
ABSTRACT
Intrinsic resistance to cytotoxic T-lymphocyte (CTL) killing limits responses to immune activating anti-cancer therapies. Here, we established that activation of the G2/M cell cycle checkpoint results in tumor cell cycle pause and protection from granzyme B-induced cell death. This was reversed with WEE1 kinase inhibition, leading to enhanced CTL killing of antigen-positive tumor cells. Similarly, but at a later time point, cell cycle pause following TNFα exposure was reversed with WEE1 kinase inhibition, leading to CTL transmembrane TNFα-dependent induction of apoptosis and necroptosis in bystander antigen-negative tumor cells. Results were reproducible in models of oral cavity carcinoma, melanoma and colon adenocarcinoma harboring variable Tp53 genomic alterations. WEE1 kinase inhibition sensitized tumors to PD-1 mAb immune checkpoint blockade in vivo, resulting in CD8+-dependent rejection of established tumors harboring antigen-positive or mixed antigen-positive and negative tumor cells. Together, these data describe activation of the G2/M cell cycle checkpoint in response to early and late CTL products as a mechanism of resistance to CTL killing, and provide pre-clinical rationale for the clinical combination of agents that inhibit cell cycle checkpoints and activate anti-tumor immunity.
Introduction
Extrinsic mechanisms of local immunosuppression within the tumor microenvironment include tumor hypoxia, T-cell anergy and effector immune cell suppression by myeloid cells and regulatory T-lymphocytes.Citation1-Citation3 Loss of highly antigenic tumor cell variants though immunoeditingCitation4, development of antigen loss variants through epitope mutation or selectionCitation5,Citation6 and disruption of antigen processing machineryCitation7,Citation8 can also lead to ineffective antigen-specific T-cell-mediated anti-tumor immunity. However, tumor cells themselves possess pro-survival mechanisms to resist death following cytotoxic or genotoxic insults.Citation9 Tumor cell intrinsic mechanisms of resistance to effector immune cell products in the face of an intact antigen-specific immune response, such as granzyme B and tumor necrosis factor receptor (TNFR) superfamily ligands, are less well studied.
Cell cycle progression is a tightly orchestrated process within cells, and cell cycle pause in response to cytotoxic/genotoxic insults to allow repair is a well-documented response to avoid replication catastrophe.Citation10-Citation12 WEE1 kinase is a druggable target that contributes to cell cycle control through post-translational control of CDK1 function at the G2/M cell cycle checkpoint.Citation13 Here, we demonstrated that AZD1775, a WEE1 kinase small molecule inhibitor, sensitizes tumor cells to early, granzyme B-dependent cytotoxic T-lymphocyte (CTL) killing of antigen-positive tumor cells through reversal of granzyme B-induced CDK1 phosphorylation and cell cycle pause. We also demonstrated enhanced TNFα-dependent bystander killing of antigen-negative tumor cells with exposure to AZD1775 through similar mechanisms. This bystander killing occurred through CTL transmembrane (TM) TNFα, resulted in apoptosis and necroptosis, and was independent of MHC class I expression. These results were replicated in a number of different cell lines with variable Tp53 status. Results were validated in vivo as combination AZD1775 and PD-1 mAb treatment induced CD8+ cell-dependent rejection of established model antigen-positive but not model antigen-negative tumors. Additionally, bystander killing of antigen-negative cells was demonstrated with rejection of mixed antigen-positive and negative tumors following combination treatment. Cumulatively, this work identifies cell cycle pause as an intrinsic mechanism of resistance to early and late CTL products and provides the pre-clinical rationale for the combination of agents that prevent cell cycle checkpoint activation and treatments that enhance anti-tumor immunity.
Results
MOC1 cells engineered to express a model antigen are susceptible to specific CTL lysis
To engineer cancer cells expressing a model antigen, we stably transduced MOC1 carcinogen-induced murine oral squamous cell carcinoma cells with a construct that encodes the far-red fluorescent protein mKate2 and the H-2Kb-restricted ovalbumin epitope SIINFEKL in the same open reading frame (M1mK2SIIN cells). In the presence of IFNγ to induce expression of MHC class I, flow cytometric analysis revealed that cells positive for mKate2 also present SIINFEKL on H-2Kb (). Using CTLs specific for SIINFEKL presented on H-2Kb generated ex vivo from OT-I mice, we assessed baseline M1mK2SIIN susceptibility to CTL lysis in a real-time impedance-based cytotoxicity assay (). M1mK2SIIN but not parental MOC1 without SIINFEKL (pMOC1) cells were susceptible to CTL lysis in a dose-dependent fashion, and CTL at an E:T ratio of 10:1 resulted in a complete loss of M1mK2SIIN target cell index without recovery.
Figure 1. AZD1775 sensitizes antigen-positive M1mK2SIIN cells to perforin/granzyme B-dependent OT-I CTL killing.
A, pMOC1 or M1mK2SIIN cells with or without IFNγ (20 ng/mL for 24 hours) were evaluated for mKate2 fluorescence and the presentation of SIINFEKL presentation via H-2Kb via flow cytometry.B, growth control or killing of M1mK2SIIN cells with OT-I CTL (added at the indicated vertical line) at varying E:T ratios. 10:1 represents the minimum E:T ratio that does not allow M1mK2SIIN cell recovery over time. Representative impedance plot on left and quantification of loss of cell index at 48 hours (after the addition of CTL) on right. OT-I CTL control of pMOC1 cells was quantified at 48 hours as a negative control.C, M1mK2SIIN cells were plated at time 0 in the presence of AZD1775 (250 nM) or DMSO (volume equivalent, control) and CTL were added at the indicated vertical line, with quantified loss of cell index at 12 and 48 hours after the addition of CTL.D, SIINFEKL presentation via H-2Kb was quantified on M1mK2SIIN cells treated with AZD1775 (250 nM) or DMSO alone via flow cytometry. Pre-treatment with IFNγ (20 ng/mL for 24 hours) was used as a positive control.E, either M1mK2SIIN tumor cells or OT-I CTL alone were exposed to AZD1775 (250 nM) or DMSO alone and CTL growth control or killing of M1mK2SIIN cells was assayed, with quantified loss of cell index at 12 and 48 hours after the addition of CTL. For tumor cells exposure alone, M1mK2SIIN cells were plated in AZD1775 and time 0 and washed three times before the addition of CTL. For OT-I CTL exposure alone, AZD1775 was added to CTL culture for 24 hours, and CTL were washed three times before addition to the impedance assay.F, OT-I CTL killing of M1mK2SIIN cells treated with AZD1775 (250 nM) or DMSO alone was assayed in the presence or absence of the perforin inhibitor concanamycin A (OT-1 CTL treated with 100 nM for 4 hours prior to addition to impedance assay), with quantification 18 hours after the addition of CTL.**, p < 0.01, ***, p < 0.001; n/s, non-significant. Representative data (mean±SD) from one of three independent assays for each experiment, each performed in technical triplicate.
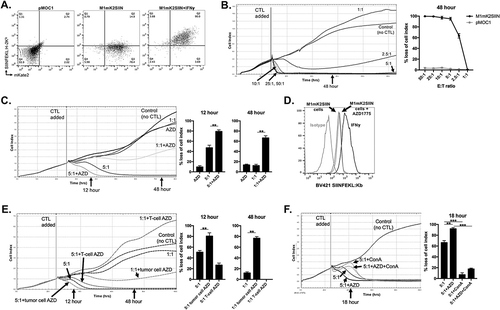
WEE1 kinase inhibition sensitizes antigen-positive target cells to early perforin/granzyme b-dependent killing
Cell cycle checkpoints serve to pause cell cycle progression to allow repair following genotoxic insults, and WEE1 kinase plays an integral role in transition through the G2/M cell cycle checkpoint.Citation14,Citation15 To determine if CTL lysis of M1mK2SIIN cells could be enhanced with WEE1 kinase inhibition, we treated M1mK2SIIN cells with AZD1775, a small molecule inhibitor of WEE1 kinase, and assessed CTL lysis at low E:T ratios. CTL lysis at 5:1 and 1:1 E:T ratios were significantly enhanced in the presence of AZD1775 at low nanomolar (physiologic) concentrations compared to control (). AZD1775 did not enhance SIINFEKL presentation on H-2Kb, indicating that this enhanced CTL activity was not simply due to enhanced antigen presentation (). Additionally, AZD1775 alone at doses up to 2,000 nM did not significantly alter the growth or viability of M1mK2SIIN cells (Supplemental Figure S1). We next selectively exposed either target M1mK2SIIN cells or OT-I CTLs to AZD1775 and assessed CTL lysis. Exposure of target M1mK2SIIN cells alone to AZD1775 but not exposure of OT-I CTLs alone enhanced CTL lysis (), indicating a tumor cell-specific mechanism of enhanced sensitivity to CTL killing. While exposure of AZD1775 to CTL alone appeared to modestly inhibit CTL killing at 5:1 and 1:1 E:T ratios, follow-up experiments indicated AZD1775 had little to no effect on T-lymphocyte expansion or IFNγ production capacity in response to APC pulsed with cognate antigen or to non-specific CD3/28 stimulation (Supplemental Figure S2A&B). Exposure of OT-I CTL to the perforin inhibitor concanamycin A to inhibit perforin reversed early (first 18 hours) baseline and AZD1775-enhanced CTL lysis (), indicating WEE1 kinase inhibition sensitizes antigen-positive target cells to early, perforin/granzyme B-dependent killing.
WEE1 kinase inhibition sensitizes antigen-negative cells to transmembrane tnfα-dependent OT-I CTL control through induction of apoptosis and necroptosis
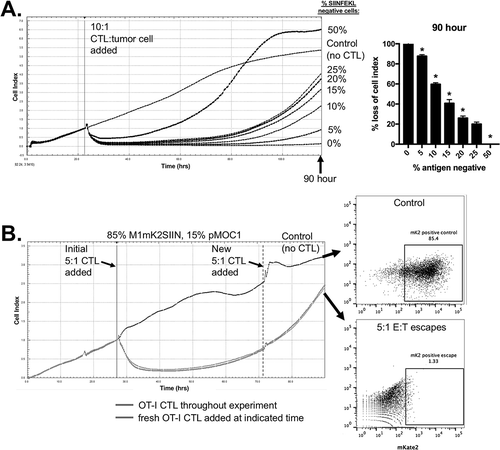
Immune escape through antigen loss leads to the outgrowth of immunoedited tumor cells that are resistant to antigen-specific killing.Citation4,Citation6 To model antigen loss, we mixed SIINFEKL-positive M1mK2SIIN cells with SIINFEKL-negative pMOC1 cells. The addition of pMOC1 cells led to a late, dose-dependent outgrowth of cells resistant to OT-I CTL control after an early period of loss of cell index (). To rule out that this was secondary to CTL exhaustion, we added freshly generated OT-I CTL to a mixture of 15% antigen-negative cells following the start of growth recovery (). These CTL did not alter target cell recovery, indicating that CTL exhaustion was not playing a role. Cells that escaped CTL growth control and CTL-naïve target cells were assayed by flow cytometry for mKate2 positivity. While many CTL-naïve target cells were mKate2 positive as expected, target cells that escaped CTL control were mKate2 negative, demonstrating the outgrowth of SIINFEKL-negative pMOC1 cells in the face of an antigen-specific CTL response and validating this system as an in vitro model of antigen escape.
Figure 2. Mixing M1mK2SIIN and pMOC1 cells allows in vitro modeling of antigen escape.
A, SIINFEKL positive (M1mK2SIIN) and SIINFEKL negative (pMOC1) cells were mixed at the ratios indicated on the right side of the plot and assayed for growth control following the addition of 10:1 CTL at the indicated vertical line, quantified at 90 hours after the addition of CTL. *, p < 0.05 in relation to the previous dilution. Representative data from one of three independent assays.B, OT-I CTL (5:1 E:T) growth control of a mixture of 15% SIINFEKL negative (pMOC1) cells was assayed, with addition of fresh OT-I CTL (5:1 E:T, dashed vertical line) after tumor cell growth rebound. Control tumor cells (no CTL exposure) and tumor cells that had escaped 5:1 CTL control were assessed for mKate2 positivity via flow cytometry. Representative data from one of two independent assays.
We next hypothesized that WEE1 kinase inhibition could enhance T-lymphocyte-dependent control of antigen-negative tumor cells within this model system. The addition of AZD1775 significantly enhanced both early and late OT-I CTL (10:1 E:T) control of a mixture of 15% SIINFEKL-negative target cells (). Given known mechanisms of late CTL killing, we repeated these experiments in the presence of TNFα, FasL or TRAIL mAbs. Whereas neutralization of TRAIL had little effect, the addition of FasL mAb modestly and TNFα mAb significantly reversed AZD1775-enhanced OT-I CTL control of antigen-negative target cells. To verify that this apparent TNFα-dependent effect was due to signaling through TNFR1 on antigen-negative target cells, we generated CRISPR/Cas9 TNFR1 knockout pMOC1 cells (Supplemental Figure S3A-C). Replacing pMOC1 antigen-negative cells with TNFR1 knockout pMOC1 cells totally reversed AZD1775-enhanced OT-I CTL control of antigen-negative target cells ().
Figure 3. AZD1775 sensitizes SIINFEKL negative cells to transmembrane TNFα-dependent control by antigen specific CTL.
A, OT-I CTL (10:1 E:T) control of 15% SIINFEKL negative pMOC1 tumor cells in the presence of AZD1775 (250 nM) or DMSO alone was assayed. Monoclonal antibodies targeting TNFα (clone XT3.11), FasL (MFL3) or TRAIL (N2B2; all 2 μg/mL) were added with CTLs to some wells (CTL and mAbs mixed before addition to target cells), with quantification 60 hours after the addition of CTL.B, OT-I CTL (10:1 E:T) control of 15% SIINFEKL negative tumor cells in the presence of AZD1775 (250 nM) or DMSO alone was assayed. In some wells, SIINFEKL negative pMOC1 cells were replaced with TNFR1 knockout pMOC1 cells, with quantification 60 hours after the addition of CTL.C, OT-I CTL (10:1 E:T) control of 15% SIINFEKL negative pMOC1 tumor cells in the presence of AZD1775 (250 nM) or DMSO alone was assayed. In some wells, TAPI-0 (10 μM) was added with or without TNFα mAb (2 μg/mL) with CTLs. When used, CTLs were exposed to TAPI-0 for 2 hours prior to being added to target cells. Quantification of loss of cell index performed 72 hours after the addition of CTLs.D, OT-I CTL (10:1 E:T) growth control or killing of 15% SIINFEKL negative pMOC1 cells in the presence of AZD1775 (250 nM) or DMSO alone was assayed via impedance analysis in the presence or absence of ZVAD (20 μM) or necrostatin (20 μM) alone or in combination, with quantification 72 hour after the addition of CTL.E, OT-I CTL (10:1 E:T) control of 15% SIINFEKL negative tumor cells in the presence of AZD1775 (250 nM) or DMSO alone was assayed. In some wells, SIINFEKL negative pMOC1 cells were replaced with β2M knockout pMOC1 cells, with quantification 72 hours after the addition of CTL.F, SIINFEKL positive M1mK2SIIN (85%) and antigen-negative β2M knockout pMOC1 cells (15%) were co-incubated for 24 hours, stimulated together with 10 ng/mL IFNγ for an additional 24 hours, and assessed for SIINFEKL presentation on H-2Kb via flow cytometry.*, p < 0.05, **, p < 0.01; ***, p < 0.001; n/s, non-significant. Representative data from one of three independent assays for each experiment.
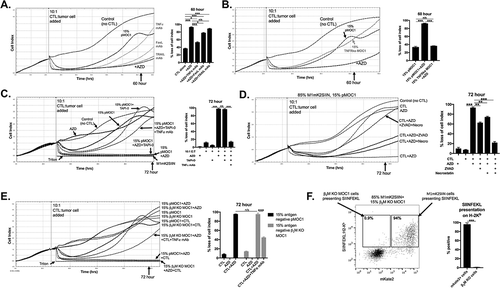
Activated CTL express transmembrane (TM) TNFα that can yield soluble TNFα through the enzymatic activity of tumor necrosis factor-alpha converting enzyme (TACE). After verifying that activated OT-I CTL express both TM and soluble TNFα (Supplemental Figure S4), we validated that inhibition of TACE with TNFα processing inhibitor-0 (TAPI-0) induced accumulation of TM TNFα and prevented release of soluble TNFα (Supplemental Figure S5). The addition of TAPI-0 did not reverse AZD1775-enhanced OT-I CTL control of antigen-negative cells, indicating that soluble TNFα is not required (). To ensure that TAPI-0 was not somehow inhibiting antigen-negative tumor cells growth, TNFα mAb was used to again subtotally reverse OT-I CTL control of antigen-negative cells in the presence of TAPI-0.
Previous work from our laboratory and others has demonstrated that tumor cell death downstream of TNFα signaling results in both apoptosis and necroptosis.Citation16,Citation17 To determine mechanisms of TNFα-dependent control of antigen-negative cells, OT-I CTLs were added to a mixture of 15% antigen-negative cells in the presence of AZD1775 with or without the pan-caspase inhibitor ZVAD or RIPK1 necroptosis inhibitor necrostatin alone or in combination (). ZVAD treatment had no effect on early OT-I killing of antigen-positive cells. Conversely, while ZVAD or necrostatin alone partially reversed AZD1775-enhanced OT-I CTL control of antigen-negative cells, the combination totally reversed this enhancement, indicating that TNFα is inducing both apoptosis and necroptosis in antigen-negative cells in the presence of AZD1775. To further validate the effects of TNFα on antigen-negative tumor cells in the presence of AZD1775, pMOC1 cells were treated with OT-I CTLs or soluble rmTNFα (Supplemental Figure S6A&B) in the presence of AZD1775 with or without ZVAD and necrostatin alone or in combination. In the presence but not absence of AZD1775, OT-I CTL or rmTNFα treatment resulted in growth control of antigen-negative tumor cells, and this effect was reversed following inhibition of apoptosis and necroptosis.
OT-I CTL control of antigen-negative cells following WEE1 kinase inhibition does not occur through uptake of released antigen or MHC cross dressing
We next performed a series of experiments to rule out other possibilities to explain OT-I CTL control of antigen-negative antigen cells. One possibility was that antigen (SIINFEKL) released from dying or dead M1mK2SIIN cells killed by OT-I CTLs was processed and presented by SIINFEKL-negative pMOC1 cells. We generated H-2Kb and H-2Db knockout pMOC1 cells through CRISPR/Cas9 deletion of β2M (Supplemental Figure S7A-C). Replacing pMOC1 cells with β2M KO pMOC1 cells did not reverse AZD1775-enhanced OT-I CTL control of SIINFEKL negative cells, demonstrating MHC class I expression on antigen-negative tumor cells is not required ().
Another possibility is that intact SIINFEKL:H-2Kb complexes were transferred from M1mK2SIIN cells to SIINFEKL-negative pMOC1 cells in a process that has been described in dendritic cells as “MHC cross dressing”.Citation18 To rule out this possibility, we used flow cytometry to demonstrate that SIINFEKL:H-2Kb complexes are not transferred to the surface of mKate2-negative, SIINFEKL-negative β2M KO pMOC1 cells after a period of incubation with M1mK2SIIN cells ().
WEE1 kinase inhibition reverses early granzyme b and late tnfα-induced G2/M cell cycle checkpoint activation
CTL induce target cell death by multiple mechanisms that are temporally regulated. Chen et al. demonstrated granzyme B-dependent loss of cell viability can be reduced by forced overexpression of WEE1 kinase over 20 years ago.Citation19 Late CTL killing mediated through TNF superfamily ligands, such as TRAIL, can be enhanced in triple negative breast cancer cells through WEE1 kinase blockade.Citation20 To determine the effects of granzyme B and TNFα on cell cycle progression and how this is altered with AZD1775, we performed 4-marker cell cycle analysis on unsynchronized pMOC1 cells. To mimic the addition of OT-I CTL 24-hours after plating cells in AZD1775, recombinant murine granzyme B (with streptolysin O to allow cytoplasmic entry) or TNFα was added to pMOC1 cells after 24 hours of WEE1 kinase inhibitor treatment. Compared to control, AZD1775 alone produced little alteration of cell cycle distribution (). Granzyme B alone induced early (2 hour) and sustained (8 hour) accumulation of cells in the G2/M gate (quantified in ). Within this G2/M gate, >95% of the cells were in G2 after treatment with granzyme B, with very few cells entering into mitosis, indicating activation of the G2/M cell cycle checkpoint. TNFα treatment induced a similar G2/M block that was more pronounced at 8 hours. This granzyme B- or TNFα-induced G2/M block was reversed with the addition of AZD1775, pushing cells into mitosis and correlating with DNA fragmentation and accumulation of cells in subG1.
Figure 4. Early granzyme B- and late TNFα-induced cell cycle pause at G2/M is reversed in the presence of AZD1775 leading to DNA damage and subG1 accumulation.
A, pMOC1 cells were treated with AZD1775 (250 nM for 24 hours) or DMSO control followed by streptolysin O (40 ng/mL)/granzyme B (1500 ng/mL) or TNFα (20 ng/mL) for 2 or 8 hours before 4 marker (DNA content, EdU, p-HH3S10 and p-γH2AXS139) flow cytometry analysis. Representative flow cytometry dot plots and p-HH3 histogram from G2/M gated cells shown.B, quantification of accumulation of cells within specific phases of the cell cycle following treatments as in A. Pooled data from three independent experiments, each performed in technical triplicate.C, quantification of p-γH2AXS139 mean fluorescence intensity (MFI) by cell cycle phase following treatments as in A. Pooled data from three independent experiments, each performed in technical triplicate.D, representative half-offset flow cytometry histograms demonstrating p-γH2AXS139 staining intensity during M-phase from selected conditions. Specific MFI for each condition listed adjacent to each histogram.E, pMOC1 cells irradiated to 8Gy were used as a positive control for cell cycle analysis. Representative dot plot shown on the left, with overlaid histogram demonstrating p-γH2AXS139 staining intensity in each phase of the cell cycle on the right.F, pMOC1 cells were treated as in A and expression and phosphorylation of select proteins was assessed via western blot analysis at the indicated time points.*, p < 0.05, **, p < 0.01; ***, p < 0.001
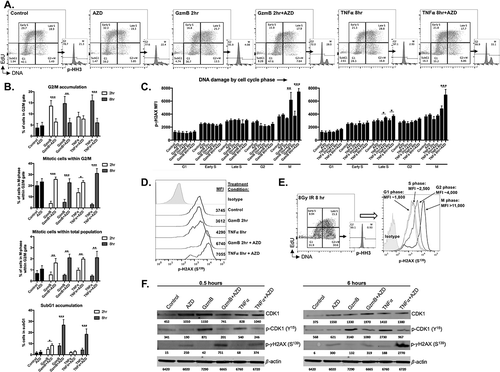
The addition of AZD1775 to granzyme B or TNFα resulted in significant accumulation of DNA damage in M-phase cells (quantified in , representative histograms in ). Notably, granzyme B or TNFα alone did not induce significant DNA damage. While the addition of AZD1775 to TNFα did modestly enhance DNA damage, significantly enhanced phosphorylation of γH2AX occurred primarily in M-phase, arguing against the presence of significant replication stress.Citation21,Citation22 As a positive control for cell cycle analysis and measurement of DNA damage, pMOC1 cells were irradiated to 8Gy and analyzed (representative data in ). By western blot analysis, G2/M checkpoint activation correlated with CDK1 phosphorylation at Y15 and early (0.5 hour) and late (6 hour) time points after granzyme B and TNFα exposure (). The addition of AZD1775 reversed CDK1 phosphorylation, validating the on-target effect of AZD1775, and correlated with phosphorylation of γH2AX at S139. Again, significant phosphorylation of γH2AX was not observed after granzyme B or TNFα alone, suggesting that the observed G2/M block is not simply a DNA damage response. Immunofluorescence of p-γH2AX revealed that the addition of AZD1775 to granzyme B or TNFα induced time-dependent induction of pan-nuclear staining (Supplemental Figure S8), indicative of replication fork collapse and mitotic catastrophe.Citation23 Both granzyme B and TNFα appeared to stabilize total CDK1 expression at early and late time points, but in contrast to results from Hamilton et al.,Citation24 this stabilized CDK1 expression appeared to have little impact on lamin A/C breakdown (Supplemental Figure S9).
Cumulatively, these results indicated that WEE1 kinase inhibition reversed the ability of cells to activate the G2/M cell cycle checkpoint in response to granzyme B and TNFα at early and late time points, respectively. Cells pushed into mitosis after exposure to WEE1 kinase inhibition accumulated significant DNA damage and pan-nuclear p-γH2AX, consistent with mitotic catastrophe, resulting in apoptosis as evidenced by DNA fragmentation.
Enhanced OT-I CTL control of antigen-positive and negative cells following WEE1 kinase inhibition occurs in cell lines with variable origins and tp53 genomic alterations
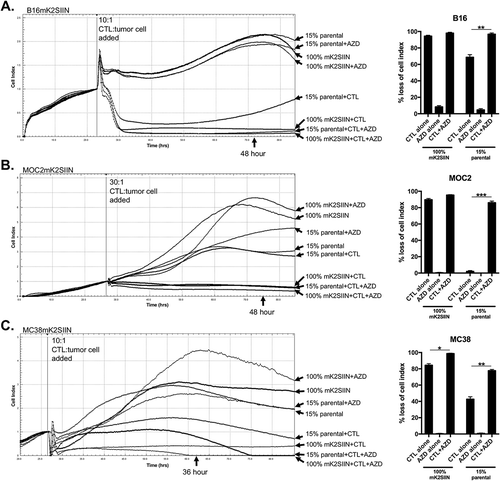
Previous work by others has demonstrated the ability of AZD1775 to enhance cytotoxicity secondary to DNA damaging agents in cells with altered Tp53.Citation13,Citation25 We wished to assess whether our findings in pMOC1 cells, which harbor a nonsynonymous Tp53 mutation with low TP53 expression, could be recapitulated in cancer cell lines with different origins and Tp53 alterations. We transduced our mKate2-SIINFEKL construct into B16 melanoma (WT Tp53 with low TP53 expressionCitation26), MOC2 oral cavity squamous cell carcinoma (nonsense Tp53 mutation with no TP53 proteinCitation27) and MC38 colon adenocarcinoma (WT Tp53 with overexpressed TP53 proteinCitation28) cells. OT-I CTLs, at E:T ratios pre-determined to kill antigen positive (mKate2-SIINFEKL) cell variants for each cell line without recovery over time, were added to 15% SIINFEKL negative parental cells for each cell line in the presence or absence of AZD1775 (). While OT-I CTL control of antigen-positive cells was variably enhanced depending upon the baseline sensitivity of each cell line, exposure to AZD1775 significantly enhanced OT-I CTL control of antigen-negative target cells in each cell line, suggesting that enhanced direct CTL lysis of antigen-positive cells and enhanced bystander control of antigen-negative cells in the presence of AZD1775 is not restricted to cell lines with specific Tp53 genomic alterations.
Figure 5. AZD1775-induced sensitization of SIINFEKL negative cells to CTL control is reproduced in an array of cancer models.
The mKate2-SIINFEKL retroviral construct was stably transduced into (A) B16 melanoma, (B) MOC2 oral cavity squamous cell carcinoma and (C) MC38 colon adenocarcinoma cell lines. Antigen positive (mK2SIIN positive variants) cells and mixtures of 15% SIINFEKL negative (parental variant) cells for each cell line, plated at time 0 in AZD1775 (250 nM) or DMSO alone (control), were assayed for growth control following the addition of CTL (at different E:T ratios) at the indicated vertical line. Quantification for each cell line at the time point indicated is shown to the right.*, p < 0.05, **, p < 0.01; ***, p < 0.001. Representative data from one of three independent assays for each experiment.
WEE1 kinase inhibition sensitizes antigen-positive and negative cells to PD-1 mab-enhanced anti-tumor immunity in vivo
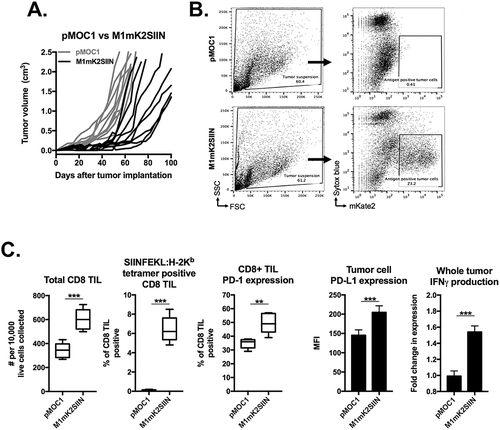
To investigate whether AZD1775 could enhance an in vivo anti-tumor immune response, we first verified that antigen-positive M1mK2SIIN establish tumors in WT B6 mice. As demonstrated in , transplantation of M1mK2SIIN cells generated tumors that engrafted but grow slower compared to pMOC1 derived tumors. Flow cytometry of tumor single cell suspensions revealed that many cells within M1mK2SIIN tumors remained positive for mKate2 (), indicating that they remain antigen-positive in the face of an endogenous WT immune response. Immune correlative analysis revealed increased infiltration of total and SIINFEKL-specific CD8+ TIL in M1mK2SIIN tumors compared to pMOC1 tumors, but also increased expression of PD-1 expression on these TIL and increased expression of PD-L1 on CD45.2−CD31− tumor cells (). Cells within M1mK2SIIN tumors produced more IFNγ, known to drive expression of PD-L1 in MOC cells.Citation29
Figure 6. M1mK2SIIN tumors engraft but escape anti-tumor immunity despite persistent antigen expression.
A, M1mK2SIIN or pMOC1 cells (5x106) were implanted subcutaneously in the flanks of WT B6 mice, and tumors were allowed to engraft and grow; plotted are individual tumor growth curves.B, established M1mK2SIIN or pMOC1 tumors were harvested, digested into a single cell suspension, and assayed for mKate2 positivity via flow cytometry. Dead cells were excluded via sytox blue staining.C, total infiltrating CD45.2+CD3+CD8+ TIL, PD-1 expression on CD8+ TIL, SIINFEKL:H-2Kb tetramer positivity and PD-L1 expression on CD45.2−CD31− cells was assessed in digested M1mK2SIIN and pMOC1 tumors via flow cytometry; total tumor IFNγ expression was determined via qRT-PCR of whole tumor lysate. Data pooled from two independent experiments.
Treatment of mice bearing established pMOC1 tumors, which are T-cell inflamed but escape anti-tumor immunity through at least expression of immune checkpoints and local immunosuppression,Citation30-Citation32 with either PD-1 mAb or AZD1775 alone resulted in slight growth delay (). Treatment with combination AZD1775 and PD-1 mAb resulted in more significant growth delay, prolonged survival and rejection of one established tumor, but most tumors progressed. Conversely, M1mK2SIIN tumors treated with either PD-1 mAb or AZD1775 resulted in the rejection of a subset of established tumors (), and treatment with combination AZD1775 and PD-1 mAb induced complete rejection of all (17/17) treated mice. Mice tolerated the treatments well without weight loss (Supplemental Figure S10). Mice that rejected established M1mK2SIIN tumors after combination treatment did not engraft new tumors when challenged with transplantation of M1mK2SIIN cells (), demonstrating the presence of immunologic memory. Combination AZD1775 and PD-1 mAb treatment of M1mK2SIIN tumors was repeated in the presence or absence of CD8+ or NK1.1+ cell depletion to determine effector immune cell requirements (). CD8+ cell but not NK1.1+ cell depletion totally abrogated M1mK2SIIN tumor rejection, and made tumors grow faster than control untreated M1mK2SIIN tumors, demonstrating that baseline immunity and rejection of M1mK2SIIN tumors following combination treatment is mediated through CD8+ cells.
Figure 7. Combination AZD1775 and PD-1 mAb treatment induces consistent CD8+ cell-dependent rejection of established SIINFEKL positive and mixed SIINFEKL negative tumors.
A, established pMOC1 tumors (5x106 cells/mouse) were treated with PD-1 mAb (clone RPM1-14, 200 μg/injection IP twice weekly for 3 weeks) or AZD1775 (60mg/kg OG 5 days on, 2 days off for 3 weeks) alone or in combination beginning at day 14; individual growth curves are shown (n = 9–10 mice/group). Timing of treatments indicated by black bar along x-axis. Control mice were treated with IP injection of rat IgG2a isotype control antibody and/or oral gavage of 0.5% methylcellulose carrier. Number of mice that rejected established tumors indicated below legends. Survival analysis displayed on right. Kaplan-Meier curves were compared using the log-rank/Mantel Cox test.B, established M1mK2SIIN tumors (5x106 cells/mouse) were treated as in A (n = 8–9 mice/group, pooled data from two independent experiments). Survival analysis displayed on right.C, mice that rejected established M1mK2SIIN tumors with combination AZD1775 and PD-1 mAb treatment or naïve WT B6 mice were challenged with M1mK2SIIN cells (3x106) injected subcutaneously into the contralateral flank and followed for tumor engraftment and growth.D, WT B6 mice were engrafted with M1mK2SIIN tumors and treated with combination AZD1775 and PD-1 mAb as in A, but with the addition of CD8+ cell depletion (clone YTS169.4, 200 μg IP twice weekly) or NK1.1+ cell depletion (clone PK136, 200 μg IP twice weekly) beginning one day prior to treatment (day 13, black arrow) and followed for tumor growth (n = 5–7 mice/group). Tumor volume at day 48 was compared to assess statistical significance via ANOVA.E, WT B6 mice were engrafted with a mixture of 85% antigen positive M1mK2SIIN cells and 15% antigen negative either pMOC1 or TNFR1 ko pMOC1 cells (5x106 cells/mouse total, n = 9–11 mice/group). Mice were treated with combination AZD1775 and PD-1 mAb as in A, and followed for tumor growth.**, p < 0.01; ***, p < 0.001.
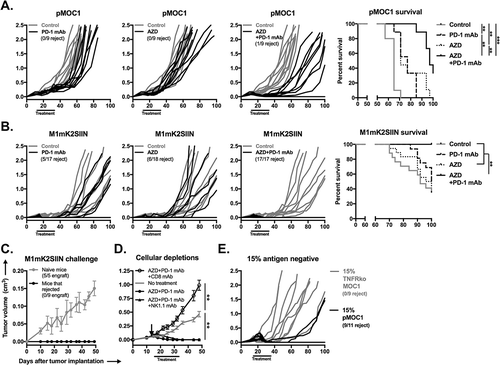
To determine if AZD1775 can sensitize subpopulations of antigen-negative cells within a largely antigen-positive tumor to an in vivo anti-tumor response in WT B6 mice, we established tumors following transplantation with 15% antigen-negative pMOC1 cells. When tumors were established with antigen-negative cells harboring an intact TNFR1, combination AZD1775 and PD-1 mAb treatment induced rejection in 82% (9/11) of treated mice (). However, treatment of tumors established with 15% TNFR1 KO antigen-negative cells resulted in tumor outgrowth and a 0% rejection rate. These results provide translational in vivo support our in vitro findings of TNFα-dependent elimination of antigen-negative cells following sensitization of tumor cells with AZD1775 in the setting of immune checkpoint blockade with PD-1 mAb.
Discussion
While many extrinsic mechanisms of resistance to immunotherapy have been clearly defined, including effector immune cell exclusion, exhaustion, and suppression within the tumor microenvironment,Citation33 tumor cell intrinsic mechanisms of resistance to immune killing are less well studied. Here, we demonstrate CDK1 phosphorylation and activation of the G2/M cell cycle checkpoint following exposure to granzyme B or TNFα. Our work clearly demonstrates reversal of CDK1 phosphorylation and cell cycle pause following exposure to a WEE1 kinase inhibitor, leading to significant DNA damage in cells pushed through the G2/M checkpoint into mitosis and subsequent apoptosis. This accumulation of DNA damage within mitosis and the presence of pan-nuclear p-γH2AX staining with combination treatment points to replication fork collapse and mitotic catastrophe as a primary mechanism of death.Citation23 Greenberg and colleagues were among the first to recognize the critical role of CDK1 activation in apoptosis,Citation34 and the first to demonstrate that constative activation of WEE1 kinase blocks apoptosis following exposure to granzyme B.Citation19 Our work is the first to demonstrate granzyme B-induced CDK1 phosphorylation and G2/M cell cycle block as a direct mechanism of resistance to CTL killing. Though this G2/M checkpoint activation was reversed following WEE1 kinase inhibition, we cannot conclude that CDK1 phosphorylation upon exposure to granzyme B and TNFα was directly due to WEE1 kinase activation, only that WEE1 kinase inhibition is sufficient to reverse this resistance mechanism. Regulation of CDK1 phosphorylation and subsequent activation is complex and involves the activity of multiple kinases (WEE1 kinase and Myt1) and phosphatases (CDC25A/B).Citation35–Citation37 Granzyme B and TNFα alone did not directly induce DNA damage at the time points investigated, suggesting the presence of a mechanism of CDK1 phosphorylation other than typical DNA damage response such as that observed after ionizing radiation or genotoxic chemotherapy.Citation13,Citation25,Citation38 Precisely how granzyme B and TNFα directly or indirectly induce CDK1 phosphorylation is being further investigated.
While we definitively demonstrated a G2/M transition block but not significant accumulation of cells within other cell cycle phases following exposure to granzyme B or TNFα, this does not rule out the possibility that granzyme B or TNFα alter other cell cycle regulatory elements. Mechanistically, WEE1 kinase inhibition alone had little measurable effects in MOC1 cells. Only after cellular insult with granzyme B or TNFα did WEE1 kinase inhibition induce M-phase DNA damage and DNA fractionation. Others have observed S-phase DNA damage indicative of replication stress when WEE1 kinase was inhibited in combination with multiple targeted therapies,Citation21,Citation22 but this was not observed when WEE1 kinase was inhibited in the presence of granzyme B. This differential accumulation of DNA damage within different phases of the cell cycle in the presence of different cellular insults suggests that WEE1 kinase inhibition may promote either replication stress and or mitotic catastrophe in a context dependent fashion.
In our experiments, WEE1 kinase inhibition was achieved with AZD1775, an orally bioavailable and specific small molecule inhibitor of WEE1 kinase that has demonstrated an acceptable safety profile in phase I studiesCitation39,Citation40 and objective responses when combined with cytotoxic chemotherapy.Citation41 Most work has focused on using AZD1775 to sensitize cells to cytotoxic chemotherapy, particularly in cells with altered TP53 function.Citation13,Citation25 Here, rather than using chemotherapy as the cytotoxic insult, we used CTL products which, when delivered in an antigen-specific fashion, provide a degree of tumor specificity not achieved with systemic chemotherapy.
Our work demonstrating details of enhanced TNFα-dependent “bystander” killing of antigen-negative cells by antigen-specific CTL following WEE1 kinase inhibition is important for several reasons. First, it demonstrates that activation of the G2/M cell cycle checkpoint occurs after exposure to TNFα similar to granzyme B, but at a later time point, mimicking kinetics of early and late mechanisms used by CTL to kill target cells. Old et al. first demonstrated TNFα-induced G2 arrest,Citation42 and others have documented enhanced sensitivity of cells in log phase growth to TNFα-induced apoptosis compared to quiescent cells.Citation43 AZD1775-induced sensitization of antigen-negative cells to TNFα suggests that both antigen-positive and negative cells could be made susceptible to CTL-derived cytotoxic insults. Our demonstration that transmembrane but not soluble TNFα is required for antigen-negative killing by CTL in the presence of AZD1775 is consistent with evidence from others indicating that bystander killing of antigen-negative cells requires direct CTL:target cell contact.Citation44,Citation45 Our results establishing that MHC class I expression on antigen negative cells is not required for bystander CTL killing not only rules out the possibility that antigen (SIINFEKL) released from dying antigen positive cells was processed and presented, it also indicates that syngeneic MHC class I expression is not required for bystander CTL killing, as observed in other studies.Citation46 While our data clearly demonstrates G2/M accumulation following treatment of cells with soluble rmTNFα, signaling through target cell TNFR1 has been shown to induce adhesion molecules that allow CTL:target cell contact,Citation47 bringing up the possibility that loss of TNFR1 in antigen negative cells lead to decreased CTL:target cell contact and subsequent loss of sensitivity to bystander CTL killing in the presence of AZD1775 by multiple mechanisms. Our results indicated only partial reversal of bystander antigen-negative cell killing with FasL blockade, but Kojima et al. demonstrated Fas:FasL-dependent CTL killing of bystander cells,Citation48 suggesting that there may be cooperative induction of apoptosis amongst TNFRs or that model-to-model variability may exist.
Our in vivo results demonstrating complete CD8+ cell-dependent rejection of all SIINFEKL positive M1mK2SIIN tumors but not SIINFEKL negative pMOC1 treated with combination AZD1775 and PD-1 mAb indicates that SIINFEKL induces strong T-lymphocytes responses in WT B6 mice, consistent with work in other models.Citation49-Citation51 Our M1mK2SIIN cells were engineered to express high levels of SIINFEKL:H-2Kb, and in vivo results with 100% M1mK2SIIN tumors serve as proof-of-principle that WEE1 kinase can sensitize highly antigenic tumors to PD-1 mAb-enhanced T-lymphocyte responses resulting in tumor rejection. Combination treatment led to enhanced tumor control and survival in antigen-negative pMOC1 tumors, but no tumors were rejected. These results are consistent with seminal work by Schreiber demonstrating that tumors uniformly expressing high but not low levels of tumor rejection antigens can be efficiently eradicated by CTL.Citation6,Citation52 We also demonstrated a high rejection rate following combination treatment in tumors harboring 15% antigen-negative pMOC1 cells with an intact TNFR1, but not when TNFR1 was genetically eliminated. This and our in vitro data strongly supports the role of CTL-derived TM TNFα in bystander tumor cell elimination. Consistent with these results, Zhang et al. demonstrated that while CTL-derived perforin/granzyme B was required for elimination of antigen-positive tumor cells, CTL-derived TNFα and IFNγ were required for elimination of antigen loss variants and tumor stromal cells to achieve tumor eradication.Citation53 One criticism of our work is that we incorporated a low percentage of antigen negative cells (15%) in our mixed tumor experiments, but again these results serve as proof-of-concept that WEE1 kinase inhibition can sensitize antigen-negative cells to TNFα-dependent bystander killing. Given that SIINFEKL is capable of generating strong T-lymphocyte responses in vivo, additional experiments in other syngeneic models may clarify whether such AZD1775-enhanced bystander killing can be achieved when antigen-positive tumor cells harbor weaker antigens.
Our results demonstrated elevated total CDK1 levels after WEE1 kinase inhibition. Hamilton et al. mechanistically linked restoration of CDK1 expression to induction of lamin A/C breakdown following exposure to TRAIL, another TNFR superfamily ligand, in human pancreatic cancer cells.Citation24 Despite increased total CDK1 levels in pMOC1 cells after WEE1 kinase inhibition, no lamin A/C breakdown was observed in pMOC1 cells. Murine granzyme B would not be expected to induce lamin A/C breakdown,Citation54 and our results indicate that TNFα alone is insufficient. Lamin breakdown is also dependent upon TP53 function,Citation55 which is lacking in pMOC1 cells.Citation27 Regardless, these data suggest that WEE1 kinase inhibition may have multiple mechanisms that sensitize tumor cells to death following exposure to TNFR superfamily ligands.
Cumulatively, these results establish G2/M cell cycle checkpoint activation in response to early and late CTL products as an additional mechanism of intrinsic tumor cell resistance to immune elimination. Given our data that the mechanism of AZD1775 enhanced sensitivity to CTL destruction is at least through reversal of tumor cell G2/M checkpoint activation, that AZD1775 appears to not significantly alter CTL function, and that AZD1775-enhanced control of antigen negative cells appears to occur in cancer cell lines with variable Tp53 status, the use of G2/M cell cycle checkpoint inhibitors may be broadly applicable as a means to sensitize tumor cells to many different forms of immunotherapy. These results strongly support the clinical investigation of WEE1 kinase or other cell cycle checkpoint inhibitors in combination with immunotherapy.
Methods
Cell lines and mice
MOC1, MOC2, B16F10 and MC38 cells were maintained as previously described,Citation56 verified to be free of murine associated pathogens and mycoplasma infection, and used at early passage number. For use in select in vitro experiments, concanamycin A, ZVAD, necrostatin and TAPI-0 (Sigma) concentrated in DMSO, TNFα (BioXCell), FasL and TRAIL (eBioscience) mAbs concentrated in PBS and rmTNFα (Biolegend) were diluted in culture media. Pharmaceutical grade AZD1775 was obtained from AstraZeneca through a cooperative research and development agreement. For in vitro experiments, AZD1775 was suspended in DMSO and volume equivalent DMSO alone was used for controls. For select experiments, streptolysin O was used as a substitute for perforin to allow cellular entry of granzyme BCitation57 and was separately verified to have no effect alone on tumor cell growth or cell cycle distribution. All animal experiments were approved by the NIDCD Animal Care and Use Committee. Wild-type C57BL/6 mice (WT B6) were purchased from Taconic. Tumor measurements and weights were obtained 2–3 times per week and volume was calculated as: (length2 x width)/2. Monoclonal antibodies used for in vivo treatments (PD-1, CD8, NK1.1) were purchased form BioXCell. In vivo AZD1775 was administered via oral gavage in 0.5% methylcellulose. Non-specific rat IgG2a antibody and oral gavage of 0.5% methylcellulose alone were used as controls for in vivo experiments.
Engineering of mkate2-siinfekl expressing cells
A retroviral construct encoding mKate2 and SIINFEKL in the same open reading frame on an MSGV2 backbone, driven by the retroviral LTR promoter, was engineered. This and the retroviral envelop plasmid VSV-G were expanded in MAX efficiency DH5α cells and isolated with an EndoFree Plasmid Maxi Kit (Qiagen). Viral packaging was performed in 293gp cells. Overnight transduction of cells was performed on retronectin (TaKaRa) coated plates pre-seeded with retrovirus via centrifugation of viral supernatant.
Generation of OT-I ctls
To generate SIINFEKL-specific CTLs, splenocytes from OT-I mice were cultured in the presence of SIINFEKL (2 μg/ml) with daily 2:1 splitting. After 72 hours in culture, >90% of remaining cells are CD8+Vα2+ cells.
Real-time impedance assay
Number of target cells plated ranged from 7.5x103 to 1 × 104 and was determined for each cell line based on growth kinetics. After adherence overnight, OT-I CTLs and the indicated E:T ratios were added to target cells plated in a 96-well E-Plate (ACEA Biosciences) and alteration of impedance was acquired using the xCELLigence Real-Time Cell Analysis (RTCA) platform per manufacturer recommendations. For each plot, y-axis is cell index and x-axis is time in hours. Triton X-100 (0.2%) was added to some wells to verify complete loss of cell index with total cell lysis, and CTLs alone were plated up to 1x106/well to verify that they do not contribute to gain of impedance. Percent loss of cell index for a given time point was calculated as: 1-(experimental cell index/control cell index).
Crispr/cas9 knockout variants
CRISPR/Cas9 plasmids containing guide RNAs for TNFR1 and β2M were purchased from Santa Cruz Biotechnology. Plasmids were transfected into cell lines with Plasmid Transfection Medium and UltraCruz Transfection Reagent (Santa Cruz). Following a 48-hour transfection period, cells were cultured for two passages, then analyzed for genetic deletion of the gene of interest.
Qrt-pcr
RNA from whole tumor lysates was purified using the RNEasy Mini Kit (Qiagen) per the manufacturer’s protocol. cDNA was synthesized utilizing a high capacity cDNA reverse transcription kit with RNase inhibitor (Applied Biosystems). A Taqman Universal PCR master mix was used to assess the relative expression of target genes compared to GAPDH on a Viia7 qPCR analyzer (Applied Biosystems). All primers were purchased from Thermo Scientific.
Tnfα ELISA
Murine TNFα ELISA kit (eBioscience) used per manufacturer recommendations
Flow cytometry
Only fresh cultured cells or tissue prepared into single cell suspensions were analyzed. The harvest and digestion of tumors into single cell suspensions was performed as previously described.Citation31 Nonspecific staining was minimized by staining with CD16/32 (FcR). Anti-mouse SIINFEKL:H-2Kb (clone 25-D1.16), CD45.2 (104), CD3 (17A2), CD8 (53–5.8), PD-1 (RMP1-14) and PD-L1 (10F.9G2) antibodies (Biolegend) and anti-mouse TNFα (MP6-XT22, eBioscience) antibodies were applied for 30–60 minutes at titrated concentrations. For tetramer staining, the CD8 clone KT15 was specifically used to identify CD8 TIL and SIINFEKL:H-2Kb tetramer was purchased from MBL (validated in OT-I transgenic mice). Dead cells excluded via 7AAD uptake and a “fluorescence-minus-one” technique was used to validate specific staining. For intracellular TNFα analysis, treated cells were exposed to brefeldin A for the final 4 hours of incubation. Fixation, permeation and staining were achieved with the intracellular fixation and permeabilization kit (eBioscience #88–8823-88) per manufacturer recommendations. All analyses were performed on a BD FACSCanto analyzer running FACSDiva software and interpreted using FlowJo (vX10.0.7r2).
Cell cycle analysis
The Click-iT EdU flow cytometry assay kit (Thermo) was used per manufacturer recommendations, with the addition of primary conjugated antibodies for p-HH3S10 (clone 11D8) and p-γH2AXS139 (clone 2F3) from Biolegend after fixation and permeabilization. As a positive control for cell cycle analysis and DNA damage, pMOC1 cells were irradiated (single fraction of 8Gy) using a 137Cs source cell irradiator (Gammacell-1000) at a dose rate of 0.74 Gy/min.
Western blot analysis
Standard lysate acquisition and blotting techniques were used. Anti-mouse CDK1, p-CDK1, p-γH2AXS139, p-p65S536, Lamin A/C, and β-actin antibodies were purchased from Cell Signaling Technologies. Blots were imaged using Image Studio software (LI-COR Biosciences).
Immunofluorescence
Staining was performed as previouslyCitation30 described using an unconjugated p-γH2AXS139 primary antibody and secondary antibody conjugated to AF594 from Life Technologies.
T-lymphocyte suppression assays
For antigen specific OT-I T-lymphocyte stimulation, OT-I splenocytes were harvested and T-lymphocytes were sorted on an autoMACS magnetic sorter (Miltenyi Biotec) using the pan T-cell negative selection kit from Miltenyi (#130–095-130) per the manufacturer’s instructions. Sorted T-lymphocytes were then CFSE labeled and combined with WT B6 splenocytes pulsed with SIINFEKL (2 μg/mL). For non-specific stimulation, WT B6 splenic T-lymphocytes were sorted, CFSE-labelled and stimulated 1:1 with anti-CD3/anti-CD28 coated dynabeads (Thermo). CFSE spread was analyzed by flow cytometry.
Statistics
Tests of significance between pairs of data are reported as p-values, derived using a Fisher’s exact test or student’s t-test with a two-tailed distribution and calculated at 95% confidence. Comparison of multiple sets of data was achieved with one or two-way analysis of variance (ANOVA). Error bars reflect standard deviation from individual experiments. Analysis was performed using GraphPad Prism v6.
Disclosure of Potential Conflicts of Interest
The authors have no financial conflicts of interest to disclose. AZD1775 was provided by AstraZeneca through a Cooperative Research and Development Agreement.
Supplemental Material
Download MS Word (3.6 MB)Acknowledgments
We thank Drs. Christian Hinrichs and Claudia Palena for their critical reviews of this manuscript.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Gajewski TF, Meng Y, Harlin H. Immune suppression in the tumor microenvironment. J Immunother. 2006;29:233–240. doi:10.1097/01.cji.0000199193.29048.56.
- Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, Lepage P, Boneca IG, Chamaillard M, Kroemer G, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. 2016;44:1255–1269. doi:10.1016/j.immuni.2016.06.001.
- Davis RJ, Van Waes C, Allen CT. Overcoming barriers to effective immunotherapy: mDSCs, TAMs, and Tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral Oncol. 2016;58:59–70. doi:10.1016/j.oraloncology.2016.05.002.
- Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi:10.1016/j.immuni.2004.07.017.
- Bai XF, Liu J, Li O, Zheng P, Liu Y. Antigenic drift as a mechanism for tumor evasion of destruction by cytolytic T lymphocytes. J Clin Invest. 2003;111:1487–1496. doi:10.1172/JCI17656.
- Leisegang M, Engels B, Schreiber K, Yew PY, Kiyotani K, Idel C, Arina A, Duraiswamy J, Weichselbaum RR, Uckert W, et al. Eradication of large solid tumors by gene therapy with a T-cell receptor targeting a single cancer-specific point mutation. Clin Cancer Res. 2016;22:2734–2743. doi:10.1158/1078-0432.CCR-15-2361.
- Zheng P, Guo Y, Niu Q, Levy DE, Dyck JA, Lu S, Sheiman LA, Liu Y. Proto-oncogene PML controls genes devoted to MHC class I antigen presentation. Nature. 1998;396:373–376. doi:10.1038/24628.
- Ferris RL, Whiteside TL, Ferrone S. Immune escape associated with functional defects in antigen-processing machinery in head and neck cancer. Clin Cancer Res. 2006;12:3890–3895. doi:10.1158/1078-0432.CCR-05-2750.
- Zeligs KP, Neuman MK, Annunziata CM. Molecular pathways: the balance between cancer and the immune system challenges the therapeutic specificity of targeting nuclear factor-KappaB signaling for cancer treatment. Clin Cancer Res. 2016;22:4302–4308. doi:10.1158/1078-0432.CCR-15-1374.
- Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–392. doi:10.1038/nrm3115.
- Allday MJ, Inman GJ, Crawford DH, Farrell PJ. DNA damage in human B cells can induce apoptosis, proceeding from G1/S when p53 is transactivation competent and G2/M when it is transactivation defective. EMBO J. 1995;14:4994–5005.
- Toledo L, Neelsen KJ, Lukas J. Replication catastrophe: when a checkpoint fails because of exhaustion. Mol Cell. 2017;66:735–749. doi:10.1016/j.molcel.2017.05.001.
- Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, et al. Small-molecule inhibition of WEE1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi:10.1158/1535-7163.MCT-09-0463.
- Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2007;19:238–245. doi:10.1016/j.ceb.2007.02.009.
- De Witt Hamer PC, Mir SE, Noske D, Van Noorden CJ, Wurdinger T. WEE1 kinase targeting combined with DNA-damaging cancer therapy catalyzes mitotic catastrophe. Clin Cancer Res. 2011;17:4200–4207. doi:10.1158/1078-0432.CCR-10-2537.
- Eytan DF, Snow GE, Carlson S, Derakhshan A, Saleh A, Schiltz S, Cheng H, Mohan S, Cornelius S, Coupar J, et al. SMAC mimetic birinapant plus radiation eradicates human head and neck cancers with genomic amplifications of cell death genes FADD and BIRC2. Cancer Res. 2016;76:5442–5454. doi:10.1158/0008-5472.CAN-15-3317.
- Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS, Tran JH, Nedospasov SA, Liu Z-G. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem. 2004;279:10822–10828. doi:10.1074/jbc.M313141200.
- Campana S, De Pasquale C, Carrega P, Ferlazzo G, Bonaccorsi I. Cross-dressing: an alternative mechanism for antigen presentation. Immunol Lett. 2015;168:349–354. doi:10.1016/j.imlet.2015.11.002.
- Chen G, Shi L, Litchfield DW, Greenberg AH. Rescue from granzyme B-induced apoptosis by WEE1 kinase. J Exp Med. 1995;181:2295–2300.
- Garimella SV, Rocca A, Lipkowitz S. WEE1 inhibition sensitizes basal breast cancer cells to TRAIL-induced apoptosis. Mol Cancer Res. 2012;10:75–85. doi:10.1158/1541-7786.MCR-11-0500.
- Tanaka N, Patel AA, Tang L, Silver NL, Lindemann A, Takahashi H, Jaksik R, Rao X, Kalu NN, Chen T-C, et al. Replication stress leading to apoptosis within the s-phase contributes to synergism between vorinostat and AZD1775 in HNSCC harboring high-risk TP53 mutation. Clin Cancer Res. 2017;23:6541–6554. doi:10.1158/1078-0432.CCR-17-0947.
- Hauge S, Naucke C, Hasvold G, Joel M, Rodland GE, Juzenas P, Stokke T, Syljuåsen RG. Combined inhibition of WEE1 and Chk1 gives synergistic DNA damage in S-phase due to distinct regulation of CDK activity and CDC45 loading. Oncotarget. 2017;8:10966–10979. doi:10.18632/oncotarget.14089.
- Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–2837. doi:10.1038/sj.onc.1207528.
- Hamilton DH, Huang B, Fernando RI, Tsang KY, Palena C. WEE1 inhibition alleviates resistance to immune attack of tumor cells undergoing epithelial-mesenchymal transition. Cancer Res. 2014;74:2510–2519. doi:10.1158/0008-5472.CAN-13-1894.
- Osman AA, Monroe MM, Ortega Alves MV, Patel AA, Katsonis P, Fitzgerald AL, Neskey DM, Frederick MJ, Woo SH, Caulin C, et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol Cancer Ther. 2015;14:608–619. doi:10.1158/1535-7163.MCT-14-0735-T.
- Melnikova VO, Bolshakov SV, Walker C, Ananthaswamy HN. Genomic alterations in spontaneous and carcinogen-induced murine melanoma cell lines. Oncogene. 2004;23:2347–2356. doi:10.1038/sj.onc.1207405.
- Onken MD, Winkler AE, Kanchi KL, Chalivendra V, Law JH, Rickert CG, Kallogjeri D, Judd NP, Dunn GP, Piccirillo JF, et al. A surprising cross-species conservation in the genomic landscape of mouse and human oral cancer identifies a transcriptional signature predicting metastatic disease. Clin Cancer Res. 2014;20:2873–2884. doi:10.1158/1078-0432.CCR-14-0205.
- Hilburger Ryan M, Abrams SI. Characterization of CD8+ cytotoxic T lymphocyte/tumor cell interactions reflecting recognition of an endogenously expressed murine wild-type p53 determinant. Cancer Immunol Immunother. 2001;49:603–612.
- Shah S, Caruso A, Cash H, Waes CV, Allen CT. Pools of programmed death-ligand within the oral cavity tumor microenvironment: variable alteration by targeted therapies. Head Neck. 2016;38:1176–1186. doi:10.1002/hed.24269.
- Clavijo PE, Moore EC, Chen J, Davis RJ, Friedman J, Kim Y, Van Waes C, Chen Z, Allen CT. Resistance to CTLA-4 checkpoint inhibition reversed through selective elimination of granulocytic myeloid cells. Oncotarget. 2017;8:55804–55820. doi:10.18632/oncotarget.18437.
- Moore E, Clavijo PE, Davis R, Cash H, Van Waes C, Kim Y, Allen C. Established T cell-inflamed tumors rejected after adaptive resistance was reversed by combination STING activation and PD-1 pathway blockade. Cancer Immunol Res. 2016. doi:10.1158/2326-6066.CIR-16-0104.
- Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, Silvin C, Van Waes C, Allen C. Anti-PD-L1 efficacy can be enhanced by inhibition of myeloid-derived suppressor cells with a selective inhibitor of PI3Kdelta/gamma. Cancer Res. 2017;77:2607–2619. doi:10.1158/0008-5472.CAN-16-2534.
- Zhao X, Subramanian S. Intrinsic resistance of solid tumors to immune checkpoint blockade therapy. Cancer Res. 2017;77:817–822. doi:10.1158/0008-5472.CAN-16-2379.
- Shi L, Nishioka WK, Th’ng J, Bradbury EM, Litchfield DW, Greenberg AH. Premature p34cdc2 activation required for apoptosis. Science. 1994;263:1143–1145.
- Booher RN, Holman PS, Fattaey A. Human Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not Cdk2 activity. J Biol Chem. 1997;272:22300–22306.
- Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955–1957.
- Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. doi:10.1038/374131a0.
- Cuneo KC, Morgan MA, Davis MA, Parcels LA, Parcels J, Karnak D, Ryan C, Liu N, Maybaum J, Lawrence TS. WEE1 kinase inhibitor AZD1775 radiosensitizes hepatocellular carcinoma regardless of TP53 mutational status through induction of replication stress. Int J Radiat Oncol Biol Phys. 2016;95:782–790. doi:10.1016/j.ijrobp.2016.01.028.
- Leijen S, Van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, Lam R, Demuth T, Rose S, Lee MA, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2016;34:4371–4380. doi:10.1200/JCO.2016.67.5991.
- Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, Collins J, Chen AP, Doroshow JH, Kummar S. Phase I study of single-agent AZD1775 (MK-1775), a WEE1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol. 2015;33:3409–3415. doi:10.1200/JCO.2014.60.4009.
- Mendez E, Rodriguez CP, Kao M, Harbison RA, Martins RG, Futran ND, Santana-Davila R, Chow LQM. A phase I clinical trial of AZD1775 in combination with neoadjuvant weekly cisplatin and docetaxel in borderline resectable head and neck squamous cell carcinoma (HNSCC). Chicago, IL: ASCO; 2017.
- Darzynkiewicz Z, Williamson B, Carswell EA, Old LJ. Cell cycle-specific effects of tumor necrosis factor. Cancer Res. 1984;44:83–90.
- Baxter GT, Kuo RC, Jupp OJ, Vandenabeele P, MacEwan DJ. Tumor necrosis factor-alpha mediates both apoptotic cell death and cell proliferation in a human hematopoietic cell line dependent on mitotic activity and receptor subtype expression. J Biol Chem. 1999;274:9539–9547.
- Fleischer B. Lysis of bystander target cells after triggering of human cytotoxic T lymphocytes. Eur J Immunol. 1986;16:1021–1024. doi:10.1002/eji.1830160826.
- Kinkhabwala M, Sehajpal P, Skolnik E, Smith D, Sharma VK, Vlassara H, Cerami A, Suthanthiran M. A novel addition to the T cell repertory. Cell surface expression of tumor necrosis factor/cachectin by activated normal human T cells. J Exp Med. 1990;171:941–946.
- Duke RC. Self recognition by T cells. I. Bystander killing of target cells bearing syngeneic MHC antigens. J Exp Med. 1989;170:59–71.
- Mackay F, Loetscher H, Stueber D, Gehr G, Lesslauer W. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J Exp Med. 1993;177:1277–1286.
- Kojima H, Eshima K, Takayama H, Sitkovsky MV. Leukocyte function-associated antigen-1-dependent lysis of Fas+ (CD95+/Apo-1+) innocent bystanders by antigen-specific CD8+ CTL. J Immunol. 1997;159:2728–2734.
- Schuler T, Blankenstein T. Cutting edge: CD8+ effector T cells reject tumors by direct antigen recognition but indirect action on host cells. J Immunol. 2003;170:4427–4431.
- Fankhauser M, Broggi MAS, Potin L, Bordry N, Jeanbart L, Lund 890 AW, Da Costa E, Hauert S, Rincon-Restrepo M, Tremblay C,et al. Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci Transl Med. 2017:9: eaal4712.
- Aranda F, Llopiz D, Diaz-Valdes N, Riezu-Boj JI, Bezunartea J, Ruiz M, Martínez M, Durantez M, Mansilla C, Prieto J, et al. Adjuvant combination and antigen targeting as a strategy to induce polyfunctional and high-avidity T-cell responses against poorly immunogenic tumors. Cancer Res. 2011;71:3214–3224. doi:10.1158/0008-5472.CAN-10-3259.
- Spiotto MT, Rowley DA, Schreiber H. Bystander elimination of antigen loss variants in established tumors. Nat Med. 2004;10:294–298. doi:10.1038/nm999.
- Zhang B, Karrison T, Rowley DA, Schreiber H. IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J Clin Invest. 2008;118:1398–1404. doi:10.1172/JCI33522.
- Zhang D, Beresford PJ, Greenberg AH, Lieberman J. Granzymes A and B directly cleave lamins and disrupt the nuclear lamina during granule-mediated cytolysis. Proc Natl Acad Sci U S A. 2001;98:5746–5751. doi:10.1073/pnas.101329598.
- Rao L, Perez D, White E. Lamin proteolysis facilitates nuclear events during apoptosis. J Cell Biol. 1996;135:1441–1455.
- Judd NP, Winkler AE, Murillo-Sauca O, Brotman JJ, Law JH, Lewis JS Jr., Dunn GP, Bui JD, Sunwoo JB, Uppaluri R. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res. 2012;72:365–374. doi:10.1158/0008-5472.CAN-11-1831.
- Browne KA, Blink E, Sutton VR, Froelich CJ, Jans DA, Trapani JA. Cytosolic delivery of granzyme B by bacterial toxins: evidence that endosomal disruption, in addition to transmembrane pore formation, is an important function of perforin. Mol Cell Biol. 1999;19:8604–8615.