
ABSTRACT
Background: B cells can function as antigen-presenting cells by presenting antigens captured by the B-cell receptor (BCR) on Class II Major Histocompatibility Complex (MHC II) to T cells. In addition, B-cells can also maintain immune homeostasis by expressing PD-L1 and suppressing T-cell activity. Epstein-Barr virus (EBV) infection can disrupt B-cell function and lead to B cell malignancies, including diffuse large B-cell lymphoma (DLBCL). Here we show that EBV-positive DLBCL (EBV+ DLBCL) has decreased expression of BCR and MHC II, but over-expressed PD-L1, which may lead to immune evasion.
Methods: An EBV+ DLBCL cohort (n = 30) and an EBV- DLBCL control cohort (n = 83) were established. Immunostaining of PD-L1, MHC II, MHC II Transactivator (CIITA) and pBTK was performed on automated stainer. H-score was used to denote the results of staining of PD-L1 and pBTK. Break apart and deletion of CIITA locus was studied by fluorescent in situ hybridization. Surface immunoglobulin mean fluorescent insensitivity (MFI) was detected by flow cytometry to demonstrate the level BCR.
Results: EBV+ DLBCL showed significantly lower expression of CIITA and MHC II compared to EBV- DLBCL. Genetic aberrations involving CIITA were also more common in EBV+ DLBCL, with 23% break apart events and 6% deletion events, comparted to 2% break apart and 0% deletion in EBV- DLBCL. In addition to the loss of antigen presentation molecule, the antigen capture receptor, BCR, was also down-regulated in EBV+ DLBCL. Accordingly, BCR signaling was also significantly decreased in EBV+ DLBCL as denoted by the respective pBTK levels.
Conclusions: EBV+ DLBCL shows over expression of the T-cell inhibitory ligand, PD-L1. Antigen capture and presentation system were disrupted, and T-cell inhibitory molecule was hijacked in EBV+ DLBCL, which may contribute to immune escape in this high risk disease. Therapies targeting these aberrations may improve the outcome of patients with EBV+ DLBCL.
Introduction
Epstein–Barr virus-positive diffuse large B-cell lymphoma (EBV+ DLBCL), recognized by the updated WHO classification of lymphoid neoplasm as a new entity, is a disease with high risk and poor prognosis.Citation1 Systemic immunosuppression or immune escapeCitation2,Citation3 in the microenvironment have been found to contribute to the pathogenesis of EBV+ DLBCL. However, the biologic mechanism resulting in the immune escape remains unknown.
B cells may modulate immune function through various means. They may function as antigen-presenting cells by capturing antigens via the BCR and presenting them to T cells through MHC II.Citation4 Antigen capturing also triggers the BCR signaling cascade to help maintain B cell survival and differentiation. In addition to antigen presentation, B cells can also express PD-L1 to suppress T cell function and maintain immune homeostasisCitation5 These immune-regulatory properties may be retained during B cell transformation and hijacked by the malignant cells as a survival mechanism. In particular, B cell infected with EBV often show disrupted function and differentiation, such as abnormal plasmacytic differentiation and aberrant expression of latent membrane protein 1 (LMP1).Citation6,Citation7 We hypothesize that EBV infection may also cause changes in the immunomodulatory activities of transformed B-cells, including antigen capture, antigen presentation, and T-cell suppression in EBV+ DLBCL, leading to the aggressive phenotype observed clinically. Here we compare tissue samples from EBV+ and EBV- DLBCL patients to determine if EBV+ DLBCL has altered BCR activity or aberrant expression of MHC II and PD-L1.
Results
Demographics and clinicopathological features
Thirty EBV+ DLBCL and 83 EBV- DLBCL patient samples were included for analysis. The clinicopathologic features of the patients are summarized in . Overall, the two groups do not have significant differences in gender, age, IPI, bulky disease, and immunosuppression status. Consistent with previous reports,Citation8–Citation11 EBV+DLBCL patients were predominantly of non-GCB subtype (27/30, 90%) compared to EBV-DLBCL (44/83, 53%, P = .001).
Table 1. Clinico-pathologic features of EBV+ and EBV- DLBCL
Antigen presentation elements were deficient in EBV+ DLBCL
Normal B cells function as antigen-presenting cells by presenting processed antigen peptide on MHC II to T cells and activate immune response. As EBV+ DLBCL show features of systemic immunosuppression, we hypothesized that the malignant cells may achieve this through down-regulation of MHC II which would undermine antigen presentation and T cell activation. As expected, MHC II protein was expressed only in 9 out of 30 EBV+ DLBCL cases (30%), compared with 49 out of 83 (59%) in EBV- DLBCL (P = .006, and ).
Figure 1. EBV+ DLBCL features deficiency in antigen presentation elements. (a) Compared to EBV- DLBCL, EBV+ DLBCL demonstrated loss expression of MHC II and its transcription activator, CIITA. Red arrow highlighted scattered macrophages positive for MHC II or CIITA in EBV+ DLBCL. (b) Representative cases of EBV- DLBCL showed CIITA gene locus deletion or break-apart
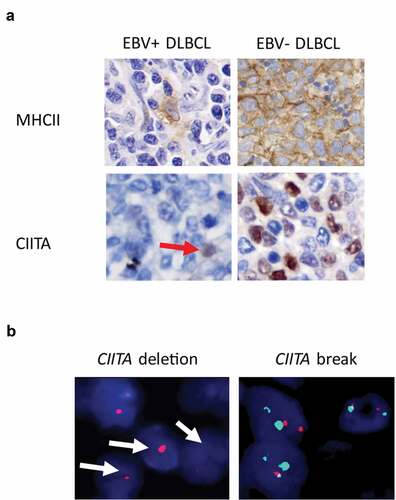
As MHC II protein expression is highly dependent on the transcriptional co-activator CIITA, we next sought to determine if CIITA is altered in EBV+DLBCL. Indeed, CIITA IHC positivity was demonstrated in fewer cases in EBV+DLBCL compared to EBV-DLBCL ( and , 43% vs. 79%, P = .001). To understand if loss of CIITA is associated with genetic alteration, we performed FISH analysis on CIITA gene. We found that CIITA genetic aberrations, including seven cases (23%) of break apart and two cases (6%) of gene deletion, were detected in EBV+ DLBCL samples ( and ). In contrast, CIITA alterations were scarcely found in EBV- DLBCL (2 out of 83 cases, 2% break apart, ). Thus MHC II protein, as well as its upstream transcription factor CIITA, are frequently disrupted in EBV+ DLBCL.
Antigen capture elements were deficient in EBV+ DLBCL
In order to present antigens on MHC II molecules, B cells must first acquire antigens through BCR in a specific manner. This will initiate the BCR signaling cascade, which activates the B cells and coordinates various cellular activities.Citation12 B-cell antigen capturing capability is partially preserved in B cell lymphomas.Citation13–Citation17 To evaluate if this feature is also altered in EBV+ DLBCL, we analyzed the level of BCR signaling activity via IHC staining for pBTK.
In the EBV- DLBCL cohort, the level of pBTK was heterogenous with a median IHC score of 85 (ranging from 0 to 300) indicating a various activated status of BCR signaling (, ). In contrast, pBTK was consistently expressed at much lower levels in the EBV+ DLBCL cohort, with a median IHC score of 5 (ranging from 0 to 60, , ).
Figure 2. EBV+ DLBCL features down-regulated antigen-capture elements. (a&b) Compared to EBV- DLBCL, EBV+ DLBCL demonstrated decreased level of B-cell receptor signaling kinase, pBTK. (c) Latent membrane protein 1 was expressed on TMD8 cells after being successfully infected by EBV. (d&e) Surface IgG was decreased after EBV infection. Representative flow results were presented in E; assays were performed in triplicate
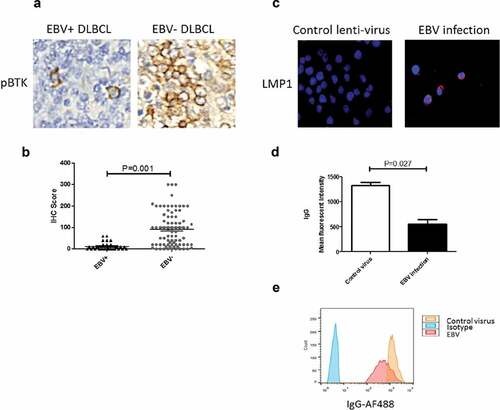
EBV infection has been found to disrupt various functions of B-cells, which could also impact the integrity of BCR. To test this hypothesis, we infected TMD8 cells with EBV, followed by flow cytometry to detect surface IgG. LMP1 immunofluorescent staining was used to confirm EBV infection status (). As expected, EBV infection resulted in significantly lower surface IgG mean fluorescent intensity (MFI) compared to cells infected with control virus (). We thus showed that EBV infection not only undermines BCR signaling but also disrupts its integrity in DLBCL.
EBV+ DLBCL features a hijacked T-cell suppression program
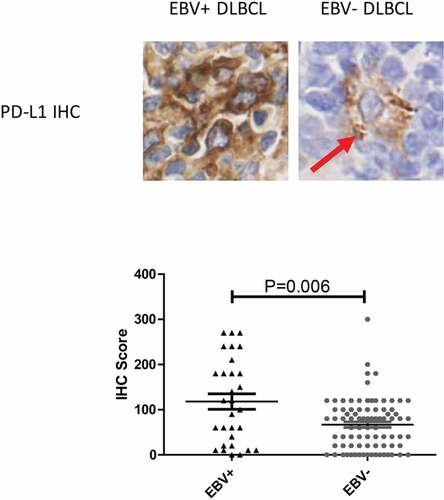
PD-L1 is normally expressed on antigen-presenting cells, including B cells, to interact with PD-l receptors on T cells and dampen their activity, this mechanism is often co-opted by malignant cells, in which PD-L1 over-express was a means of immune escape. A portion of DLBCL cases were also found to express high level of PD-L1. To determine if PD-L1 expression is also altered in EBV+ DLBCL, we analyzed PD-L1 IHC level in our patient cohort. Interestingly, although PD-L1 was expressed heterogeneously in both EBV- and EBV+ DLBCL, the level of PD-L1 in EBV+ DLBCL was much higher than that in EBV- DLBCL ( and , median IHC score 110 vs. 80, P = .006), indicating that PD-L1 expression may be hijacked by EBV to induce T-cell suppression.
Figure 3. PD-L1 was over-expressed in EBV+ DLBCL. Red arrow highlighted scattered macrophages express PD-L1
Discussion
EBV infection-associated lymphoproliferative diseases represent a spectrum of heterogenous diseases from self-limiting conditions to overt malignancies. Considering that younger EBV+ DLBCL patients show better prognosis than old patients, the strategy shown in this study might generate better results when age was stratified in two groups. We indeed tried performing the subgroup analysis, but the total sample size of EBV+ cohort was small, undermining the statistic power. In addition, although some EBV infection-associated lymphoproliferative diseases initially present as self-limiting or indolent, they may potentially progress to aggressive malignancies. EBV is not only contribute to disease progression but also associated with more aggressive behaviors in lymphomas, as EBV-positive lymphomas tend to be more aggressive than their EBV-negative counterpartsCitation18 This may be achieved through immune disruption at the local tumor environment or at a systemic level. However, the detailed mechanisms responsible for EBV-induced immune-suppression phenotype are largely unknown.
As B-cells can function as antigen-presenting cells and regulate T-cells, we hypothesized that the aberrant host response against EBV-associated lymphomas may be driven by abnormal B-cell antigen capture, presentation, and T-cell interaction. DLBCL, NOS, an aggressive B-cell malignancy has been shown to have deficient antigen presentation through loss of MHC II or its transcriptional co-activator CIITA. Here we show that in EBV+ DLBCL, an EBV infected and clonally driven DLBCL, showed significantly more disruption in antigen-presenting elements as most EBV+ DLBCL lost expression of both MHC II (negativity in 70% cases) and CIITA (negativity in 57% cases). While CIITA alteration may be directly linked to MHC II loss, there are likely additional mechanisms that suppress MHC II expression in EBV+ DLBCL, since CIITA expression is unchanged in a portion of MHCII negative cases. We also found multiple EBV+ DLBCL cases that show CIITA genetic aberration including break-part and gene locus deletion, though further studies would be needed to determine the cause of CIITA loss in cases without genetic alteration. What’s more, there are some recently studies showed that EBV+ DLBCL do not appear to demonstrate CIITA mutations at significant levels, but exhibited a genetic profile distinct from EBV negative one, characterized by frequent TET2 and DNMT3A mutations and the paucity of CD79B, MYD88, CDKN2A, and FAS alterationsCitation19 These results will provide a new insight for further research of cases without CIITA genetic alteration.
In addition to deficient antigen presentation machinery in EBV+ DLBCL, we found that BCR structure and signaling activity are also significantly disrupted compared to EBV- DLBCL. Surprisingly, even though EBV+ DLBCL are predominantly of non-GCB, or ABC immunophenotype, the malignant cells demonstrated very low level of BCR signaling as denoted by pBTK level, which suggests that BCR signaling inhibitor such as ibrutinib would be ineffective against this particular non-GCB disease. Early studies showed that EBV infection can induce B-cell abnormal differentiation toward and down-regulate BCR signalingCitation6 Additionally, the latent membrane protein 1/2A (LMP 1/2A) coded by the EBV genome may transmit truncated BCR downstream signaling as a BCR surrogateCitation20 Consistent with the down-regulation of BCR signaling, the level of surface immunoglobulin (repaent of BCR) is also down-regulated by EBV infection, as the membrane conjugated immunoglobulin (BCR) program is shifted toward secretory immunoglobulin (antibody) during EBV-driven plasma cell differentiation.Citation21
Finally, we found that EBV infection is associated with over-expression of PD-L1 in our cohort. Similar findings have also been observed in EBV+ DLBCL cohorts reported by othersCitation22 as well as EBV infection-associated solid tumorsCitation23 We speculate that the high level of PD-L1 expression on lymphoma cells will result in T cell anergy, rendering the massive infiltration of T cells in EBV+ DLBCL ineffective and even harmful to the host, and PD-L1 or PD-1 inhibitors may potentially benefit patient with this particular type of DLBCL. The connection between PD-L1 overexpression and EBV infection is still unclear. Human papillomavirus (HPV) infection has been reported to induce PD-L1 overexpression by disrupting PD-L1 negative regulation element in the untranslated region,Citation24 whether EBV alters PD-L1 expression through similar mechanisms remain to be explored. There is also hypothesis that it would appear PD-L1 genetic alterations are key in EBV+ lymphomas, so it would be of interest to see their occurrence in this cohort. A high frequency of PD-L1/PD-L2-involving genetic aberrations was observed in EBV-positive diffuse large B-cell lymphoma (DLBCL, 19%)Citation19 However, one study by another group from our department shows that PD-L1 genetic alteration is a rare event even in the EBV+ positive subgroup.Citation25 In our EBV+ positive cohort, no PD-L1 genetic alteration was identified either.
In conclusion, we show evidence of immune disruption in EBV+ DLBCL, including deficient antigen capture and presentation machinery, as well as over-expression of PD-L1, which may contribute to immune escape in this high-risk disease. Therapies targeting these aberrations, such as immune checkpoint inhibitors may improve the outcome of patients with EBV+ DLBCL.
Materials and methods
Case selection
We retrospectively reviewed archives maintained at the Department of Pathology, Fudan University Shanghai Cancer Centre from 2010 to 2013. All cases included in current work received histological confirmation as de novo DLBCL, not otherwise specified (DLBCL, NOS) by two independent pathologists (X-N J and X-Q L) in compliance with WHO classification. EBV infection status was determined with EBV-encoded RNA (EBER) in situ hybridization. The use of human samples was approved by the ethical committee of Fudan University Shanghai Cancer Centre.
Morphology and immunohistochemistry
Formalin-fixed, paraffin-embedded (FFPE) tissues were obtained and cut into 4-μm sections for morphological examination via H&E staining. PD-L1 antibody (clone number: 28–8, Abcam, Cambridge, MA; 1:200 dilution), MHC II antibody (clone number: CR3/43, Abcam, Cambridge, MA; 1:500 dilution) and CIITA (clone number: 7-1H, Santa Cruz Biotechnology, Dallas, TX; 1:250 dilution) were applied on BOND-III automated immunostainer (Leica Biosystems, Melbourne, Australia). The following antibodies (Ventana Medical Systems, Tucson, Arizona, USA) were applied on BenchMark XT automated immunostainer (Ventana Medical Systems, Tucson, Arizona, USA) with Cell Conditioning 1 heat retrieval solution (Ventana Medical Systems, Tucson, Arizona, USA): CD10, BCL6, and MUM1. Antibody of phospho (pY223) BTK (Abcam, Cambridge, MA, USA) in 1:100 dilution was applied manually using Envision Method (Dako, Glostrup, Denmark) according to the manufacturer’s protocol. For all stainings, tonsils with reactive hyperplasia were served as external controls, and the reactive lymphocytes as internal controls. The cutoff value of positivity for CIITA and MHC II was 40%, and cutoff value of positivity for CD10, BCL6, and MUM1 was set at 30%. IHC results of pBTK and PD-L1 were calculated as IHC score by multiply the percentage of positive cells (0 to 100, recorded in the increment by 5%) with mean intensity (0, no staining; 1, weak staining; 2, moderate staining; 3, strong staining), and given a range from 0 to 300. Cases were designated as GCB or non-GCB, using the algorithm specified by Hans et al.Citation26 Two pathologists (X-N J and B-H Y) were independently responsible for evaluating the morphological and IHC results.
Genetics
Interphase FISH technique was used to detect CIITA gene breaks, and CIITA gene deletion was performed with CIITA Break Apart FISH Probe (Empire Genomics, Buffalo, NY, USA) and CIITA FISH Probe (Empire Genomics, Buffalo, NY, USA), respectively, using FISH-Tissue Implementation Kit (ZytoVision, Bremerhaven, Germany) according to the protocol specified by the manufacturer. Fifty interphase nuclei were counted for each tested probe. Break apart signal in >15% nuclei was defined as translocation. Less than 2 CIITA signal in >30% nuclei was defined as deletion.
Cell lines and EBV infection
TMD8 DLBCL cell line was used in this study. TMD8 was kindly provided by Dr. Lynn Y. Wang (University of Chicago, Chicago, IL, USA). Cells were maintained at 37 C° in a 5% CO2 incubator and grown in 1640 medium (Gibco, Grand Island, NE, USA) supplemented with 10% FBS (Gibco, Grand Island, NE, USA). EBV infection was performed according to the published protocols.Citation27 Briefly, 2*106/ml TMD8 DLBCL cells were infected with EBV with a multiple of infection (MOI) of 100, or with blank control lentivirus with a MOI of 20. Immunofluorescent staining of LMP1 (Abcam, clone number: CS 1–4; Cambridge, MA; 1:300 dilution) was performed to confirm the successful infection and latent infection status of EBV, according to the assay recommended by the manufacturer.
Flow cytometry analysis of surface igg
DLBCL cells were stained with Anti-PD-L1-Alexa Fluor 488 antibody without any permeabilization on ice for 20 min and analyzed by flow cytometry using Accuri C6 (BD, Biosciences, San Jose, CA, USA). Data were analyzed using FlowJo software (Tree Star). Experiments were performed in triplicate.
Statistical analyses
Comparisons of continuous variables were performed using unpaired t-test or Mann–Whitney test. Linear correlation and linear regression were used to reveal the relationship between IHC or ISH markers. A P value below 0.05 was considered significant. All analyses were performed using Stata program (V11.0, StataCorp LP, College Station, TX, USA).
Availability of data and material
The datasets used during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare that they have no competing interests.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Authors’ contributions
Formal analysis: XNJ and BHY, investigation: XNJ, methodology: BHY and WHY, project administration: XYZ, resources: XQL, software: XNJ, writing, review &editing: XNJ JL XQL.
Additional information
Funding
References
- Castillo JJ, Beltran BE, Miranda RN, Miranda RN, Young KH, Chavez JC, Sotomayor EM. EBV-positive diffuse large B-cell lymphoma of the elderly: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:529–7. doi:10.1002/ajh.24370.
- Wu L, Ehlin-Henriksson B, Zhou X, Zhu H, Ernberg I, Kis LL, Klein G. Epstein-Barr virus (EBV) provides survival factors to EBV(+) diffuse large B-cell lymphoma (DLBCL) lines and modulates cytokine induced specific chemotaxis in EBV(+) DLBCL. Immunology. 2017;152:562–573. doi:10.1111/imm.12792.
- Healy JA, Dave SS. The role of EBV in the pathogenesis of diffuse large B cell Lymphoma. Curr Top Microbiol Immunol. 2015;390:315–337. doi:10.1007/978-3-319-22822-8_13.
- Chen X, Jensen PE. The role of B lymphocytes as antigen-presenting cells. Arch Immunol Ther Exp (Warsz). 2008;56:77–83. doi:10.1007/s00005-008-0014-5.
- Khan AR, Hams E, Floudas A, Sparwasser T, Weaver CT, Fallon PG. PD-L1hi B cells are critical regulators of humoral immunity. Nat Commun. 2015;6:5997. doi:10.1038/ncomms6997.
- Styles CT, Bazot Q, Parker GA, White RE, Paschos K, Allday MJ. EBV epigenetically suppresses the B cell-to-plasma cell differentiation pathway while establishing long-term latency. Plos Biol. 2017;15:e2001992. doi:10.1371/journal.pbio.2001992.
- Vrzalikova K, Vockerodt M, Leonard S, Bell A, Wei W, Schrader A, Wright KL, Kube D, Rowe M, Woodman CB, et al. Down-regulation of BLIMP1alpha by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood. 2011;117:5907–5917. doi:10.1182/blood-2010-09-307710.
- Oyama T, Ichimura K, Suzuki R, Suzumiya J, Ohshima K, Yatabe Y, Yokoi T, Kojima M, Kamiya Y, Taji H, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16–26. doi:10.1097/00000478-200301000-00003.
- Oyama T, Yamamoto K, Asano N, Oshiro A, Suzuki R, Kagami Y, Morishima Y, Takeuchi K, Izumo T, Mori S, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinctclinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124–5132. doi:10.1158/1078-0432.CCR-06-2823.
- Nicolae A, Pittaluga S, Abdullah S, Steinberg SM, Pham TA, Davies-Hill T, Xi L, Raffeld M, Jaffe ES. EBV-positive large B-cell lymphomas in young patients: a nodal lymphoma with evidence for a tolerogenic immune environment. Blood. 2015;126:863–872. doi:10.1182/blood-2015-02-630632.
- Dojcinov SD, Venkataraman G, Pittaluga S, Wlodarska I, Schrager JA, Raffeld M, Hills RK, Jaffe ES. Age-related EBV-associated lymphoproliferative disorders in the Western population: a spectrum of reactive lymphoid hyperplasia and lymphoma. Blood. 2011;117:4726–4735. doi:10.1182/blood-2010-12-323238.
- Kenkre VP, Kahl BS. The future of B-cell lymphoma therapy: the B-cell receptor and its downstream pathways. Curr Hematol Malig Rep. 2012;7:216–220. doi:10.1007/s11899-012-0127-0.
- Fichtner M, Spies E, Seismann H, Riecken K, Engels N, Gosch B, Dierlamm J, Gerull H, Nollau P, Klapper W, et al. Complementarity determining region-independent recognition of a superantigen by B-cell antigen receptors of mantle cell lymphoma. Haematologica. 2016;101:e378–381. doi:10.3324/haematol.2016.141929.
- Corso J, Pan, K.T., Walter, R., Doebele, C., Mohr, S., Bohnenberger, H., Ströbel, P., Lenz, C., Slabicki, M., Hüllein, J., et al. Elucidation of tonic and activated B-cell receptor signaling in Burkitt’s lymphoma provides insights into regulation of cell survival. Proc Natl Acad Sci U S A. 2016;113:5688–5693. doi:10.1073/pnas.1601053113.
- Young RM, Wu T, Schmitz R, Dawood M, Xiao W, Phelan JD, Xu W, Menard L, Meffre E, Chan WCC, et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proc Natl Acad Sci U S A. 2015;112:13447–13454. doi:10.1073/pnas.1514944112.
- Cha S-C, Qin H, Kannan S, Rawal S, Watkins LS, Baio FE, Wu W, Ong J, Wei J, Kwak B, et al. Nonstereotyped lymphoma B cell receptors recognize vimentin as a shared autoantigen. J Immunol. 2013;190:4887–4898. doi:10.4049/jimmunol.1300179.
- Sachen KL, Strohman MJ, Singletary J, Alizadeh AA, Kattah NH, Lossos C, Mellins ED, Levy S, Levy R. Self-antigen recognition by follicular lymphoma B-cell receptors. Blood. 2012;120:4182–4190. doi:10.1182/blood-2012-05-427534.
- Battle-Lopez A, Gonzalez de Villambrosia S, Nuñez J, Cagigal M-L, Montes-Moreno S, Conde E, Piris MA. Epstein-Barr virus-associated diffuse large B-cell lymphoma: diagnosis, difficulties and therapeutic options. Expert Rev Anticancer Ther. 2016;16:411–421. doi:10.1586/14737140.2016.1149065.
- Keisuke K, Miyoshi H, Sakata S, Dobashi A, Couronné L, Kogure Y, Sato Y, Nishida K, Gion Y, Shiraishi Y, et al. Frequent structural variations involving programmed death ligands in Epstein-Barr virus-associated lymphomas. Leukemia. 2019;33:1687–1699. doi:10.1038/s41375-019-0380-5.
- Engels N, Yigit G, Emmerich CH, Czesnik D, Schild D, Wienands J. Epstein-Barr virus LMP2A signaling in statu nascendi mimics a B cell antigen receptor-like activation signal. Cell Commun Signal. 2012;10:9. doi:10.1186/1478-811X-10-9.
- Seremetis S, Inghirami G, Ferrero D, Newcomb E, Knowles D, Dotto G, Dalla-Favera R. Transformation and plasmacytoid differentiation of EBV-infected human B lymphoblasts by ras oncogenes. Science. 1989;243:660–663. doi:10.1126/science.2536954.
- Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MGM, Xu ML, Yu H, Fletcher CDM, Freeman GJ, Shipp MA, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res. 2013;19:3462–3473. doi:10.1158/1078-0432.CCR-13-0855.
- Fang W, Zhang J, Hong S, Zhan J, Chen N, Qin T, Tang Y, Zhang Y, Kang S, Zhou T, et al. EBV-driven LMP1 and IFN-γ up-regulate PD-L1 in nasopharyngeal carcinoma: implications for oncotargeted therapy. Oncotarget. 2014;5:12189–12202. doi:10.18632/oncotarget.v5i23.
- Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, Maeda T, Nagata Y, Kitanaka A, Mizuno S, et al. Aberrant PD-L1 expression through 3ʹ-UTR disruption in multiple cancers. Nature. 2016;534:402–406. doi:10.1038/nature18294.
- Sun C, Jia Y, Wang W, Bi R, Wu L, Bai Q, Zhou X. Integrative analysis of PD‐L1 DNA status, mRNA status and protein status, and their clinicopathological correlation, in diffuse large B‐cell lymphoma. Histopathology. 2018;74(4):618–628. doi:10.1111/his.13765.
- Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, Müller-Hermelink HK, Campo E, Braziel RM, Jaffe ES, Pan Z, Farinha P, Smith LM, Falini B, Banham AH, Rosenwald A, Staudt LM, Connors JM, Armitage JO, Chan WC. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103:275–282. doi:10.1182/blood-2003-05-1545.
- Hui-Yuen J, McAllister S, Koganti S, Hill E, Bhaduri-McIntosh S. Establishment of Epstein-Barr virus growth-transformed lymphoblastoid cell lines. J Vis Exp. 2011;57:3321. doi:10.3791/3321.