
ABSTRACT
In spite of the success of PD-1 blocking antibodies in the clinic their benefits are still restricted to a small fraction of patients. Immune-desert tumors and/or the highly immunosuppressive tumor milieu might hamper the success of PD-1/PD-L1 blocking therapies into a broader range of cancer patients. Although still under debate, there is a cumulative body of evidence that indicates B tumor-infiltrating lymphocytes are a good prognostic marker in most types of cancer, especially in those that form ectopic lymphoid tissue structures. Taking this into account, we reason that the adoptive transfer of activated B lymphoblasts (ABL) in the tumor could be a feasible therapeutic approach to shift the immunosuppressive tumor microenvironment into an immune-permissive one. In this work we show the antitumor effect of ABL therapy in two different tumor models: colon carcinoma (CT26) and melanoma (B16/F10). The ABL transfer in the most relevant non-immunogenic B16/F10 melanoma model depicts synergism with anti-PD-1 antibody therapy. Furthermore, systemic antitumor immunity was detected in mice treated with PD-1 antibody/ABL combination which was able to reach distal metastatic lesions.
KEYWORDS:
Introduction
Immune-checkpoint blockade antibodies, especially PD-1 blockade axis has shown unprecedented results in clinical trials; however, the effect still seems to be restricted to a small fraction of patients.Citation1 Searching for new combinations with PD-1 blockade that could raise the number of patients that respond to the treatment seems urgent.Citation2-Citation3 Lack of tumor antigenicity and active mechanisms of immunosuppression creates tumor milieus devoid of T lymphocytes or with very limited antitumor activity.Citation4 Tumor-specific infiltrating T lymphocytes have the potential to eradicate the tumor. Unfortunately, these lymphocytes are present in a quite immunosuppressive milieu and they lack costimulatory signals. In this environment the lymphocytes are not only unable to get activated but they become anergic, which could be considered an immune escape mechanism by which the tumor can discard reactive T lymphocytes.Citation5 Providing artificial costimulatory ligands in the tumor may overcome this limitation as has been demonstrated in a plethora of preclinical studies. That can be accomplished by using exogenous artificial agonistic molecules against costimulatory receptors (4-1BB, OX40, etc)Citation6-Citation7 or by directly inducing the expression of the costimulatory ligands in the tumor (ICOSL, B7, etc).Citation8-Citation10 Costimulatory ligands are expressed on activated antigen-presenting cells (APC) such as Dendritic Cells (DCs), macrophages, and B lymphocytes (BL) among others. In fact, B-cell infiltration in the tumor has been associated with better prognosis in a lot of different types of tumors.Citation11-Citation14 BL in the tumor can be organized in ectopic lymphoid tissue structures with even a formation of alike germinal centers in which there are B lymphoblasts under high proliferation rate.Citation15-Citation16 Activated B lymphocytes in the tumor have been documented to be in close proximity to CD8T cells which have also been associated with induction of CD8T-cell response and with longer overall survival in cancer patients.Citation12,Citation17 The role of the tumor infiltrated B lymphocytes is not completely understood and needs to be elucidated. We reason that the activated B lymphocytes that infiltrate the tumor could be a potential source of costimulatory signals to the tumor resident T lymphocytes that might contribute to the maintenance and expansion of the tumor immunity. In order to prove this hypothesis we decided to investigate whether the adoptive transfer of activated B lymphoblast (ABL) in the tumor could improve the immune response outcome. In fact, herein we show that ABL intratumoral inoculation in combination with PD-1 blocking antibodies is a feasible immunotherapeutic approach to enhance local and systemic tumor immunity. The local transfer of ABLs in the tumor elicits a systemic immune response able to target disseminated distal tumor lesions.
Results
ABLs express costimulatory ligands and elicit efficient T-cell activation in vitro
The design workflow to test the immunostimulatory activity of ABLs is shown in . ABLs are generated after a three-day stimulus of B lymphocytes with LPS and Dextran ex vivo; B lymphocytes in this condition proliferate very efficiently getting a large amount of ABLs after three-day incubation. Cells are washed twice with a large volume of PBS to remove any trace of LPS (non-traces of LPS were detected by high sensitivity detection kit (< 0.01EU/ml)) (Fig. S1). The microscopy morphology of ABL is shown in . These ABLs were later characterized in vitro and used for intratumoral tumor injection in different models.
Figure 1. ABLs express costimulatory ligands and are very efficient activators of T-immune responses. A) Rationale of ABL therapy. B lymphocytes are expanded and activated in vitro with Dextran and LPS. ABL are assessed for the expression of costimulatory ligands and then inoculated intratumorally. ABLs persist in the tumor for a long period of time providing costimulatory signals to tumor-infiltrating lymphocytes, promoting a further grade of expansion and triggering a more efficient antitumor immune response. B) Flow cytometry analysis of different costimulatory ligands expressed in ABLs. C) Microscopic image of ABLs at 20X. D) IFN-γ ELISPOT performed on isolated CD8 lymphocytes from OT-I transgenic mice stimulated with 0.5 µg/ml of SIINFEKL and activated B lymphoblast (ABL) or B lymphocytes (BL). E) Dots representing the grade of activated CD8 lymphocytes that secrete IFN-γ for each condition are shown at the top of the panel, as represented on panel D (shown as triplicates of the experiment). F) Same as in C, but measuring IFN-γ by ELISA in supernatant. G) Proliferation of CD8 OT-I lymphocytes measured by 3H thymidine after three days' incubation.
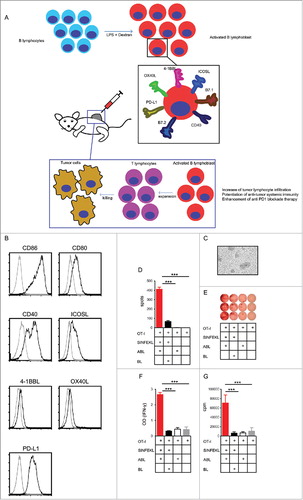
Activated B lymphocytes are known to be efficient antigen-presenting cells (APC), up-regulating molecules related to T-cell activation. The expression on ABLs of costimulatory ligands (CD86, CD80, CD40, ICOSL, 4-1BBL, and OX40L) and PD-L1 negative immune-checkpoint ligands were measured by flow cytometry. There is clear expression of immune-promoting costimulatory ligands: CD86, CD80, CD40 and ICOSL, while 4-1BBL and OX40L are barely expressed (). Expression of costimulatory ligands on BL is reduced (Fig. S2), while maturated bone marrow derived dendritic cells (BMDC) show a slightly higher expression of costimulatory ligands as compared to ABL (Fig. S3) On the other hand, PD-L1 - which is an immunosuppressive receptor - is also expressed on ABL. The co-expression of immune-activating and immune-suppressive receptors opens the debate as to whether these cells could actually promote tumor immunity or impede it. In order to have a deeper insight of the immune function of these cells, we performed an in vitro assay with ovalbumin reactive T lymphocytes (OT-I). Isolated CD8 OT-I lymphocytes were cultured with ABLs or non-activated B lymphocytes with a suboptimal amount of the ovalbumin determinant antigen (SIINFEKL). Stimulation of CD8 OT-I lymphocytes was measured by IFN-γ ELISPOT (, ), the CD8 OT-I cells incubated with ABL shown a 4 fold increase in the amount of spots. These results were also corroborated by IFN-γ ELISA () in the supernatant and proliferation by 3H thymidine incorporation (). In all cases we observed a higher activation of CD8 OT-I lymphocytes when cultured with ABLs. These results indicate that, in spite of PD-L1 expression on ABLs, these cells have an overall immune-activated function.
Intratumoral injection of syngeneic or allogeneic ABLs reduces tumor growth and enhances tumor rejection in immunogenic tumor (CT26)
As described in , ABLs expressed higher levels of costimulatory receptors but also PD-L1. That leads to the possibility that ABLs, in vivo, could induce immunosuppression through PD-1 signaling. To address which mechanism (immunostimulation or immunosuppression) might prevail we decided to test the effect of direct intratumoral injection of ABLs in a highly immunogenic tumor model (CT26). Mice were treated following the schedule depicted in . Mice that were intratumorally injected with ABLs ten days after tumor implantation have a significantly reduction on tumor growth () as compared with control group. Mice treated with the ABLs as monotherapy showed nearly 75% of long-term survivors () with similar rate of tumor rejection (). Most of the mice were free of tumor 20 days after ABL therapy.
Figure 2. Local intratumoral injection of ABLs as monotherapy in CT26 induces a potent antitumor response. A) Schedule of treatment. Balb/c mice were inoculated subcutaneously with CT26 colon carcinoma cells. Tumor bearing mice were injected intratumorally with syngeneic or allogeneic ABLs at day 10, 13 and 16 after tumor inoculation. B) Tumor growth kinetics was monitor by caliber measurement every other day (8 mice per group). C) Tumor rejection depicted by time and mice in each group (8 mice per group). D) Overall mice survival of the same groups as in C (8 mice per group). The experiments were repeated twice with similar outcomes.
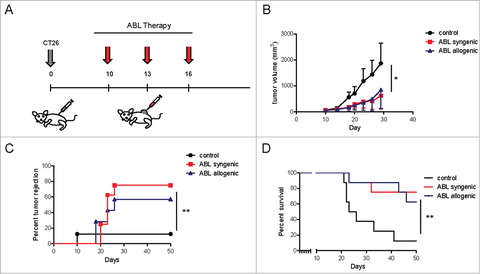
The mechanism of action might depend on interaction of costimulatory ligands of the ABLs with the costimulatory receptor expressed in the tumor-infiltrating lymphocytes. However, there is also the possibility that the ABLs could function as antigen-presenting cells (APCs) that enabled presentation of tumor antigens and primed of a new set of tumor-reactive T-lymphocytes. In order to elucidate which mechanism is responsible for tumor rejection, we treated the mice with allogeneic ABLs that do not share MHC-I/II with the tumor-bearing mice, but they do express the costimulatory ligands. As it is shown in , the antitumor effect elicited with allogeneic ABLs was similar to the treatment with syngeneic ABLs indicating that it does not depend on antigen presentation. These results still need to be taken with caution: an allogenic immune response could be partially obscuring the antitumor outcome. Further therapy experiments with ABL derived from ß2 microglobulin knockdown mice will ultimately confirm this observation.
Another plausible explanation might be associated with the expression of T-cell attractiving chemokines, as in the characterization of the cytokine profile produced by ABLs we observed a 15 and 10-fold upregulation of mRNA encoding for the chemokine CXCL11 and CCL21 (Fig. S4). These two cytokines are important in the recruitment of activated CD8 lymphocytes to the tumor.Citation18-Citation21
Intratumoral injection of B lymphoblasts in combination with systemic PD-1-blockade therapy reduces tumor growth in aggressive, poorly immunogenic tumor (B16/F10)
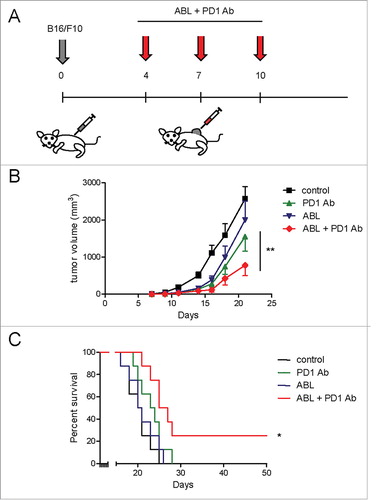
Based on the results obtained in the CT26 tumor model we desired to test this approach in the more aggressive, poorly immunogenic tumor (B16/F10 melanoma) model. In this model, the ABLs as monotherapy following treatment schedule depicted in , did not show any significant antitumor effect (data not shown). Therefore, we considered blocking the PD-1/PD-L1 axis as well. The blockade antibody PD-1 has shown unprecedented results in melanoma patients, yet far from optimal. We treated melanoma B16/F10-bearing mice with PD-1 blocking antibody intraperitoneally and ABLs locally intratumorally as shown in . ABLs can persist in the tumor for several days after intratumoral injection as shown by immunohistochemistry (Fig. S5). As expected, nearly no B220+ B lymphocytes in PD-1 antibody-treated tumors were detected;Citation22 B220+ B cells tend to accumulate in clusters imitating ectopic tissue architectures (Fig. S5). Mice that were treated with ABLs as monotherapy did not show any significant delay in tumor growth, while PD-1-blockade antibody as monotherapy showed a slight reduction in tumor growth () with a modest effect on overall survival (). The combination of both treatments displayed a synergistic effect on tumor growth () with a direct impact on overall survival () in a 50 days follow up, achieving curative responses in approximately 20% of mice.
Figure 3. ABLs enhance the antitumor response of PD-1-blockade therapy in B16/F10 melanoma tumors. A) Treatment schedule. B16/F10 melanoma tumor cells were inoculated subcutaneously in C57BL6 mice. On days 4, 7 and 10 after tumor inoculation PD-1 blocking antibody was injected intraperitoneally in combination with ABL therapy injected intratumorally. B) Tumor growth kinetics was monitored by measurement with calibers every other day (8 mice per group). C) Overall survival of mice (8 mice per group). Experiment was repeated twice with similar results.
Intratumoral injection of B lymphoblast and PD-1-blocking antibody increases tumor T-cell infiltration and promotes systemic immunity
An important evidence of the existence of tumor-mediated immunity is the presence of T-lymphocyte infiltrates in the tumor milieu. In order to determine the degree of T-cell tumor infiltration, we performed anti-CD3 immunohistochemistry on sections of paraffin embedded tumor tissue obtained from mice that underwent PD-1-blockade antibody and intratumoral ABL therapy. We noticed higher infiltration of CD3+ T lymphocytes (depicted in red) () in tumors of mice treated with the combination therapy, compare to the control groups. Performing an image area count analysis of the CD3+ T cells in the combination therapy, mice double the amount CD3 infiltration when compare to controls (). Similar analyses were performed with CD8+ T-lymphocytes staining to corroborate the increase infiltration of cytotoxic T lymphocytes (), in this case there is a threefold increase in CD8 infiltration over the controls (). By immunofluorescence staining with anti-CD8 antibody and anti-B220 there is evidence of proximity and some interaction of CD8 lymphocytes and ABL (Fig. S6).
Figure 4. PD-1 blockade in combination with intratumoral injection of ABLs elicits a potent local and systemic antitumor immune response. A) Representative immunohistochemistry images of anti-CD3 antibody stained B16 tumor samples, with their corresponding treatment types. CD3 positive cells are stained red with Stay Red dye. B) CD3 positive cells were quantified as shown in materials and methods, and analysis in each group was conducted on three independent tumor samples (3 mice per group). C) Same as in A, but with anti-CD8a antibody. D) Analysis of CD8a positive cells infiltrating the tumor area calculated as in B. E) IFN-γ ELISPOT was performed with isolated splenocytes obtained from B16/F10 tumor-bearing mice treated with anti-PD-1 antibody intraperitoneally at days 4, 7 and 10 with or without intratumoral injection of ABLs. Splenocytes were cultivated with B16/F10 irradiated tumor cells as indicated in materials and methods (3 mice per group). F) Representative image of the amount of spots detected by ELISPOT of E. G) Induction of T-cell activation by combination of PD-1 blockade and intratumoral injection of ABL, confirmed by the presence of IFN-γ detected via ELISA on the supernatant of the cultured cells. Experiments were repeated twice with similar results.
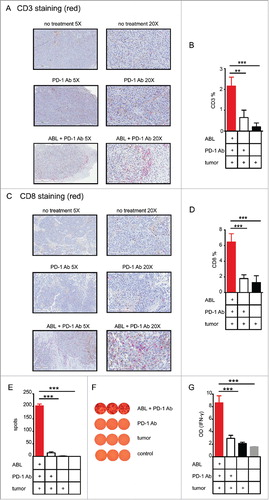
To confirm that tumor suppression is in fact mediated by the induction of a systemic immune response we performed IFN-γ ELISPOT and ELISA experiments on B16/F10 melanoma tumor-bearing mice treated with anti-PD-1 blocking antibody and intratumoral injection of ABLs. We chose to evaluate the immune response of treatment groups that displayed an antitumor effect on B16/F10 tumors (namely PD-1 antibody and the combination PD-1 antibody/ABLs); ABLs as monotherapy had no effect on B16/F10 tumors. Treated mice were sacrificed 4 days after finishing the scheduled treatment (delivered at days 4, 7 and 10 after tumor inoculation) and lymphocytes were isolated from the tumor's draining lymph nodes. B16/F10 irradiated tumor cells were used as target cells in mixed culture with the isolated T lymphocytes. After overnight incubation, the number of tumor-reactive T lymphocytes was estimated by quantifying the number of spots in IFN-γ ELISPOT assays and the amount of IFN-γ production was estimated by ELISA. Mice treated with the combination of PD-1-blockade antibody and intratumoral ABLs displayed a stronger INF-γ mediated T-cell immune response against the tumor cells compared to the control groups with a ten to fivefold increase, determined both as number of spots (, ) as well as the overall IFN-γ production ().
Intratumoral injection of ABLs in combination with PD-1-blockade antibody displays an antitumor effect in distal metastasis (lung and brain metastasis models)
Primary tumors usually metastasize to other distal tissues and organs and constitute a major cause of cancer mortality. One of the main advantages of cancer immunotherapy over other types of therapies is the high specificity of the adaptive immune response upon mobilization of tumor immunity. The development of a therapeutic strategy that would only succeed in a local tumor would have limited translation into the clinic. It is important to pursue a therapeutic approach that would favor the awakening of potent tumor-specific systemic immunity that could target any disseminated tumor lesion. Melanoma usually metastasizes to the lungs and sometimes in the brain,Citation23 so we tested the efficacy of ABL therapy in combination with PD-1 blockade in these two varied, orthotopic-like, metastatic models. First, we mimicked a melanoma lung metastasis model by intravenous injection of B16/F10 melanoma cells, engrafting the tumor cells directly in the lung. The same mice were also implanted with a subcutaneous B16/F10 tumor in the flank. Mice bearing B16/F10 melanoma in the lungs and flank were treated with PD-1 antibody intraperitoneally and with ABLs intratumorally only in the accessible lesions on the skin in the subcutaneous tumor. The treatment schedule is shown in . When the first mice in the control group showed signs of discomfort (fatigue and sickness) all the mice were sacrificed to examine the melanoma-tumor burden in the lungs. Importantly, the lung of mice treated with PD-1 blocking antibody together with intratumoral injection of ABLs in the subcutaneous tumors, have significantly lower weights compared to control ones (). PD-1 antibody as monotherapy did not have any significant antitumor effect on the lungs in this aggressive model.
Figure 5. Immune response induced by PD-1 blockade and ABLs target distal tumor metastasis. A) To establish an orthotopic lung metastasis model of melanoma; C57/BL6 mice were implanted with B16/F10 tumor cells subcutaneously in the flank and intraperitoneally. At days 4, 7 and 10 after tumor implantation, they were treated with anti-PD-1 antibody intraperitoneally and ABL therapy intratumorally at the subcutaneous tumor site only. B) Mice were sacrificed when the size of the tumor in the control group reached 15 mm in diameter, and lungs were extracted to examine tumor load by overall weight. Normal weight of healthy age-controlled lung is indicated with the gray bar. C) To establish an orthotopic, brain metastasis model of B16/F10 melanoma, C57/BL6 mice were subcutaneously injected with B16/F10 tumor cells at day 0, and intrathecally at day 9 after subcutaneous implantation. At days 4, 7 and 10 after implantation, mice were treated with anti-PD-1 intraperitoneally antibody and ABL therapy, intratumorally at the site of the subcutaneous tumor only (10 mice per group). D) Mice were sacrificed when the size of the tumor of the control group reached 15 mm in diameter, and brain was extracted to determine CD3-infiltration in the brain-tumor lesions by immunohistochemistry. Representative images of one mouse per group are shown. CD3-positive cells are shown in red (Stay Red Dye). E) Analysis of infiltration of CD3-positive cells in each group is represented as the percentage of positive CD3 (red) in the whole tumor area (10 mice per group). F), G) Same as in D) and E) respectively, but for CD8a-positive cell infiltrates in the brain tumor lesions (10 mice per group).
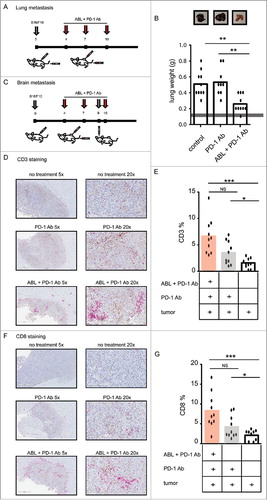
The second orthotopic metastatic model was established by intracranial implantation of B16/F10 tumors as described in . As the intracranial tumor growth of B16/F10 is very aggressive, with an overall survival of less than 15 days, we had to delay brain-tumor implantation to day 9 after subcutaneous tumor inoculation. Mice started receiving immunotherapy treatment with PD-1 antibody intraperitoneally and ABLs intratumorally at days 4, 7 and 10 (). When flank-tumors on control group mice reached a size of 15 mm in diameter, all the mice were sacrificed and their brains were extracted to measure tumor-lymphocyte infiltration with immunohistochemistry staining with anti-CD3 and CD8 antibodies. The group of mice treated with both PD-1 blocking antibody and ABLs in the distal tumor showed a higher infiltration rate of CD3 positive cells in the tumor lesion implanted in the brain (). Analyzing the CD3 positive in the brain tumors we detect a nearly twofold increase of CD3 infiltration in the ABL and PD-1 antibody treated mice over the control groups (). The same analysis was also performed with an anti-CD8a antibody. In a similar manner mice treated with both PD-1 blockade and ABLs in a subcutaneous tumor, displayed a much higher infiltrate of CD8a cell in brain tumors (twofold) compared to the controls ( and ).
To further confirm the indispensable role of CD8 lymphocytes in this therapeutic approach we performed an in vivo CD8 depletion experiment in B16/F10 tumor-bearing mice treated with ABL and PD1 blockade antibody. The group of mice that underwent CD8 lymphocyte depletion were unable to control tumor growth (Fig. S7).
Materials & methods
Cells, buffer and media
B16/F10 and CT26 cell were acquired from ATCC and cultured according to their guides. Spleens were processed using PBS buffer; PBS without MgCl2 and CaCl2 (Gibco from Life Technologies, Paisley, UK) containing EDTA 2 mM (Invitrogen from Life Technologies, Grand Island, NY, USA) and supplemented with 0.5% of Bovine serum albumin (Sigma-Aldrich, St. Louis, MO, USA). ABLs were cultured in ABL medium; RPMI medium (Gibco from Life Technologies, Paisley, UK) containing β-Mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA) 50 μM supplemented with 10% of Fetal Bovine Serum (Biochrom, Berlin, Germany), 1% of Penicillin/Streptomycin (Gibco from Life Technologies, Paisley, UK), 1% of L-Glutamine (Gibco from Life Technologies, Paisley, UK), 1% of Non-essential amino acids (Gibco from Life Technologies, Paisley, UK), 1% of HEPES (Gibco, from Life Technologies, NY, USA) and 1% of Sodium pyruvate (Gibco from Life Technologies, Paisley, UK).
Generation of activated B lymphoblasts (ABLs)
Spleens were harvested from naïve C57BL/6J or BALBc mice and processed in PBS buffer, they were transferred to 50 ml conical tube through the strainer using a sterile plunger from a 5 ml syringe. Splenocytes were spun down and washed with ABL medium. Cells were counted and the cell density adjusted to 1.5 × 106 cell/ml and cultured in T-75 (Corning, NY, USA) flasks with 30 ml of ABL media containing 5 μg/ml of LPS (Sigma-Aldrich, St. Louis, MO, USA) and 7 μg/ml of Dextran sulphate (Sigma-Aldrich, St. Louis, MO, USA) for 3 days under standard conditions of oxygen, CO2 and temperature. After incubation, cells were washed twice with 50 ml of PBS; after the two washes, the lack of traces of LPS was verified by ToxinSensor Endotoxin Detection System with a sensitivity of 0.01 EU/ml (GenScript, Piscataway, New Jersey, USA).
Characterization of ABL
5 × 105 ABLs were spun down and washed with PBS buffer. Cells were incubated with α-PD-L1 (Clone 10F.9G2), α-OX40L (Clone TKS-1), α-ICOSL (Clone HK5.3), α-CD86 (Clone GL-1), α-4-1BBL (Clone RM1343), APC-labeled α-CD40 (Clone 3/23)and FITC-labeled α-CD80 (Clone 16-10A1) all purchased from Biolegend (San Diego, CA, USA). The expression level of these markers was measured using BD FACSCaliburTM (BD Bioscience, San Diego, CA, USA) and analyzed using FlowJo vX.0.7. BMDC were used as a reference of expression of costimulatory ligands isolated and expanded as previously described, and maturated with Poly I:C (20 µg/mL) and LPS (0,5 µg/mL) at 37°C for 18 hours.Citation24
Cytokine and chemokine expression on ABLs over non-activated lymphocytes were conducted by qRT-PCR. RNA was extracted by was extracted with RNAsye kit (Qiagen). 2 μg of total RNA was retrotranscribed and qPCR was performed using the following primers: CXCL9 (Fwd CGAGGCACGATCCACTACAA, Rev GAGTCCGGATCTAGGCAGGT); CXCL11 (Fwd GGCTTCCTTATGTTCAAACAGGG, Rev GCCGTTACTCGGGTAAATTACA). CXCL13 (Fwd GGCCACGGTATTCTGGAAGC, Rev GGGCGTAACTTGAATCCGATCTA); CCL21 (Fwd GTGATGGAGGGGGTCAGGA, Rev GGGATGGGACAGCCTAAACT); lymphotoxin (Fwd GCAGTGCCTATCACTGTCC, Rev CAAGATGCACGACGGTTTGC); IFN-γ (Fwd GCCACGGCACAGTCATTGA, Rev TGCTGATGGCCTGATTGTCTT); IL12 (Fwd CAATCACGCTACCTCCTCTTTT, Rev CAGCAGTGCAGGAATAATGTTTC); TNF-α (Fwd CCCTCACACTCAGATCATCTTCT, Rev GCTACGACGTGGGCTACAG)
3H Thymidine proliferation assay
1 × 105 CD8 lymphocytes purified from spleens of OT-I transgenic mice were incubated with 0.5 µg/ml of SIINFEKL in the presence of 1 × 104 ABLs or non-activated B lymphocytes for 72 h in 96-well plates. Non-activated B lymphocytes were isolated in fresh from C56/BL6 mice spleen with B-cell negative selection fraction kit (Miltenyi Biotec, Dergish Gladbach, Germani). After 72 hours, 0.5 μCi of 3H thymidine (Perkin Elmer, Boston, MA, USA) was added overnight and measured by scintillation.
IFN-γ ELISPOT
4 × 105 CD8 lymphocytes purified from spleen of OT-I transgenic mice were incubated with 0.5 µg/ml of SIINFEKL in the presence of 1 × 104 ABLs or non-activated B lymphocytes for 24 h in 96 well plates. As previously mentioned, non-activated B lymphocytes were purified using Miltenyi beads. The experiment was performed following manufacturer's instructions (BD Bioscience, San Diego, CA, USA). Assessment of IFN-γ-expressing cells was measured and analyzed with CTL ImmunoSpot® Analyzer (CTL-Europe GmbH, Bonn, Germany).
IFN-γ ELISA
1 × 105 CD8 lymphocytes purified from spleen of OT-I transgenic mice were incubated with 0.5 µg/ml of SIINFEKL in the presence of 1 × 104 ABLs or non-activated B lymphocytes for 72 h in 96-well plates. The supernatant was collected to perform ELISA experiments. IFN-γ was measured by ELISA (BD Bioscience, San Diego, CA, USA) following manufacturer's instructions. ELISA was quantified by TECAN SunriseTM (TECAN, Männedorf, Switzerland) and analyzed with MagellanTM (TECAN, Männedorf, Switzerland).
Immunohistochemistry and immunofluorescence
B16/F10 tumor bearing C57/BL6 mice treated with αPD-1 antibody and ABL therapy at days 4, 7 and 10 after tumor inoculation were sacrificed at day 15 after tumor inoculation. For immunohistochemistry studies tumors were first processed, embedded paraformaldehyde, fixed paraffin and antigen retrieval was performed for 30 min at 95 °C in 0,01 M Tris-1mM EDTA buffer (pH = 9) in a Pascal pressure chamber (Dako, Carpinteria, CA, USA).
Rabbit monoclonal α-CD3 antibody clone SP7, was used as primary antibody at dilution 1:300 (Thermo Fischer Scientific, Fremont, CA, USA). Rabbit monoclonal α-CD8a antibody clone D4W2Z was used as primary antibody at dilution 1:300 (Cell Signaling, Danvers, MA, USA). MACH 2 rabbit AP-polymer (Biocare Medicals, Pacheco, CA, USA) was used as detection system and further developed with Stay Red (Abcam, Cambridge, UK).
For B-infiltration analysis, tumor were extracted 10 days after the last treatment and rat monoclonal anti-mouse α-B220 antibody clone RA3-6B2 (BD Pharmingen, San Diego, CA, USA) was used as primary antibody at dilution 1:20,000. A rabbit α-rat antibody at dilution 1:200 (Vector Laboratories, Burlingame, CA, USA) was used as detection system followed by MACH 2 rabbit AP-Polymer to be further developed with Stay Red. Images were taken with Aperio CS2 (Leica, Wetzlar, Germany) using the program Aperio ImageScope (Leica, Wetzlar, Germany). Image quantification was performed using Fiji and the values were obtained as the percentage of area occupied by positive cells with respect to the tumor tissue area.
For immunofluorescence studies, tumor-embedded paraffin slices were stained with rabbit monoclonal α-CD8a antibody clone D4W2Z as a primary antibody at dilution 1:300 (Cell Signaling, Danvers, MA, USA). Donkey anti-rat antibody labeled with Alexa 488 (Thermo Fisher Scientific, Freemont, CA, USA ref A21208) was used as secondary antibody at 1:200 dilution. Anti-mouse α-B220 antibody clone RA3-6B2 (BD Pharmingen, San Diego, CA, USA) was used as primary antibody at dilution 1:20,000 and Donkey anti-rabbit Alexa Fluor 555 (Thermo Fisher Scientific, Freemont, CA, USA ref A31572) was used as a secondary antibody at 1:200 dilution. Fluorescent images were recorded at 40x with Axioimager M1 microscope with Zen software and Axiocam MRm camera (Zeiss, Oberkochen, Germany).
In vivo tumor model studies
Ethical approval for animal studies was granted by the Animal Ethical Committee of the University of Navarra (CEEA; Comité Etico de Experimentación Animal under the protocol number CEEA/069-13 for orthotopic intracranial metastasis model and CEEA/051-14 for the subcutaneous and orthotopic lung metastasis model. All animal studies were conducted in the veterinary facilities of the Center for Applied Medical Research (CIMA) in accordance with institutional, regional, and national laws and ethical guidelines for experimental animal care. For subcutaneous models, animals were monitored on a daily basis and were euthanized when tumors reached more than 15 mm of diameter. The animals were sacrificed with CO2 inhalation.
Subcutaneous colon carcinoma animal model
1.5 × 105 CT26 colon carcinoma cells were inoculated in the right flank of the animals were allocated into two different groups: those treated with 100 μl of PBS as negative control or treated with 8 × 106 ABLs in 100 μl of PBS. Treatments were intratumorally administered on days 10, 13 and 16 after tumor inoculation. Tumor volume was monitored every other day by caliber measurement and tumor volume was calculated as (large diameter × short diameter × short diameter)/2.
Subcutaneous skin melanoma animal model
1.5 × 105 B16/F10 skin melanoma cells were inoculated in the right flank of the animals and mice were stratified into four treatment groups: those treated with 100 μl of PBS as negative control, treated with 50 μg in 100 µl of InVivoMab antibody α-PD-1, with clone RMP1-14 (Bioxcell, West Lebanon, NH, USA), 8 × 106 ABLs in 200 µl, or with the combination of α-PD-1 and ABLs. α-PD-1 was systemically intraperitoneally administered and ABLs were administered intratumorally both: on days 4, 7 and 10 after tumor inoculation. Tumor volume was monitored by caliber measurement every other day.
Orthotopic, lung metastasis melanoma model
1.5 × 105 B16/F10 skin melanoma cells were inoculated in the right flank of the animals The same day 1.5 × 105 B16/F10 skin melanoma cells were intravenously administered to mimic lung metastasis. Mice were separated into three treatment groups: those treated with 100 μl of PBS as negative control, those treated with 50 μg per 100 µl of InVivoMab antibody α-PD-1, (clone RMP1-14 (Bioxcell, West Lebanon, NH, USA)), or with the combination of α-PD-1 and ABLs in 200 µl. α-PD-1 was systemically intraperitoneally administered and ABLs intratumorally, both on days 4, 7 and 10 after tumor implantation. Afterwards, when subcutaneous tumor reach 15 mm in diameter mice were euthanized and lungs were removed and weighed to assess the lung tumor load.
Orthotopic, brain metastasis melanoma model
1.5 × 105 murine B16/F10 skin melanoma cells were inoculated into the right flank of C57/BL6 mice (Harlan, Barcelona, Spain). Mice were allocated into three treatment groups as described above: treated with PBS as negative control, treated with α-PD-1, and treated with the combination of α-PD-1 and ABLs. At day 9 after tumor inoculation 5 × 104 B16 skin melanoma cells were engrafted into the caudate nucleus of C57/BL6 mice using a guide-screw system. On days 4, 7, and 10 after implantation of the subcutaneous tumor, animals were intratumorally treated on subcutaneous lesions with injections of 8 × 106 ABLs in 200 µl and 50 µg 100 µl of injected intraperitoneally. Animals were sacrificed when control tumor reached over 15 mm diameter. Brains were removed, fixed in 4% formalin, and embedded in paraffin.
CD8 lymphocyte depletion
C57/BL6 mice were injected i.p. at day 1, 4, 7, 14 after tumor implantation (B16/F10) with 100 µg per injection of monoclonal antibody anti CD8b 53-58 control group was treated with isotype antibody both purchased from Bio X Cell. CD8 lymphocyte depletion was confirmed in blood by flow cytometry at day 10.
Monitoring immune responses
IFN-γ ELISA
1 × 105 B16/F10 skin melanoma irradiated cells were cultured in a 96-well plate with 1 × 104 leukocytes extracted from draining lymph nodes of tumor-bearing mice at day 14 after the treatment with αPD-1 antibody and ABL therapy at day 4, 7, 10. They were incubated for 72 hours in 200 μl of ABL medium. The supernatant was collected to perform ELISA experiments. IFN-γ was measured by ELISA (BD Bioscience, San Diego, CA, USA) following manufacturer's instructions. ELISA was quantified by TECAN SunriseTM (TECAN, Männedorf, Switzerland) and analyzed with MagellanTM (TECAN, Männedorf, Switzerland).
IFN-γ ELISPOT
For the ELISPOT assay, leukocytes were extracted from draining lymph nodes of tumor-bearing mice at day 14 after the treatment with PD-1 antibody and ABL at day 4, 7 and 10. 4 × 105 leukocytes were cultured for 24 hours with 1 × 106 B16/F10 skin melanoma irradiated cells at 30 K rads. The experiment was performed following manufacturer's instructions. IFN-γ-expressing cells were quantified and analyzed with CTL ImmunoSpot® Analyzer (CTL-Europe GmbH, Bonn, Germany).
Statistics
One way ANOVA was used to assess the best treatment followed by post-hoc tests. Long-rank test on Kaplan meir curves was conducted for survival experiment. Data are presented as mean ± SEM using GraphPad Prism 5.
Discussion
In this study we are showing that B activated lymphocytes can be used in adoptive therapy to enhance anti tumor immunity as monotherapy in CT26 colon carcinoma model and in the context of PD-1 immune checkpoint blockade therapy in a variety of B16/F10 melanoma aggressive tumor models (subcutaneous, lung and brain orthotopic models). This is a simple and clinically feasible approach that could be conducted in most GMP facilities to elicit a systemic antitumor immune response by providing multiple costimulatory ligands in the tumor through intratumoral inoculation of ABLs.
The rate of B-cell infiltration in tumors has been underscored as a good prognostic marker in several types of cancer.Citation11,Citation15 However, its role in antitumor immunity has been under debate. On the one hand B-cell deficient mice (BCDM) show a reduction in tumor growth.Citation25-Citation26 On the other hand, mice that are purged of B cells via antibody mediated depletion increased tumor growth.Citation12,Citation27 These apparently contradictory results might indicate that B cells have a pro-tumorigenic effect in early stages of tumor development maybe hampering the induction of a robust antitumor immunity. Meanwhile, once the tumor is established and an antitumor immune response has been initiated, B cells in the tumor could be important players in the maintenance of immunity. Another possibility is that BCDM display other alterations in the ontogenesis of T lymphocytes that affect at the induction of tumor immunity.Citation28 One cytokine that is produced by activated B cells is IL-10, a molecule that has been highlighted as a potent immunosuppressive cytokine. Recent literature indicates that IL-10 might play the immunosuppressive role mostly during immunity induction (via vaccination) and not necessarily establishing an immunosuppressive tumor microenvimmet.Citation29 In fact, IL10 has been shown to have an important role in immune surveillance in the tumor milieu.Citation30 In this study we have not focused much on the role of IL-10. Hence, further studies should be carried out to address the role of IL-10 in this therapeutic strategy, and whether its blockade could enhance and further extend the therapeutic benefits of this novel approach.
We have basically focused on PD-L1/PD-1 blockade, which is highly expressed in B lymphoblasts together with other important costimulatory ligands such as CD80, CD86, CD40, ICOSL, etc. Herein we show that the blockade of PD-1 and the intratumoral injection of ABLs have a synergic antitumor effect.
APCs account for several types of cells (DC, BL, Monocytes, etc.) with BL not having gained popularity in cancer immunotherapy. Tumor-antigen loaded DCs have been extensively used as vaccine platforms and some promising results have also been documented with intratumoral injection of DCs or modified DCs to promote antitumor immunity.Citation31-Citation32 DCs are very efficient antigen-presenting cells. DCs specialize in uptake of antigens and, once activated, migrate to secondary lymphoid organs where they initiate a de novo immune response.Citation33-Citation35 Therefore, the persistence of activated DC in the tumor will likely be limited. On the other hand, activated B lymphocytes can persist and proliferate in the tumor milieu.Citation16 In light of the experiments performed in the present study, it is possible that activated B-lymphocytes are actually playing an important role in T cell recruitment as well as in potentiating and maintaining tumor immune responses, and not so much in the initial induction of de novo tumor immunity as attributed to DC.
We observed that ABLs could persist in the tumor microenvironment for more than five days after tumor injection, forming B-cell cluster structures (). It is hard to predict whether these structures will actually organize themselves into lymphoid ectopic tissue with formation of germinal centers as it occurs in some sets of cancer patients.Citation16 The generation of such organized ectopic tissue requires longer periods of time that are unavailable and limited by the aggressive tumor progression exhibited in these artificial mouse tumor models. Supporting this hypothesis we have observed that ABLs express higher levels of mRNAs encoding for two chemokines (CXCL11 and CCL21) previously underscored for recruiting activated T lymphocytes to the tumor milieuCitation18-Citation21 (Fig. S7). It is plausible that another major function of B lymphocytes infiltration is to provide costimulatory signals to infiltrating T tumor lymphocytes, promoting a higher expansion and a more robust antitumor immune response, but further studies will need to be conducted to confirm this possibility.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
supp_data.zip
Download Zip (48.5 MB)Additional information
Funding
References
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. PMID:22658127.
- Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–84. PMID:26228759.
- Melero I, Berman DM, Aznar MA, Korman AJ, Perez Gracia JL, Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer. 2015;15:457–72. PMID:26205340.
- Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–30. PMID:28102259.
- Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. 2013;25:214–21. PMID:23298609.
- Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, Mittler RS, Chen L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3:682–5. PMID:9176498.
- Linch SN, McNamara MJ, Redmond WL. OX40 Agonists and combination Immunotherapy: Putting the Pedal to the metal. Front Oncol. 2015;5:34. PMID:25763356.
- Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J Exp Med. 2014;211:715–25. PMID:24687957.
- Townsend SE, Allison JP. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science (New York, NY. 1993;259:368–70.
- Zamarin D, Holmgaard RB, Ricca J, Plitt T, Palese P, Sharma P, Merghoub T, Wolchok JD, Allison JP. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat Commun. 2017;8:14340. PMID:28194010.
- Iglesia MD, Vincent BG, Parker JS, Hoadley KA, Carey LA, Perou CM, Serody JS. Prognostic B-cell signatures using mRNA-seq in patients with subtype-specific breast and ovarian cancer. Clin Cancer Res. 2014;20:3818–29. PMID:24916698.
- Garnelo M, Tan A, Her Z, Yeong J, Lim CJ, Chen J, et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut. 2017;66:342–51. PMID:26669617.
- Sakimura C, Tanaka H, Okuno T, Hiramatsu S, Muguruma K, Hirakawa K, Wanibuchi H, Ohira M. B cells in tertiary lymphoid structures are associated with favorable prognosis in gastric cancer. J Surg Res. 2017;215:74–82. PMID:28688665.
- Meshcheryakova A, Tamandl D, Bajna E, Stift J, Mittlboeck M, Svoboda M, Heiden D, Stremitzer S, Jensen-Jarolim E, Grünberger T. B cells and ectopic follicular structures: novel players in anti-tumor programming with prognostic power for patients with metastatic colorectal cancer. PLoS One. 2014;9:e99008. PMID:24905750.
- Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, Rabbe N, Laurans L, Tartour E, de Chaisemartin L. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol. 2008;26:4410–7. PMID:18802153.
- Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, Lepelley A, Becht E, Katsahian S, Bizouard G. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med. 2014;189:832–44. PMID:24484236.
- Nielsen JS, Sahota RA, Milne K, Kost SE, Nesslinger NJ, Watson PH, Nelson BH. CD20+ tumor-infiltrating lymphocytes have an atypical CD27- memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin Cancer Res. 2012;18:3281–92. PMID:22553348.
- Hensbergen PJ, Wijnands PG, Schreurs MW, Scheper RJ, Willemze R, Tensen CP. The CXCR3 targeting chemokine CXCL11 has potent antitumor activity in vivo involving attraction of CD8+ T lymphocytes but not inhibition of angiogenesis. J Immunother. 2005;28:343–51. PMID:16000952.
- Moon EK, Wang L-CS, Bekdache K, Lynn RC, Lo A, Thorne SH, et al. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology. 2018;7:e1395997. PMID:29399394.
- Lin Y, Sharma S, John MS. CCL21 Cancer immunotherapy. Cancers (Basel). 2014;6:1098–110. PMID:24810425.
- Kar UK, Srivastava MK, Andersson A, Baratelli F, Huang M, Kickhoefer VA, Dubinett SM, Rome LH, Sharma S. Novel CCL21-vault nanocapsule intratumoral delivery inhibits lung cancer growth. PLoS One. 2011;6:e18758. PMID:21559281.
- Coffman RL, Weissman IL. B220: a B cell-specific member of th T200 glycoprotein family. Nature. 1981;289:681–3. PMID:6970340.
- Sandru A, Voinea S, Panaitescu E, Blidaru A. Survival rates of patients with metastatic malignant melanoma. J Med Life. 2014;7:572–6. PMID:25713625.
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–702. PMID:1460426.
- Shah S, Divekar AA, Hilchey SP, Cho HM, Newman CL, Shin SU, et al. Increased rejection of primary tumors in mice lacking B cells: inhibition of anti-tumor CTL and TH1 cytokine responses by B cells. Int J Cancer. 2005;117:574–86. PMID:15912532.
- Zhang Y, Eliav Y, Shin SU, Schreiber TH, Podack ER, Tadmor T, Rosenblatt JD. B lymphocyte inhibition of anti-tumor response depends on expansion of Treg but is independent of B-cell IL-10 secretion. Cancer Immunol Immunother. 2013;62:87–99. PMID:22772949.
- DiLillo DJ, Yanaba K, Tedder TF. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol. 2010;184:4006–16. PMID:20194720.
- Yang X, Brunham RC. Gene knockout B cell-deficient mice demonstrate that B cells play an important role in the initiation of T cell responses to Chlamydia trachomatis (mouse pneumonitis) lung infection. J Immunol. 1998;161:1439–46. PMID:9686609.
- Llopiz D, Aranda F, Diaz-Valdes N, Ruiz M, Infante S, Belsue V, et al. Vaccine-induced but not tumor-derived Interleukin-10 dictates the efficacy of Interleukin-10 blockade in therapeutic vaccination. Oncoimmunology. 2016;5:e1075113. PMID:27057445.
- Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, Blaisdell S, Basham B, Dai J, Grein J. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell. 2011;20:781–96. PMID:22172723.
- Saji H, Song W, Furumoto K, Kato H, Engleman EG. Systemic antitumor effect of intratumoral injection of dendritic cells in combination with local photodynamic therapy. Clin Cancer Res. 2006;12:2568–74. PMID:16638867.
- Melero I, Duarte M, Ruiz J, Sangro B, Galofre J, Mazzolini G, Bustos M, Qian C, Prieto J. Intratumoral injection of bone-marrow derived dendritic cells engineered to produce interleukin-12 induces complete regression of established murine transplantable colon adenocarcinomas. Gene Ther. 1999;6:1779–84. PMID:10516729.
- Grayson MH, Ramos MS, Rohlfing MM, Kitchens R, Wang HD, Gould A, Agapov E, Holtzman MJ. Controls for lung dendritic cell maturation and migration during respiratory viral infection. J Immunol. 2007;179:1438–48. PMID:17641009.
- Lappin MB, Weiss JM, Delattre V, Mai B, Dittmar H, Maier C, Manke K, Grabbe S, Martin S, Simon JC. Analysis of mouse dendritic cell migration in vivo upon subcutaneous and intravenous injection. Immunology. 1999;98:181–8. PMID:10540216.
- Tomura M, Hata A, Matsuoka S, Shand FH, Nakanishi Y, Ikebuchi R, Ueha S, Tsutsui H, Inaba K, Matsushima K. Tracking and quantification of dendritic cell migration and antigen trafficking between the skin and lymph nodes. Sci Rep. 2014;4:6030. PMID:25112380.