic Cells Expressing Polypeptides That Link MHC-I Presentation to Constitutive TLR4 Activation Confer Tumor Immunity. Mol Ther. 2015;23(8):1391–400. doi:10.1038/mt.2015.90. PMID:25997427.
- Sharbi-Yunger A, Grees M, Tzehoval E, Utikal J, Umansky V, Eisenbach L. mRNA-based dendritic cell immunization improves survival in ret transgenic mouse melanoma model. Oncoimmunology. 2016;5(6):e1160183. doi:10.1080/2162402X.2016.1160183. PMID:27471629.
- Kersten K, de Visser KE, van Miltenburg MH, Jonkers J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol Med. 2017;9(2):137–53. doi:10.15252/emmm.201606857. PMID:28028012.
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–52. doi:10.1038/ng.356. PMID:19282848.
- Kato M, Takahashi M, Akhand AA, Liu W, Dai Y, Shimizu S, Iwamoto T, Suzuki H, Nakashima I. Transgenic mouse model for skin malignant melanoma. Oncogene. 1998;17(14):1885–8. doi:10.1038/sj.onc.1202077. PMID:9778055.
- Meyer C, Sevko A, Ramacher M, Bazhin AV, Falk CS, Osen W, Borrello I, Kato M, Schadendorf D, Baniyash M, et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc Natl Acad Sci U S A. 2011;108(41):17111–6. doi:10.1073/pnas.1108121108. PMID:21969559.
- Sevko A, Michels T, Vrohlings M, Umansky L, Beckhove P, Kato M, Shurin G V, Shurin MR, Umansky V. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J Immunol. 2013;190(5):2464–71. doi:10.4049/jimmunol.1202781. PMID:23359505.
- Dougan SK, Dougan M, Kim J, Turner JA, Ogata S, Cho H-I, Jaenisch R, Celis E, Ploegh HL. Transnuclear TRP1-specific CD8 T cells with high or low affinity TCRs show equivalent antitumor activity. Cancer Immunol Res. 2013;1(2):99–111. doi:10.1158/2326-6066.CIR-13-0047. PMID:24459675.
- Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI : database for MHC ligands and peptide motifs. Immunogenetics. 1999;50(3–4):213–9. doi:10.1007/s002510050595. PMID:10602881.
- Cafri G, Amram E, Rinott G, Koifman G, Fishman S, Keisari Y, Tzehoval E, Margalit A, Eisenbach L, Gross G. Coupling presentation of MHC class I peptides to constitutive activation of antigen-presenting cells through the product of a single gene. Int Immunol. 2011;23(7):453–61. doi:10.1093/intimm/dxr033. PMID:21652516.
- Fujiwara Y, Okada K, Omori T, Sugimura K, Miyata H, Ohue M, Kobayashi S, Takahashi H, Nakano H, Mochizuki C, et al. Multiple therapeutic peptide vaccines for patients with advanced gastric cancer. Int J Oncol. 2017;50(5):1655–62. doi:10.3892/ijo.2017.3955. PMID:28393243.
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–21. doi:10.1038/nature22991. PMID:28678778.
- Cho H-I, Celis E. Optimized peptide vaccines eliciting extensive CD8 T-cell responses with therapeutic antitumor effects. Cancer Res. 2009;69(23):9012–9. doi:10.1158/0008-5472.CAN-09-2019. PMID:19903852.
- Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res. 2017;5(1):3–8. doi:10.1158/2326-6066.CIR-16-0297. PMID:28052991.
- Callahan MK, Wolchok JD. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol. 2013;94(1):41–53. doi:10.1189/jlb.1212631. PMID:23667165.
- Mellman I. Dendritic cells: master regulators of the immune response. Cancer Immunol Res. 2013;1(3):145–9. doi:10.1158/2326-6066.CIR-13-0102. PMID:24777676.
- Bender C, Hassel JC, Enk A. Immunotherapy of Melanoma. Oncol Res Treat. 2016;39(6):369–76. doi:10.1159/000446716. PMID:27259558.
- Garg AD, Vara Perez M, Schaaf M, Agostinis P, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: Dendritic cell-based anticancer immunotherapy. Oncoimmunology. 2017;6(7):e1328341. doi:10.1080/2162402X.2017.1328341. PMID:28811970.
- Fridman WH, Zitvogel L, Sautès–Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14(12):717–34. doi:10.1038/nrclinonc.2017.101. PMID:28741618.
- Wilgenhof S, Corthals J, Van Nuffel AMT, Benteyn D, Heirman C, Bonehill A, Thielemans K, Neyns B. Long-term clinical outcome of melanoma patients treated with messenger RNA-electroporated dendritic cell therapy following complete resection of metastases. Cancer Immunol Immunother. 2015;64(3):381–8. doi:10.1007/s00262-014-1642-8. PMID:25548092.
- Anguille S, Smits EL, Bryant C, Van Acker HH, Goossens H, Lion E, Fromm PD, Hart DN, Van Tendeloo VF, Berneman ZN. Dendritic Cells as Pharmacological Tools for Cancer Immunotherapy. Pharmacol Rev. 2015;67(4):731–53. doi:10.1124/pr.114.009456. PMID:26240218.
- Kramps T, Probst J. Messenger RNA-based vaccines: progress, challenges, applications. Wiley Interdiscip Rev RNA. 2013;4(6):737–49. doi:10.1002/wrna.1189. PMID:23893949.
- Eisenächer K, Steinberg C, Reindl W, Krug A. The role of viral nucleic acid recognition in dendritic cells for innate and adaptive antiviral immunity. Immunobiology. 2007;212(9–10):701–14. doi:10.1016/j.imbio.2007.09.007. PMID:18086372.
- Van Lint S, Goyvaerts C, Maenhout S, Goethals L, Disy A, Benteyn D, Pen J, Bonehill A, Heirman C, Breckpot K, et al. Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res. 2012;72(7):1661–71. doi:10.1158/0008-5472.CAN-11-2957. PMID:22337996.
- Wilgenhof S, Van Nuffel AMT, Benteyn D, Corthals J, Aerts C, Heirman C, Van Riet I, Bonehill A, Thielemans K, Neyns B. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann Oncol. 2013;24(10):2686–93. doi:10.1093/annonc/mdt245. PMID:23904461.
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–6. doi:10.1038/nature14001. PMID:25428506.
- Marabelle A, Tselikas L, de Baere T, Houot R. Intratumoral immunotherapy: using the tumor as the remedy. Ann Oncol. 2017;28(suppl_12):xii33−43. doi:10.1093/annonc/mdx683. PMID:29253115.
- Overwijk WW. Cancer vaccines in the era of checkpoint blockade: the magic is in the adjuvant. Curr Opin Immunol. 2017;47:103–9. doi:10.1016/j.coi.2017.07.015. PMID:28806603.
- Kumai T, Kobayashi H, Harabuchi Y, Celis E. Peptide vaccines in cancer-old concept revisited. Curr Opin Immunol. 2017;45:1–7. doi:10.1016/j.coi.2016.11.001. PMID:27940327.
- van Stipdonk MJB, Hardenberg G, Bijker MS, Lemmens EE, Droin NM, Green DR, Schoenberger SP. Dynamic programming of CD8+ T lymphocyte responses. Nat Immunol. 2003;4(4):361–5. doi:10.1038/ni912. PMID:12640451.
- Kannengiesser C, Spatz A, Michiels S, Eychène A, Dessen P, Lazar V, Winnepenninckx V, Lesueur F, Druillennec S, Robert C, et al. Gene expression signature associated with BRAF mutations in human primary cutaneous melanomas. Mol Oncol. 2008;1(4):425–30. doi:10.1016/j.molonc.2008.01.002. PMID:19383316.
- Polkowska M, Czepielewska E, Kozłowska-Wojciechowska M. Drug Combinations as the New Standard for Melanoma Treatment. Curr Treat Options Oncol. 2016;17(12):61. doi:10.1007/s11864-016-0436-y. PMID:27766547.
- Liechtenstein T, Perez-Janices N, Gato M, Caliendo F, Kochan G, Blanco-Luquin I, Van der Jeught K, Arce F, Guerrero-Setas D, Fernandez-Irigoyen J, et al. A highly efficient tumor-infiltrating MDSC differentiation system for discovery of anti-neoplastic targets, which circumvents the need for tumor establishment in mice. Oncotarget. 2014;5(17):7843–57. doi:10.18632/oncotarget.2279. PMID:25151659.
- Wu X, Feng QM, Wang Y, Shi J, Ge HL, Di W. The immunologic aspects in advanced ovarian cancer patients treated with paclitaxel and carboplatin chemotherapy. Cancer Immunol Immunother. 2010;59(2):279–91. doi:10.1007/s00262-009-0749-9. PMID:19727719.
- Sevko A, Kremer V, Falk C, Umansky L, Shurin MR, Shurin G V, Umansky V. Application of paclitaxel in low non-cytotoxic doses supports vaccination with melanoma antigens in normal mice. J Immunotoxicol. 2012;9(3):275–81. doi:10.3109/1547691X.2012.655343. PMID:22449053.
- Gonda K, Shibata M, Ohtake T, Matsumoto Y, Tachibana K, Abe N, Ohto H, Sakurai K, Takenoshita S. Myeloid-derived suppressor cells are increased and correlated with type 2 immune responses, malnutrition, inflammation, and poor prognosis in patients with breast cancer. Oncol Lett. 2017;14(2):1766–74. doi:10.3892/ol.2017.6305. PMID:28789407.
- Jiang H, Gebhardt C, Umansky L, Beckhove P, Schulze TJ, Utikal J, Umansky V. Elevated chronic inflammatory factors and myeloid-derived suppressor cells indicate poor prognosis in advanced melanoma patients. Int J Cancer. 2015;136(10):2352–60. doi:10.1002/ijc.29297. PMID:25353097.
- Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, Dransart E, Sandoval F, Riquet M, Rance B, et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun. 2017;8:15221. doi:10.1038/ncomms15221. PMID:28537262.
- van Duikeren S, Fransen MF, Redeker A, Wieles B, Platenburg G, Krebber W-J, Ossendorp F, Melief CJ, Arens R. Vaccine-induced effector-memory CD8+ T cell responses predict therapeutic efficacy against tumors. J Immunol. 2012;189(7):3397–403. doi:10.4049/jimmunol.1201540. PMID:22914049.
- Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7(283):283ra52. doi:10.1126/scitranslmed.aaa4306. PMID:25877890.
- Ali OA, Lewin SA, Dranoff G, Mooney DJ. Vaccines Combined with Immune Checkpoint Antibodies Promote Cytotoxic T-cell Activity and Tumor Eradication. Cancer Immunol Res. 2016;4(2):95–100. doi:10.1158/2326-6066.CIR-14-0126. PMID:26669718.
- Wang W, Lau R, Yu D, Zhu W, Korman A, Weber J. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+ CD25(Hi) regulatory T cells. Int Immunol. 2009;21(9):1065–77. doi:10.1093/intimm/dxp072. PMID:19651643.
- Liu CY, Wang YM, Wang CL, Feng PH, Ko HW, Liu YH, Wu YC, Chu Y, Chung F-T, Kuo CH, et al. Population alterations of L-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14−/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol. 2010;136(1):35–45. doi:10.1007/s00432-009-0634-0. PMID:19572148.
- Lutz MB, Kukutsch N, Ogilvie AL, Roßner S, Koch F, Romani N, Schuler G, Rössner S. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223(1):77–92. doi:10.1016/S0022-1759(98)00204-X. PMID:10037236.