
ABSTRACT
Adoptive cell therapy (ACT) using in vitro expanded tumor infiltrating T lymphocytes (TILs) from biopsy material represents a highly promising treatment of disseminated cancer. A crucial prerequisite for successful ACT is sufficient recruitment of transferred lymphocytes to the tumor site; however, despite infusion of billions of lymphocytes, T cell infiltration into the tumor post ACT is limited. By PCR and Luminex analyses we found that a majority of malignant melanoma (MM) cell lines expressed chemokines CXCL1/Groα, CXCL8/IL-8, CXCL12/SDF-1 and CCL2. Concerning expression of the corresponding receptors on T cells, only the IL-8 receptor, CXCR2, was not expressed on T cells. CXCR2 was therefore expressed in T cells by lentiviral transduction, and shown to lead to ligand specific transwell migration of engineered T cells, as well as increased migration towards MM conditioned medium. In vivo homing was assessed in a xenograft NOG mouse model. Mice with subcutaneous human melanoma were treated with MAGE-A3 specific T cells transduced with either CXCR2 or MOCK. Transducing T cells carrying the MAGE-A3a3a high affinity T cell receptor with CXCR2 increased tumor infiltration. Flow cytometry analysis 7 days after ACT showed a doubling in CD3+ T cells in tumor digest of mice receiving CXCR2 transduced T cells compared to MOCK treated mice, a finding confirmed by immunohistochemistry. In conclusion, our CXCR2 transduced T cells are functional in vitro and transduction with CXCR2 increases in vivo homing of T cells to tumor site.
Introduction
Within the last decade, treatment of metastatic melanoma (MM) has been revolutionized by a wide array of immunotherapies including checkpoint inhibitors against CTLA-4Citation1 and PD-1/PD-L1Citation2,Citation3 as well as adoptive cell therapy (ACT).Citation4-Citation6 However, despite great advances, treatment responses are restricted to a fraction of patients.
While tumor-specific T cells are readily found in the blood of patients with MM, adequate homing of tumor-reactive T cells to tumor site be it either spontaneous elicited or administered by ACT, is a prerequisite for immune mediated tumor control. To this end, presence of tumor infiltrating lymphocytes (TILs) has been shown to correlate with better prognosis of several solid cancers including MM,Citation7 colon carcinoma,Citation8 ovarian cancerCitation9 and breast cancer.Citation10 This is supported by accumulating data linking response to immune therapy with a brisk infiltration of T cells in the tumor.Citation7,Citation11,Citation12
ACT using in vitro expanded TILs from tumor biopsies has been a great success in the treatment of melanoma. Contrary to ACT using chimeric antigen receptors (CAR) or T cell receptor (TCR) engineered T cells targeting a single antigen, TILs represent a treatment modality which targets a wide array of tumor antigens, including patient specific neo-antigens.Citation13 However, only a small fraction of in vitro expanded TILs migrate to the tumor site after i.v. administration.Citation14,Citation15 This limitation may be a result of sub-optimal expression of appropriate chemokine receptors after in vitro expansion.
Immune cell localization and migration is orchestrated by a redundant system of chemokines and chemokine receptors. In homeostasis, CCR7 expression by T cells induces cycling to lymph nodes and migration through high endothelial venules (HEVs) via CCL19 and CCL21 in the search for cognate antigen. Upon recognition of cognate antigen and activation by dendritic cells in the lymph nodes (LNs), T cells downregulate CCR7 and upregulate a set of chemokine receptors, determined by the type of stimulus and anatomical site of the LNs. Among the common pro-inflammatory chemokine receptors are CCR5 and CXCR3, whereas site specific chemokine receptors such as CCR9 or CCR10 are upregulated on T cells primed in the mesenteric or skin draining LNs for subsequent homing to either gut or skin respectively.Citation16,Citation17
Data from recent studies have linked the presence of TILs to a pro-inflammatory chemokine profile in MM lesions. Among these chemokines, CCL2, CCL3, CCL4, CCL5, CXCL9, and CXCL10 were preferentially expressed in lesions characterized by immune cell infiltration, including T cells.Citation18 However other chemokines are expressed in the tumor microenvironment of melanoma, such as CXCL8/IL-8, CXCL12/SDF1,Citation18 and CCL22,Citation19 which have been associated with promoting tumor growth through induction of angiogenesis, metastasis and recruitment of immune regulatory cell subsets, such as myeloid derived suppressor cells (MDSC) and regulatory T cells (Tregs).Citation19,Citation20
To identify the homing potential of in vitro expanded TILs, we analyzed the expression of 11 selected chemokines from 20 MM cell lines by PCR and multiplex chemokine analyses, and the expression of corresponding chemokine receptors on TILs from 10 MM lymph node metastases. We hypothesize that matching the chemokine receptor expression on TILs to the tumor microenvironment by genetic engineering, will improve homing of TILs to tumor site and ultimately clinical response to ACT.
Results
Characterization of chemokine/chemokine receptor profile in metastatic melanoma
To identify the chemokine/chemokine receptor axes present in metastatic melanoma (MM), we analyzed the mRNA expression of select chemokines CCL2/MCP-1, CXCL2/IL-8 and CXCL12/SDF-1 and found that the majority of metastatic melanoma (MM) was positive by standard PCR (). To assess the level of expression and secretion of these, we analysed the supernatants for CCL2, CXCL8, CXCL12 and additionally 8 other chemokines (CCL5, CCL17, CCL22, CXCL1, CXCL9, CXCL10, CXCL11, and CXCL16) by Bio-Plex™ analysis 48 h after seeding 5 × 104 cells in 2 mL wells (). We found high levels of CXCR2 ligands CXCL1/Groα and CXCL8/IL-8 across all tested cell lines (median 312.7 range 19.9–2409.1 and median 160.8 range 14.4–2288.3, respectively). However, CCL2 and CXCL12 were present at low (median 10.4, range 0.1–3401.5 pg/mL) and very low concentrations (median 17.3, range 7.0–27.4 pg/mL), respectively, despite being secreted from the majority of the tested cell lines. We found little or no expression of CCL5, CCL17, CCL22, CXCL9, CXCL10, CXCL11 and CXCL16.
Figure 1. Chemokine profile of MM, and corresponding chemokine receptor expression on MM tumor infiltrating T cells. Initial analyses of mRNA expression in 20 melanoma cell lines of 3 select chemokines CCL2, CXCL8/IL-8 and CXCL12/SDF-1 by standard PCR (A), and subsequent luminex analyses of 11 select chemokines secreted into the supernatant of 22 MM cell lines after 48 h (B). Expression of corresponding chemokine receptors on CD4+ and CD8+ TIL either ex vivo, in young or REPd cultures analysed by 8-color flow cytometry (C). MM = Melanoma cell lines, n = 20 (PCR), 22 (Luminex); TIL = Tumor infiltrating lymphocytes, n = 10 Green = CCL2/CCR2; Blue = CXCL8/CXCR2; Red = CXCL12/CXCR4. Horizontal bars designate mean, error bars designate standard error of the mean.
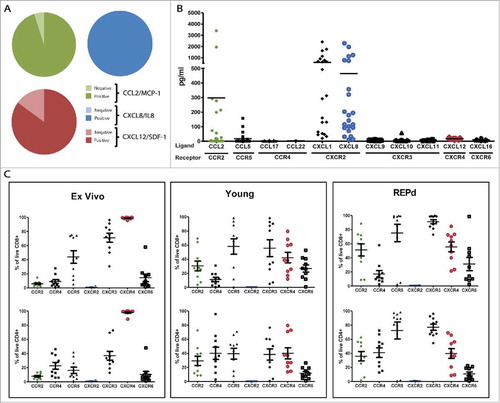
We examined the expression of corresponding chemokine receptors on TILs from MM biopsies either directly ex vivo, after initial expansion (Young TIL) or rapid expansion (REPd TIL, corresponding to the infusion product used for ACT of MM at our departmentCitation25) (gating strategy, suppl. Fig. 1). We found that TILs generally expressed an activated chemokine receptor patternCitation17 including high frequencies of CCR5+, CXCR3+ and CXCR4+ (). CCR5 and CXCR3 expression were augmented by in vitro activation and expansion, increasing expression from approx. 43.9%-75.2% (P = 0.05) and 71.1%-91.2% (P = 0.009), respectively, on REPd CD8+ TILs (, suppl. Fig. 2). Other chemokine receptors were also upregulated upon activation and expansion of TILs, among others CCR2 (from 5.6–51.18% P<0.001), CCR4 (from 22.6% to 41.0% on CD4+ T cells only, P < 0.05) and CXCR6 (14.3% to 31.2%, P = ns). In contrast, CXCR4 expression decreased from 98.9% to 43.4% (P < 0.0001) expression. Chemokine receptor expression patterns on CD8+ and CD4+ T cells were similar, though chemokine receptors were generally expressed at lower frequencies on CD4+ T cells (). Among the tested chemokine receptors, CXCR2 was not expressed on TILs neither ex vivo, in the young TIL nor REPd TIL cultures. Data from is re-presented in suppl. Fig. 2 to highlight changes in expression of individual receptors on CD4+ and CD8+ T cells including statistics.
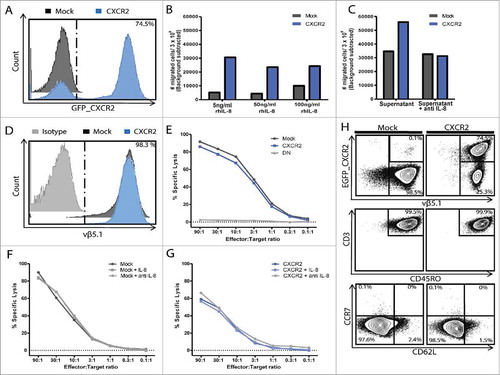
Figure 2. In vitro functionality of T cells transduced with MAGE-A3a3a and CXCR2. Purity of transduced T cell cultures after sorting and in vitro expansion was assessed by flow cytometry (A+D). Transwell® migration of CXCR2 or Mock transduced T cells towards 0.5 ng/ml, 1 ng/ml, 5 ng/ml, 50 ng/ml and 100 ng/ml rh-IL8 (B), or MM supernatant (FM82) with or without 2 μg/ml neutralizing anti-IL-8 (C). Functional ability of tumor killing of MM cell line FM82 were assessed by 51Cr-release cytotoxicity assay, using decreasing effector:target ratios (from 90:1 to 0.1:1). (E) Light grey = untransduced control T cells, Dark grey = Mock transduced T cells; Blue = CXCR2 transduced T cells, DN = Double negative (untransduced). Effect of IL-8 signalling on tumor killing of FM82 was assessed by individual 51Cr-release of Mock (F) or CXCR2 (G) transduced T cells in combination with FM82 alone, FM82 + 5 ng/ml IL-8 or FM82 + 2 μg/ml anti-IL8. Effector phenotype of CXCR2 and Mock transduced T cells gating on EGFP_CXCR2 /vβ5.1 (MAGE-A3a3a TCR), CD3 /CD45RO, CD62 L /CCR7 (H).
CXCR2 transduced T cells gain in vitro migration towards IL-8 while retaining their tumor killing potential
In order to assess the effect of CXCR2 transduction on migration, tumor recognition and -killing, we generated a tumor specific T cell culture by lentiviral transduction with a high affinity MAGE-A3a3a TCR.Citation24 MAGE-A3a3a TCR T cells were subsequently transduced with CXCR2, and sorted by combined tetramer and antibody staining into a MAGE-A3a3a TCR+ CXCR2_GFPpos or MAGE-A3a3a TCR+ CXCR2_GFPneg fraction (from here designated “CXCR2” and “Mock”) (suppl. Fig. 1). Purity of the cultures was assessed by flow cytometry. Approximately 75–90% of the CXCR2 culture expressed CXCR2 compared to 0.1% of the Mock (). We have previously shown that IL-8 binding of the CXCR2 receptor (transcript variant 1) mediates ligand specific signal transduction in genetically engineered MM TILs using a Ca2+-influx assay.Citation23
Migration of CXCR2 transduced T cells towards rh-IL8 or MM supernatant was assessed in a transwell® system. CXCR2 transduction of T cells specifically induced migration towards as little as 0.5 ng/mL rh-IL8 compared to mock transduced T cells, though a level of 5 ng/mL rh-IL8 was needed to induce maximal migration as this migration could not be increased further by increasing the concentrations of rh-IL8 (50 and 100 ng/mL respectively) in the bottom well ().
Not only did rhIL-8 induce migration of CXCR2 transduced T cells, CXCR2 transduction almost doubled the migration of T cells towards supernatant from a MM cell line FM82. Addition of IL-8 neutralizing mAb completely abolished the CXCR2 induced migration towards MM supernatant ().
To evaluate whether transduction with and signaling through CXCR2 in T cells affected the T cell reactivity, killing of tumor cell line FM82 was assessed in standard 4 h 51Cr-release assays. Expression of MAGE-A3a3a high affinity TCR was comparable on CXCR2 and Mock transduced T cells, as well was the killing of FM82 (), suggesting that CXCR2 transduction alone does not affect the recognition and killing of tumor cells. To test whether signaling through the CXCR2 might affect tumor killing, killing was assessed in the presence of FM82 cells alone; FM82 cells + 5 ng/mL rh-IL8; or FM82 + 2 ug/mL neutralizing mAb to IL-8. Tumor killing by CXCR2 and Mock transduced T cells was unaffected by the addition or blocking of IL-8 (). Both CXCR2 and Mock transduced T cells were primarily of a CD45RO+ CD62 L− CCR7− effector memory phenotype ().
CXCR2 transduction improves homing of T cells to tumor in vivo
To assess whether CXCR2 transduction was sufficient to improve in vivo homing of T cells to the tumor in vivo, immune deficient NOG mice, were inoculated with human MM FM82 tumors (30mm2) s.c. on the flank. FM82 secreted intermediate (but not high) levels of chemokine IL-8, and high levels of CXCL1/Gro-α (see and suppl. table 1), the ligands for CXCR2, and thus seemed a good candidate for attracting CXCR2 transduced T cells in vivo. In addition, FM82 express the HLA-A1, A2 molecules, as well as MAGE-A3 cancer testis antigen, allowing us to work with the HLA-A1 restricted high-affinity MAGE-A3a3a TCR.
Animals were treated with ACT of Mock or CXCR2 transduced MAGE-A3a3a TCR, while a control group received iv. PBS. Tumors were excised 7 days post ACT, which to allow for the time dependent accumulation at tumor site (optimal between 5–7 days post ACT, data not shown) and tumor infiltration was assessed by flow cytometry and IHC analyses. Flow cytometry of tumor single cell suspensions revealed that CXCR2 transduction of T cells doubled the fraction of CD3+ T cells in the tumor (n = 11, mean 2.8 ± 1.3%) compared to mice treated with Mock transduced T cells (n = 13, 1.3 ± 0.5%, p = 0.0018) (, representative gating, suppl. Fig. 3). No difference in T cell frequencies was observed among circulating T cells (blood and spleen), whereas differential localization to lung and bone marrow favoring CXCR2 or mock T cells respectively (suppl. Fig. 4). Flow data presented in and suppl. Fig. 2 represents data from 2 independent experiments, each individually significant (Experiment 1: group sizes of n = 5 mice pr. Group, P = 0.036. Experiment 2: n = 10 mice pr. group, 2 mock and 4 CXCR2 mice excluded by acquisition threshold, P = 0.014). To adjust for the pooling of data, a P-value of <0.025 was required for the data to be considered significant (P = 0.0018).
Figure 3. In vivo homing of CXCR2 transduced T cells. CXCR2 expression of ACT infusion product was assessed by flow cytometry after sorting and expansion (A). Representative tumor growth curve (tumor surface mm2) measured by calipers. Red arrow indicated initiation of treatment (B). Frequencies of tumor infiltrating T cells were measured by flow cytometry in (C) or IHC (D) 7 days after ACT. Enumeration of tumor infiltration was performed by counting CD3+ 10 HPF at 600x magnification IHC (D-E). Circles in Fig. E highlight the representative IHC sections of PBS, CXCR2 or Mock T cell treated tumors depicted in D. Flow data (C) represents data from two independent experiments, each separately significant (P = 0.036 and P = 0.014), and together P = 0.0018. Light grey = PBS treated control mice, Dark grey = Mock transduced T cell treated, n = 13; Blue = CXCR2 transduced T cell treated, n = 11. Horizontal bars designate mean, and error bars the standard error of the mean. Unpaired t-test, *P < 0.05; **P < 0.01.
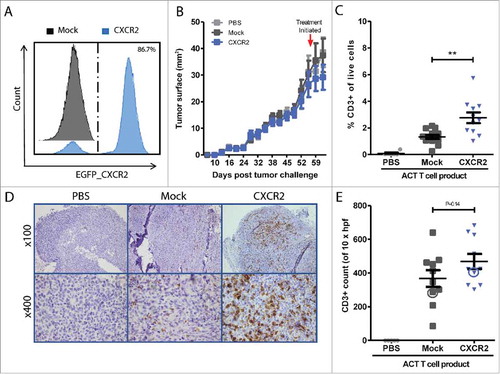
Immunohistochemistry of formalin-fixed paraffin-embedded (FFPE) sections of tumors (Experiment 2: n = 10 Mock, n = 10 CXCR2, n = 6 PBS treated) show that CD3+ T cells infiltrated deep into the tumors, and not just the tumor periphery (, suppl. Fig. 4). Quantification of T cell infiltration into tumors by counting CD3+ cells pr. 10 HPF (magnification x600), though not significant, show a similar trend, that CXCR2 transduction of T cells improves homing to the tumors in xenograft MM NOG mouse model ().
Discussion
Despite the successful application of immune therapy representing a breakthrough in the treatment of disseminated melanoma, more than half of the patients still fail to respond to therapy, or relapse upon initial response. A limiting factor in immune therapy is the insufficient homing of T cells to the tumor site. Many of the new immune therapies either require the presence of pre-existing T cells in the tumor,Citation12,Citation26,Citation27 or rely on sufficient tumor homing of tumor specific T cells after peripheral or in vitro activation.Citation14,Citation28
Contrary to the “pro-inflammatory” chemokines, found in a subset of patients with pre-existing TIL infiltration,Citation18 chemokines responsible for tumor progression through induction of angiogenesis, metastasis and recruitment of immune suppressive cell subsets such as CXCL8/IL8, CXCL12/SDF-1 and CCL22 are widely expressed in the tumor microenvironment.Citation18,Citation19
We hypothesized that equipping T cells with chemokine receptors matching the chemokines in the microenvironment of metastatic melanoma, could improve homing to the tumor site.
By PCR and luminex analyses we found, that metastatic melanoma cell lines expressed high levels of CXCR2 ligand CXCL1 and CXCL8/IL8, intermediate expression of CCL2 and low expression of CXCL12/SDF-1. The remaining 7 chemokines analyzed by luminex (CCL5, CCL17, CCL22, CXCL9, CXCL10, CXCL11 and CXCL16) were found at negligible levels after 48 h. This is most likely due to secretion of cytokines, growth factors and chemokines by stromal cells in the tumor microenvironment, which are not represented in our luminex assay, rather than by cancer cells themselves. This is in line with findings by others, that while IHC sections or RT-PCR analyses of tumor biopsies show a wide expression of these chemokine,Citation18,Citation29,Citation30 the majority of melanoma cell lines only secretes a limited variety of chemokines, among which CXCL8/IL8 are consistently secreted at high levels.Citation18,Citation30 It does however limit our ability to test a combination of chemokine receptors beyond CXCR2, by ACT of xenograft tumors, as the model would lack the cognate chemokine ligands.
Chemokine receptor expression on TILs resembled a pro-inflammatory pattern, including expression of CCR5 and CXCR3 expression on the majority of TILs; an expression that increased to 80–90% with in vitro activation and expansion. Expression of CCR5 and CXCR3 are important for appropriate recruitment and migration of T cells to sites of inflammation, and have recently been shown to correlate with likelihood and degree of response to ACT in patients with metastatic melanoma.Citation29 Mouse models investigating the mode of action suggests that CXCR3 is crucial for the firm adhesion of rolling T cells and transmigration across the vascular endothelium.Citation31 As both CCR5 and CXCR3 are highly expressed on in vitro activated and expanded TILs, trying to augment expression of these in a therapeutic setting is of little relevance. One might instead speculate whether it is possible to boost tumor secretion of the respective ligands, e.g. increase secretion of CXCR3 ligands, CXCL9, CXCL10 and CXCL11 via IFNγ stimulation.Citation32
CCR2 expression increased, while CXCR4 decreased to approx. 60% with expansion. Despite little secretion of the corresponding ligands (CCL2 and CXCL12/SDF-1 respectively) by melanoma cell lines, these chemokines are widely secreted by stromal cells in the tumor microenvironment of melanomas.Citation18,Citation33 Thus, efforts to augment expression of CCR2 and CXCR4 on TILs could be a strategy to increase the tumor specific homing of TILs in a setting of ACT.
Common for all TIL cultures, i.e. ex vivo, young and REP, was the lack of CXCR2 expression despite significant secretion of CXCR2 ligands CXCL1 and CXCL8/IL-8 by all tested melanoma cell lines. CXCR2 is thus an ideal model candidate to evaluate improved homing of T cells to the tumor site, as there is no endogenous expression of the receptor to muddle the readout. Others have pursued CXCR2 as a candidate for improving homing of T cells after ACT. Kershaw and colleagues showed that CXCR2 transduction of human TILs was feasible several years ago, improving in vitro homing while retaining functionality.Citation34 A strategy the same group later pursued in a genetically modified B16 mouse model of homing of T cells, engineered to express murine CXCR2, towards murine CXCL1.Citation35 We are the first to show, that transduction with human CXCR2 significantly increases homing and infiltration of human T cells into s.c. human tumors in a xenograft model of melanoma.
Our data showed that as little as 0.5 ng/mL rhIL-8 could increase the migration of CXCR2 transduced T cells, and thus IL-8 seems to be a very potent inducer of migration. In addition, we found that CXCR2 transduced T cells migrated maximally at 5 ng/mL rhIL-8, corresponding well with ED50 of 0.5–2.5 ng/mL supplied by the manufacturer. Though migration of CXCR2 transduced T cells seems to drop at higher concentrations, this may simply reflect that concentrations above 5 ng/ml saturate the CXCR2 receptors on the T cells, or even abolish the gradient by diffusing into the upper well faster than the T cells can migrate.
Despite observing a doubling in the frequencies of T cells infiltrating into the tumors in vivo, the T cell homing is rather modest. The FM82 cell line used to establish s.c. tumors in this study was not among the highest CXCR2-ligand producers of the 20 tested cell lines, thus one might speculate, that the levels secreted were insufficient to induce a brisk tumor infiltration. However, in light of the very low IL-8 levels required to increase migration in vitro, it is more likely due to xenograft model specific limitations. While IL-8 may induce recruitment of T cells, T cells' extravasation and migration into peripheral tissues rely on selectin and integrin binding in concert with chemokine signaling to mediate rolling and firm adhesion to the endothelial wall.Citation17,Citation36 Our xenograft model relies on stromal chemokines secreted by mouse endothelial cells and fibroblasts, and despite some sequence similarity and evolutionary conserved regions, there is a greater disparity, and these may not necessarily support T cell adhesion, extravasation and tumor infiltration.Citation37 In line with this, HEVs in human melanoma lesions have been shown to be major gateways for tumor infiltrating lymphocytes,Citation38 however lack of host immune cells in the NOG mouse model, and thus no orchestrating chemokines and cytokines, HEVs are most likely not produced in this model. One could thus speculate that human-specific adhesion molecules may need to be expressed for optimal human immune cell migration and structural organization in the mouse model.Citation39
Conclusion and future perspectives
Despite these obvious limitations in the in vivo xenograft model, our pre-clinical data, in concert with others, supports the feasibility of chemokine receptor engineering of tumor specific cells as a strategy to improve homing to tumor site. CCR2,Citation40 CXCR1,Citation30 CXCR2Citation34,Citation35 and CXCR3Citation41 transduction of T and NK cells have been pursued in pre-clinical models, showing increased homing to tumor site, and as a result improved tumor control, setting the stage for human trials. A phase I/II study of lymphodepletion plus ACT with CXCR2 and Nerve Growth Factor Receptor (NGFR) transduced TILs are currently being investigated in patients with MM (NCT01740557).
In addition to CXCR2, we identified chemokine receptors CCR2 and CXCR4 as potential candidates for chemokine receptor engineering. We propose that mixing and matching the expression of chemokine receptors on TILs to that of the patient specific chemokine profile may further improve tumor homing and ultimately response to therapy with ACT. Improvements to our in vivo xenograft model are needed before increased homing of mixed and matched chemokine receptor engineered T cells can be verified.
Materials and methods
Patient material/samples
Biopsies from patients with stage III or IV melanoma (MM) was collected from 2006–2013. Biopsy material was cut into small fragments (1–2 mm2) and kept overnight (ON) in a humidified 37° CO2 incubator. The following day, tumor fragments and cells were resuspended, washed twice in 1xPBS (Lonza) by centrifugation for 5 minutes at 1500 RPM (from here referred to as wash) and cryopreserved in fetal bovine serum (FBS, Gibco) + 10% DMSO (Sigma Aldrich). All biopsies originated from LN metastases removed under palliative surgery.
Peripheral blood mononuclear cells (PBMC) from buffycoats were isolated from healthy donors provided by the blood bank at Copenhagen State University Hospital (Rigshospitalet), Denmark. In short, buffycoats were diluted in PBS and PBMC were isolated by density gradient centrifugation on a layer of Lymphoprep™ (Fresenius Krabi) for 30 minutes at 1200 RPM without breaks. The isolated PBMC were cryopreserved in FBS + 10% DMSO (Sigma Aldrich).
Collection and use of melanoma biopsies and healthy donor PBMC were approved by the Danish Ethics Committee of Human Experimentation and informed consent was obtained from all patients and healthy donors before inclusion.
Cell lines
Malignant melanoma cell lines (25 cell lines, originally established from melanoma biopsies at the Danish Cancer Society, of which 22 are available from ESTDAB, see suppl. table 1) were cultured in RPMI 1640 (Gibco) + 10% FBS (from here referred to as R10). Cells were pelleted (3 × 106 cells/vial) and kept at −20°C until mRNA isolation. Standard PCR was run using TaqMan polymerase (Thermo Fischer Scientific), 40 cycles. CCL2 primers CCL2_217 cgcgagctatagaagaatcacc and CCL2_408 gggaaagctaggggaaaataag(213 bases), CXCL8 primers IL-8_219 tactccaaacctttccaccc and IL8_354 ttctccacaaccctctgca (155 bases), and CXCL12_169 tgagctacagatgcccatgc CXCL12_462 cacacacacctggtcctcatg (314 bases).
For isolation of cell line supernatant, 5 × 104 cancer cells/well were seeded in a 24-well plate containing 2 mL R10 medium. Supernatant was harvested 48 h later. Cellular debris was removed by centrifugation (5 minutes at 1500 RPM) and the supernatant aliquoted and kept at −20°C until use.
Luminex
Supernatant from 22 MM cell lines were analyzed for CCL2/MCP-1, CCL17, CCL22, CXCL1/Gro-α, CXCL8/IL-8, CXCL9, CXCL10, CXCL11 and CXCL12/SDF-1 using a multiplex Bio-Plex Pro™ Human Chemokine assay, and CCL5/RANTES CCL5 Bio-Plex Pro Human Cytokine Assay (Bio-Rad). Samples were prepared according to manufacturer's recommendations. Samples were acquired on a Bio-Plex 200 system and analyzed with Bio-Plex Manager™ v.6 software.
TIL isolation protocol and rapid expansion
TILs were expanded from biopsy material using 6000 U/ml rh-IL2 (Proleukine, Novartis). In short, tumor fragment suspensions were thawed in 37°C pre-warmed RPMI 1640, and washed twice by centrifugation. Dead cells were removed from tumor fragment suspensions with poor viability using Dead Cell Removal Kit (Miltenyi Biotech). 2 × 105 cells from each tumor suspension was analyzed by flow cytometry prior to culturing with IL-2 (“ex vivo” TIL). A cell concentration of 1 × 106 cells/mL were plated in 2 mL or 200 uL in a 24 well plate or 96 well plate, respectively, in 37°C pre-warmed culture medium (X-vivo (Lonza), 5% Human AB serum (HS, Sigma Aldrich)) + 6000U/ml rh-IL2 (Proleukine, Novartis). Cells were left for the initial 5 days in a 37°C humidified CO2 incubator, after which, culture medium was changed every 2–3 days by replacing half the culture medium with fresh IL-2 containing medium, without resuspending the cells. When T cells reached confluence, the cells were split 1:2 into adjacent wells. When cell counts of >50 × 106 T cells were reached (10 cultures, expansion time-range; 22–65 days in culture), from here designated “Young” TILs.Citation21 Flow cytometry was performed on 2 × 105 cells from each Young TIL culture. The remaining Young TILs were cryopreserved according to protocol described above.
TIL cultures were expanded in a rapid expansion protocol (REP) adapted from Dudley et al,Citation22 and described previously.Citation23 In short, TILs were co-cultured with irradiated (40 Gy) feeder cells at a 1:200 ratio in culture medium + 30 ng/mL anti-CD3 (OKT-3, eBioscience) and 6000 U/mL rhIL-2. REP cultures were incubated undisturbed for an initial 5 days after which, half of the cell culture medium was changed every 2–3 days. On day 14 of REP, the cells were harvested and 2 × 105 cells of each culture were analyzed by flow cytometry. Remaining cells were cryopreserved in FBS + 10% DMSO, in a gradual freezing device, and kept at −150°C until use.
Lentiviral transduction
T cell cultures were established from PBMC of healthy donor BC259 by plating 0.5 × 106 cells/mL in 2 mL culture medium + 1000U/mL rhIL-2 in a 24-well plate. Medium was replaced every 2–3 days, and cells were divided into neighboring wells when reaching confluence. At day 10 in culture, 1 × 106 T cells were used for lentiviral transduction. Remaining T cells were phenotyped by flow cytometry analysis and cryopreserved according to the protocol above.
cDNA encoding CXCR2 (accession nr. NM_001557) was synthesized and cloned into the lentiviral vector pTRP-EGFP (generously provided along with packaging and envelope plasmids (pTRP-RSV.Rev, pTRP-GAG-Pol and pTRP-VSVg) by Dr. James L. Riley, University of Pennsylvania, Philadelphia, PA) using 5`AvrII/3’SalI restriction sites (GeneArt/Thermo Fisher Scientific, Regensburg, Germany) generating pTRP-EGFP_CXCR2. This vector permits dual expression of EGFP and CXCR2 from a single RNA transcript, separated by the self-cleaving T2 A linker sequence.
Lentiviral vector containing high affinity MAGE-A3a3a TCRCitation24 and corresponding packaging and envelope plasmids (VSVG, REV and gag/pol) was generously provided under MTA.
Lentivirus was produced after transfection of 293 T human embryonic kidney cells cultured in DMEM (BioWhittaker, Rockville MD, USA), 10% FBS. 293 T cells were seeded at 5 × 105 per well in a 6-well plate 24 h before transfection. For transfection, 1 µg of pMAGE-A3a3a or pTRP-EGFP_CXCR2 and 0.5 µg of corresponding packaging and envelope plasmids were used together with TurboFect Transfection Reagent (Thermo Fisher Scientific). Cells were cultured at humidified atmosphere with 5% CO2 for 48 hours before harvesting the viral supernatant. For transduction of T cells, cells were incubated with lentivirus filtered supernatant + 1000U/mL rhIL-2 for 72 hours. MAGE-A3a3a transduction was immediately followed by reseeding of unsorted MAGE-A3a3a TCR transduced cells for subsequent transduction with GFP_CXCR2. T cells were sorted using a FACSAria cell sorter (BD Biosciences, San Jose CA, USA), sorting the EGFP_CXCR2pos (designated “CXCR2”) or EGFP_CXCR2neg (designated “Mock”) MAGE-A3 tetramer (HLA-A1; EVDPIGHLY) positive CD3+ T cells. Routine assays for gene expression via flow cytometry and for mycoplasma were conducted, and the transduced T cells were used for experimental analysis as indicated. MAGE-A3a3a CXCR2 and -Mock transduced T cells were sorted and put into a rapid expansion protocol (REP) according to the REP protocol described above.
Flow cytometry and cell sorting
Multicolor flow cytometry analyses were performed to characterize chemokine receptor expression on human TILs ex vivo, or young and REP TIL cultures, as well as chemokine receptor expression, purity and migration of transduced T cell cultures. Additionally, flow cytometry was performed on single cell suspension of human xenograft FM82 tumors, lungs, spleens, blood, and bone marrows from mice treated with ACT.
For characterization of chemokine receptor expression, samples were stained with mAbs to CD3-BV421 (Biolegend), CD8-FITC (BD Biosciences), CD4-PE-Cy7 (Biolegend). Chemokine receptor expression was assessed by labeling cells with a cocktail of either CXCR2-APC (Biolegend), CXCR3-BV510 (Biolegend), CXCR4-PE (Biolegend), or CCR2-APC (Biolegend), CCR4-BV510 (Biolegend) and CCR5-PE (Biolegend). CD8-APC (BD Biosciences) and CCR2-PerCP-Cy5.5 (Biolegend) were used in a minimally altered panel when assessing chemokine receptor expression of EGFP_CXCR2 or Mock transduced T cells. Appropriate fluorochrome conjugated isotype controls were included for all chemokine receptors, while FMOs were used to initially assess spill-over between the channels. Staining was performed for 20–30 minutes protected from light and on ice. Cells were washed twice prior to and twice post mAb staining by centrifugation with 1x PBS + 2% FBS (FACS buffer).
To assess purity and differentiation of transduced T cell cultures, the cells were stained with either mAbs CD3-BV421 (Biolegend), CD4-PE-Cy7 (Biolegend) and CXCR2-APC (Biolegend), vβ5.1-APC (eBioscience, Affyrmetrix) or CD8-APC (BD Bioscience) respectively, or CD8-APC (BD), CD3-BV421 (Biolegend), CCR7-PE-Cy7 (eBioscience, Affyrmetrix), CD45RA-PE (BD) CD45RO-PerCP-Cy5.5 (BD), CD62 L-BV510 (BD) according to the above procedure.
All phenotypic characterization was performed on a BD FACS Canto II (BD Biosciences) within 1 h of finishing staining procedure and all data was analyzed with BD FACSdiva software version 8.0.1. At least 100.000 live cells were recorded to increase sensitivity and quality of the data.
For sorting, transduced T cell cultures were first stained with 20 ng/1 × 106 T cells of each of PE and APC conjugated human HLA-A1 tetramers to MAGE-A3 (EVDPIGHLY) in 50 µL 1x PBS, 0.5% bovine serum albumin (BSA, Sigma Aldrich) and 2 mM EDTA for 30 minutes at 37°C. After tetramer staining, 50 µL mAb-mix to CXCR2-APC (Biolegend) and CD3-BV421 (Biolegend) was added directly into the tetramer/cell mix and incubated for 20–30 minutes, dark and on ice. Lentiviral transduced MAGE-A3a3a tetramer positive T cells co-expressing GFP and CXCR2 were sorted (designated “CXCR2”). GFP− CXCR2− double negative MAGE-A3a3a tetramer positive T cells were sorted as the “non-transduced” control T cells (designated “Mock”). All antibodies, buffers and procedures were kept under sterile conditions to ensure aseptic sorting of cells for further culturing. Cell sorting was performed on a BD FACS aria cell sorter (BD Biosciences).
All samples were added a near-infrared fixable dead cell stain (Life technologies) for dead cell exclusion. Setup of the instruments, quality control and compensation settings were done according to instrument requirements. Flow cytometry data was analysed using BD FACSdiva software v. 8.0.1.
Transwell migration assay
Migration of transduced T cells was assessed using a Transwell® system. The upper chamber containing a 3 μm polycarbonate membrane (Corning) was added 3 × 105 T cells /200 uL serum free medium (x-vivo), while the lower was assed either 500 μL x-vivo + 0.5–100 ng/mL rhIL-8 (208-IL, R&D systems) or 500 μL MM cell line supernatant either alone or added 2 ug/mL neutralizing mAb to IL-8 (MAB208, R&D systems). Migration was assessed after 4 h at 37°C. 1 well pr. T cell culture was used as a control for background migration (minimum migration) with serum and chemokine free medium in the lower chamber, while cells were placed directly into the lower chamber (without membrane) of another well to represent the maximum migration. The number of migrated cells in the lower chamber was counted by flow cytometry. In short, transwell® inserts were discarded and the cells in the bottom well were resuspended and transferred to 5 mL Falcon® tubes (BD). The wells were washed by 1 mL 1x PBS + 2% FBS to ensure recovery of all migrated cells. Cells were spun once and all of the supernatant removed, before resuspending the cells in exactly 100 μL 1x PBS + 2% FBS per tube. Immediately prior to acquisition on a BD FACS canto II flow cytometer, all samples were added 2 uL 7-AAD (BD biosciences) for dead cell exclusion. The samples were acquired using an automated carousel, at high flow rate, for precisely 60 seconds each, generating a value of number (#) of migrated cells / 3 × 105 cells/ minute.
51Cr release cytotoxicity assay
Conventional 51Cr release assays for cell mediated cytotoxicity were used to evaluate the killing capacity of CXCR2 or Mock transduced T cells against IL-8 secreting melanoma cell line FM82 alone or in combination with either 5 ng/mL IL-8, or 2 ug/mL neutralizing mAb to IL-8. In short, 5 × 105 target cells (FM82) were labeled with 100 μCi 51Cr (Perkin Elmer) in 100 µL RPMI + 10% FBS for 60 min. Washed target cells were subsequently plated 5 × 103 cells/well in round bottomed 96-well plates with T cells at decreasing effector:target ratios, and incubated 4 h at 37°C. For quantification of T cell-mediated killing of target cells 100 μL supernatant was aspirated and 51Cr-release was measured using a gamma counter (Perkin Elmer Wallac Wizard 1470 Automatic gamma counter). Separate wells were used to determine maximum and spontaneous 51Cr release. Wells designated maximum 51Cr release were added 100 µL 10% TritonX-100 (Sigma-Aldrich) while wells designated spontaneous 51Cr release, target cells were added 100 μL RPMI 1640 + 10% FBS only. Specific lysis was calculated using the following equation; ((sample release – spontaneous release)/(maximum release – spontaneous release))*100%.
In vivo model
Immune deficient NOG mice (NOD.Cg-PrkdcScidIL2rgtm1sug/JicTac, Taconic) were housed in individually ventilated cages at University of Copenhagen, and supplied only autoclaved or sterilized water, food pellets and nesting material. The study was approved by local ethics committee.
NOG mice were inoculated with 2.5 × 106 cells of the human melanoma cell line, FM82 (ESTDAB-27), subcutaneous (sc.) on the left flank. Tumor growth was measured with calipers twice a week, and represented as tumor surface (mm2). For studies of tumor homing, animals were treated with tail vein administration of 15 × 106 MAGE-A3 specific Mock or CXCR2 transduced T cells when tumor size reached approx. 30 mm2. ACT was combined with intra peritoneal (ip.) injection of 6 × 105 IU rh-IL2 at the day of ACT and twice daily for the subsequent two days. For study of tumor homing, blood was collected from the mandibular vein immediately prior to culling by cervical dislocation 7 days after ACT. Tumor, spleen, lungs and bone marrow were resected and kept in RPMI 1640 + 1% penicillin/streptomycin (Gibco) on ice until processing.
Immunohistochemistry
Tumors were initially cut into 3 fragments. One fragment from each tumor was fixed in formalin for minimum 24 h. Formalin fixed tumors were paraffin embedded, cut and stained at the Department of Pathology, Herlev University Hospital, Denmark. Tumors were stained with H&E and monoclonal mouse anti-human CD3 (clone F7.2.38, Dako Agilent Technologies at the Dako Autostainer-Link platform). The melanoma markers anti-S100, anti-Melan A and anti-HMB-45 (all from Dako Agilent Technologies) were also applied. Tumor T cell infiltration was quantified as number of CD3+ cells / 10 High Power Fields (HPF) at 600x magnification using an Olympus BX50 light microscope. The CD3+ cells were defined as cells with a clear immunoreaction and a visible nucleus. The cells were counted in hotspots. Photos were shot using the Olympus Cell Sens Entry software.
Tumor/organ preparation
Tumors and lungs were cut into small fragments (1–2 mm2) and digested in 4 mL enzyme mix, consisting of RPMI 1640, 1% penicillin/streptomycin, 10 U/mL Pulmozyme (Dornase alfa, Roche), and 1 mg/mL Collagenase type I (Gibco) for 4 h at 37°C. Tumor and lung digests were passed through 70 μm filters (Falcon, BD) and washed twice in 1x PBS and stained according to flow protocol above.
Spleens, blood and bone marrows were passed through 70 μm filters and red blood cells lysed in 5 mL RBC lysis solution (Qiagen) for 5 minutes, washed twice in 1x PBS and stained according to flow cytometry staining protocol above.
Single cell suspensions from xenograft tumors and mouse organs were stained with CD3-PE-Cy7 and CD3-BV21 as well as CXCR2-APC in 50 µL 1x PBS, 0.5% bovine serum albumin (BSA, Sigma Aldrich) otherwise according to above procedure. An acquisition threshold of >100.000 live cells was set to secure the reliability of CD3+ T cell frequency data from mouse organs and tumors.
Financial support
This work was supported by the Danish Council for Independent Research under grant no. DFF-1331-00095B; and Danish Cancer Society under grant no. R72-A4396-13-S2.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
supp_data.zip
Download Zip (1.7 MB)Acknowledgments
We would like to acknowledge Dr. Andrew Gerry and Dr. Bent Jakobsen of Adaptimmune Ltd. (Oxfordshire, UK) for supplying us with lentiviral vectors for high affinity MAGE-A3a3a TCR transduction, intellectual input from Hjalte List Larsen and Joost van den Berg in the very early stages of the study, Evelina Martinenaite for technical assistance with running the BioPlex™ system and technician Kirsten Nikolajsen and the FACS core facility for invaluable help with the cell sorting. Last but not least, we thank Tobias Wirenfeldt Klausen for consulting on the statistical analyses.
Additional information
Funding
References
- Hodi, FS, O'Day, SJ, McDermott, DF, Weber, RW, Sosman, JA, Haanen, JB, Gonzalez, R, Robert, C, Schadendorf, D, Hassel, JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363:711–23. doi:10.1056/NEJMoa1003466. PMID:20525992.
- Topalian, S, Hodi, F. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. Engl. J. 2012;366:2443–54. doi:10.1056/NEJMoa1200690.
- Brahmer, JR, Tykodi, SS, Chow, LQ, Hwu, WJ, Topalian, SL, Hwu, P, Drake, CG, Camacho, LH, Kauh, J, Odunsi, K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012;366:2455–65. doi:10.1056/NEJMoa1200694. PMID:22658128.
- Rosenberg, SA, Dudley, ME. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 2004;101(Suppl):14639–45. doi:10.1073/pnas.0405730101. PMID:15381769.
- Ellebaek, E, Iversen, TZ, Junker, N, Donia, M, Engell-Noerregaard, L, Met, Ö, Hölmich, LR, Andersen, RS, Hadrup, SR, Andersen, MH, et al. Adoptive cell therapy with autologous tumor infiltrating lymphocytes and low-dose Interleukin-2 in metastatic melanoma patients. J. Transl. Med. 2012;10:169. doi:10.1186/1479-5876-10-169. PMID:22909342.
- Andersen, R, Donia, M, Ellebaek, E, Borch, TH, Kongsted, P, Iversen, TZ, Hölmich, LR, Hendel, HW, Met, Ö, Andersen, MH, et al. Long-Lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated il2 regimen. Clin. Cancer Res. 2016;22:3734–45. doi:10.1158/1078-0432.CCR-15-1879. PMID:27006492.
- Weiss, SA, Han, SW, Lui, K, Tchack, J, Shapiro, R, Berman, R, Zhong, J, Krogsgaard, M, Osman, I, Darvishian, F. Immunologic heterogeneity of tumor-infiltrating lymphocyte composition in primary melanoma. Hum. Pathol. 2016;57:116–25. doi:10.1016/j.humpath.2016.07.008. PMID:27473267.
- Galon, J, Costes, A, Sanchez-Cabo, F, Kirilovsky, A, Mlecnik, B, Lagorce-Pagès, C, Tosolini, M, Camus, M, Berger, A, Wind, P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi:10.1126/science.1129139. PMID:17008531.
- Zhang, L, Conejo-Garcia, JR, Katsaros, D, Gimotty, PA, Massobrio, M, Regnani, G, Makrigiannakis, A, Gray, H, Schlienger, K, Liebman, MN, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003;348:203–13. doi:10.1056/NEJMoa020177. PMID:12529460.
- Denkert, C, Wienert, S, Poterie, A, Loibl, S, Budczies, J, Badve, S, Bago-Horvath, Z, Bane, A, Bedri, S, Brock, J, et al. Standardized evaluation of tumor-infiltrating lymphocytes in breast cancer: results of the ring studies of the international immuno-oncology biomarker working group. Mod. Pathol. 2016;29:1155–64. doi:10.1038/modpathol.2016.109. PMID:27363491.
- Fridman, WH, Pagès, F, Sautès-Fridman, C, Galon, J. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer. 2012;12:298–306. doi:10.1038/nrc3245. PMID:22419253.
- Tumeh, PC, Harview, CL, Yearley, JH, Shintaku, IP, Taylor, EJ, Robert, L, Chmielowski, B, Spasic, M, Henry, G, Ciobanu, V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71. doi:10.1038/nature13954. PMID:25428505.
- Verdegaal, EME, de Miranda, NF, Visser, M, Harryvan, T, van Buuren, MM, Andersen, RS, Hadrup, SR, van der Minne, CE, Schotte, R, Spits, H, et al. Neoantigen landscape dynamics during human melanoma–T cell interactions. Nature. 2016;536:91–95. doi:10.1038/nature18945. PMID:27350335.
- Pockaj, BA, Sherry, RM, Wei, JP, Yannelli, JR, Carter, CS, Leitman, SF, Carasquillo, JA, Steinberg, SM, Rosenberg, SA, Yang, JC. Localization of 111indium-labeled tumor infiltrating lymphocytes to tumor in patients receiving adoptive immunotherapy. Augmentation with cyclophosphamide and correlation with response. Cancer. 1994;73:1731–7. doi:10.1002/1097-0142(19940315)73:6%3c1731::AID-CNCR2820730630%3e3.0.CO;2-H. PMID:8156501.
- Fisher, B, Packard, BS, Read, EJ, Carrasquillo, JA, Carter, CS, Topalian, SL, Yang, JC, Yolles, P, Larson, SM, Rosenberg, SA. Tumor localization of adoptively transferred indium-111 labeled tumor infiltrating lymphocytes in patients with metastatic melanoma. J. Clin. Oncol. 1989;7:250–61. doi:10.1200/JCO.1989.7.2.250. PMID:2644399.
- Zlotnik, A, Burkhardt, AM, Homey, B. Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol. 2011;11:597–606. doi:10.1038/nri3049. PMID:21866172.
- Griffith, JW, Sokol, CL, Luster, AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014;32:659–702. doi:10.1146/annurev-immunol-032713-120145. PMID:24655300.
- Harlin, H, Meng, Y, Peterson, AC, Zha, Y, Tretiakova, M, Slingluff, C, McKee, M, Gajewski, TF. Chemokine expression in melanoma metastases associated with CD8 + T-CeII recruitment. Cancer Res. 2009;69:3077–85. doi:10.1158/0008-5472.CAN-08-2281. PMID:19293190.
- Klarquist, J, Tobin, K, Farhangi Oskuei, P, Henning, SW, Fernandez, MF, Dellacecca, ER, Navarro, FC, Eby, JM, Chatterjee, S, Mehrotra, S, et al. Ccl22 diverts T regulatory cells and controls the growth of melanoma. Cancer Res. 2016;76:6230–40. doi:10.1158/0008-5472.CAN-16-0618. PMID:27634754.
- Chow, MT, Luster, AD. Chemokines in Cancer. Cancer Immunol. Res. 2014;2:1125–31. doi:10.1158/2326-6066.CIR-14-0160. PMID:25480554.
- Tran, KQ, Zhou, J, Durflinger, KH, Langhan, MM, Shelton, TE, Wunderlich, JR, Robbins, PF, Rosenberg, SA, Dudley, ME. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J. Immunother. 2008;31:742–51. doi:10.1097/CJI.0b013e31818403d5. PMID:18779745.
- Dudley, ME, Wunderlich, JR, Shelton, TE, Even, J, Rosenberg, SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2008;26:332–42. doi:10.1097/00002371-200307000-00005.
- Idorn, M, Thor Straten, P, Svane, IM, Met, Ö. Transfection of Tumor-Infiltrating T Cells with mRNA Encoding CXCR2. Methods Mol. Biol. 2016;1428:261–76. doi:10.1007/978-1-4939-3625-0_17. PMID:27236805.
- Cameron, BeryAB, Dukes, J, Harper, JV, Kannan, V, Bianchi, FC, Grand, F, Brewer, JE, Gupta, M, Plesa, G, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013;5:197ra103. doi:10.1126/scitranslmed.3006034. PMID:23926201.
- Donia, M, Junker, N, Ellebaek, E, Andersen, MH, Straten, PT, Svane, IM. Characterization and comparison of ‘Standard’ and ‘Young’ tumor infiltrating lymphocytes for adoptive cell therapy at a Danish Translational Research Institution. Scand. J. Immunol. 2011;75:157–67.
- Ji, RR, Chasalow, SD, Wang, L, Hamid, O, Schmidt, H, Cogswell, J, Alaparthy, S, Berman, D, Jure-Kunkel, M, Siemers, NO, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012;61:1019–31. doi:10.1007/s00262-011-1172-6. PMID:22146893.
- Vasaturo, A, Halilovic, A, Bol, KF, Verweij, DI, Blokx, WA, Punt, CJ, Groenen, PJ, van Krieken, JH, Textor, J, de Vries, IJ, et al. T-cell landscape in a primary melanoma predicts the survival of patients with metastatic disease after their treatment with dendritic cell vaccines. Cancer Res. 2016;76:3496–506. doi:10.1158/0008-5472.CAN-15-3211. PMID:27197179.
- Hamid, O, Schmidt, H, Nissan, A, Ridolfi, L, Aamdal, S, Hansson, J, Guida, M, Hyams, DM, Gómez, H, Bastholt, L, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J. Transl. Med. 2011;9:204. doi:10.1186/1479-5876-9-204. PMID:22123319.
- Bedognetti, D, Spivey, TL, Zhao, Y, Uccellini, L, Tomei, S, Dudley, ME, Ascierto, ML, De Giorgi V, LQ, Delogu, LG, et al. CXCR3/CCR5 pathways in metastatic melanoma patients treated with adoptive therapy and interleukin-2. Br. J. Cancer. 2013;109:2412–23. doi:10.1038/bjc.2013.557. PMID:24129241.
- Sapoznik, S, Ortenberg, R, Galore-Haskel, G, Kozlovski, S, Levy, D, Avivi, C, Barshack, I, Cohen, CJ, Besser, MJ, Schachter, J, et al. CXCR1 as a novel target for directing reactive T cells toward melanoma: Implications for adoptive cell transfer immunotherapy. Cancer Immunol. Immunother. 2012;61:1833–47. doi:10.1007/s00262-012-1245-1. PMID:22441657.
- Mikucki, ME, Fisher, DT, Matsuzaki, J, Skitzki, JJ, Gaulin, NB, Muhitch, JB, Ku, AW, Frelinger, JG, Odunsi, K, Gajewski, TF, et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015;6:7458. doi:10.1038/ncomms8458. PMID:26109379.
- Mikucki, ME, Skitzki, JJ, Frelinger, JG, Odunsi, K, Gajewski, TF, Luster, AD, Evans, SS. Unlocking Tumor Vascular Barriers with CXCR3: Implications for Cancer Immunotherapy. Oncoimmunology. 2016;5:00–00. doi:10.1080/2162402X.2015.1116675.
- Viola, A, Sarukhan, A, Bronte, V, Molon, B. The pros and cons of chemokines in tumor immunology. Trends Immunol. 2012;33:496–504. doi:10.1016/j.it.2012.05.007. PMID:22726608.
- Kershaw, MH, Wang, G, Westwood, JA, Pachynski, RK, Tiffany, HL, Marincola, FM, Wang, E, Young, HA, Murphy, PM, Hwu, P. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum. Gene Ther. 2002;13:1971–80. doi:10.1089/10430340260355374. PMID:12427307.
- Peng, W, Ye, Y, Rabinovich, BA, Liu, C, Lou, Y, Zhang, M, Whittington, M, Yang, Y, Overwijk, WW, Lizée, G, et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin. Cancer Res. 2010;16:5458–68. doi:10.1158/1078-0432.CCR-10-0712. PMID:20889916.
- Sackstein, R, Schatton, T, Barthel, SR. T-lymphocyte homing: an underappreciated yet critical hurdle for successful cancer immunotherapy. Lab Investig. 2017;97:669–97. doi:10.1038/labinvest.2017.25. PMID:28346400.
- Rongvaux, A, Takizawa, H, Strowig, T, Willinger, T, Eynon, EE, Flavell, RA, Manz, MG. Human hemato-lymphoid system mice: Current use and future potential for medicine. Annual review of immunology. 2013;37:635–74. doi:10.1146/annurev-immunol-032712-095921. PMID:23330956.
- Martinet, L, Le Guellec, S, Filleron, T, Lamant, L, Meyer, N, Rochaix, P, Garrido, I, Girard, JP. High endothelial venules (HEVs) in human melanoma lesions: Major gateways for tumor-infiltrating lymphocytes. Oncoimmunology. 2012;1:829–39. doi:10.4161/onci.20492. PMID:23162750.
- Brehm, MA, Shultz, LD, Luban, J, Greiner, DL. Overcoming current limitations in humanized mouse research. J. Infect. Dis. 2013;208(Suppl):S125–30. doi:10.1093/infdis/jit319. PMID:24151318.
- Moon, EK, Carpenito, C, Sun, J, Wang, LC, Kapoor, V, Predina, J, Powell, DJ Jr, Riley, JL, June, CH, Albelda, SM. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011;17:4719–30. doi:10.1158/1078-0432.CCR-11-0351. PMID:21610146.
- Wennerberg, E, Kremer, V, Childs, R, Lundqvist, A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol. Immunother. 2014;64:225–35. doi:10.1007/s00262-014-1629-5. PMID:25344904.
- Thor Straten, P, Kirkin, AF, Seremet, T, Zeuthen, J. Expression of transporter associated with antigen processing 1 and 2 (TAP1/2) in malignant melanoma cell lines. Int J cancer. 1997;70:582–6. doi:10.1002/(SICI)1097-0215(19970304)70:5%3c582::AID-IJC15%3e3.0.CO;2-F. PMID:9052759.