
ABSTRACT
Inhibitor of apoptosis protein (IAP) antagonists have shown activity in preclinical models of head and neck squamous cell carcinoma (HNSCC), and work across several cancer types has demonstrated diverse immune stimulatory effects including enhancement of T cell, NK cell, and dendritic cell function. However, tumor-cell-intrinsic mechanisms for this immune upregulation have been largely unexplored. In this study, we show that ASTX660, an antagonist of cIAP1/2 and XIAP, induces expression of immunogenic cell death (ICD) markers in sensitive HNSCC cell lines in vitro. Experiments in syngeneic mouse models of HNSCC showed that ASTX660 can also enhance radiation-induced ICD in vivo. On a functional level, ASTX660 also enhanced killing of multiple murine cell lines by cytotoxic tumor-infiltrating lymphocytes, and when combined with XRT, stimulated clonal expansion of antigen-specific T lymphocytes and expression of MHC class I on the surface of tumor cells. Flow cytometry experiments in several human HNSCC cell lines showed that MHC class I (HLA-A,B,C) was reliably upregulated in response to ASTX660 + TNFα, while increases in other antigen processing machinery (APM) components were variable among different cell lines. These findings suggest that ASTX660 may enhance anti-tumor immunity both by promoting ICD and by enhancing antigen processing and presentation.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide, with more than 600,000 new cases and 330,000 deaths annually.Citation1 Caused by carcinogen exposure and/or the human papillomavirus (HPV), HNSCC has been traditionally treated with surgery and/or chemoradiation.Citation2 Platinum chemotherapy agents, while effective and commonly used in HNSCC, have significant adverse side effect profiles, with toxicities in numerous organ systems.Citation3 Recent advances with immune checkpoint inhibitors have produced promising results, but not all patients benefit from anti-PD-1 therapy.Citation4,Citation5 As a result, there is a tremendous need for more effective and less toxic systemic therapies that further enhance radiotherapy and immunotherapies such as immune checkpoint blockade.
Recently, The Cancer Genome Atlas (TCGA) analysis of 279 HNSCCs revealed genomic alterations in cell death pathways, with 30% of patients expressing amplications of Fas-associated death domain (FADD), with or without Baculovirus Inhibitor of Apoptosis repeat containing (BIRC2/3) genes that encode for cellular Inhibitor of Apoptosis Proteins 1/2 (cIAP1/2).Citation6–Citation8 Both FADD and cIAP1/2 play critical roles in the Tumor Necrosis Factor (TNF) Receptor family signaling pathways that determine cell death or survival.Citation9–Citation12 Increased expression of IAPs seen in HNSCC acts on these pathways by inhibiting both caspase-mediated apoptosis and/or RIP-mediated necroptosis, and enhancing pro-survival signaling.Citation12–Citation15 As a result, IAP antagonists, also called SMAC (second mitochondria-derived activator of caspases) mimetics have been developed to counter these effects and restore a pro-apoptotic response.Citation7,Citation16–Citation19 In addition to their pro-apoptotic properties, emerging data now suggests that IAP antagonists may have beneficial effects on multiple aspects of anti-tumor immunity.Citation8,Citation20–Citation26
ASTX660 is a synthetic small molecule antagonist of IAPs, leading to degradation of cIAP1/2 and inhibition of XIAP.Citation27 Our prior work in the mouse oral cancer 1 (MOC1) syngeneic mouse model showed ASTX660 to be highly efficacious when paired with radiation and/or anti-PD-1 immune checkpoint blockade.Citation8 Further experiments showed that the efficacy of ASTX660 + radiation was highly dependent on CD8 + T cells, natural killer (NK) cells, and TNF-α; dendritic cell numbers and activation were also enhanced.Citation8 In the present study, we further investigated the anti-tumor immune effects of ASTX660, focusing on early changes in the tumor cells that promote these immune responses.
Based on the prior finding of increased dendritic cell numbers and activation,Citation8 we hypothesized that ASTX660-induced changes in tumor cells may be promoting immunogenic cell death (ICD). ICD is a process by which tumor cells, when exposed to select stressors, release damage-associated molecular patterns (DAMPs) as they are dying that markedly increase their immunogenicity, leading to subsequent CD8+ dendritic cell activation and a more robust anti-tumor adaptive immune response.Citation28–Citation30 Classically described DAMPs include tumor surface expression of calreticulin (CRT) along with release of both high-mobility group box 1 protein (HMGB1) and adenosine-5ʹ-triphosphate (ATP) from the intracellular environment.Citation28,Citation29,Citation31 Other DAMPs include surface exposure of heat shock proteins (HSPs) 70/90 as well as release of intracellular CXCL10.Citation28,Citation29 During ICD, type 1 interferon (IFN) is also produced by the tumor and/or immune cells.Citation32,Citation33
Finally, given that ASTX660 is able to enhance T-cell killing of tumor cells in the absence of professional antigen-presenting cells (APCs),Citation8 we hypothesized that this drug may be enhancing the ability of tumor cells to process and present tumor antigens, in part by increasing the expression of MHC class I and other antigen processing machinery (APM) components. We used multiple human HNSCC cell lines and syngeneic mouse models to investigate whether ASTX660 is able to promote ICD and enhance APM expression in vitro and in vivo.
Results
ASTX660 in the presence of TNFα promotes ICD in vitro in a subset of sensitive tumor cell lines
Prior work from our group suggests that combination treatment with ASTX660 and radiation (XRT) in the mouse oral cancer 1 (MOC1) syngeneic mouse model enhances dendritic cell number and function in the spleen.Citation8 As a result, we hypothesized that ASTX660 mediates immune stimulatory effects in part through dendritic-cell-dependent immunogenic cell death (ICD). Because ASTX660 alone does not induce cell death, we combined it with a low dose of TNFα for in vitro experiments. We treated UMSCC-46 (HPV-) and UMSCC-47 (HPV+) human HNSCC cell lines with the known ICD inducer mitoxantrone (MTX, positive control),Citation34 TNFα only, ASTX660 only, or ASTX660 + TNFα for 24–48 hours and analyzed surface expression of CRT and HSP70 by flow cytometry.Citation35 UMSCC-47 cells were treated for 48 hours compared to 24 hours for UMSCC-46 due to cell line differences in sensitivity and timing of cell death. We found that both UMSCC-46 and UMSCC-47 cells expressed significant increases in surface CRT and HSP70 in response to treatment with ASTX660 + TNFα (,). These changes occurred early, when treated cells were just entering early apoptosis (Suppl. Figure S1,). For the UMSCC-46 cells, which are quite sensitive to ASTX660 due to FADD overexpression,Citation7 these changes were noted as early as 12 hours (Suppl. Figure S3).
Figure 1. ASTX660 combined with TNFα induces surface expression of CRT/HSP70 and release of HMGB1. UMSCC-46 (HPV-) and UMSCC-47 (HPV+) were treated with mitoxantrone (MTX, 0.25 μg/mL for UMSCC-46 and 1 μg/mL for UMSCC-47, positive control), TNFα (20 ng/mL), ASTX660 (500 nM for UMSCC-46 and 1μM for UMSCC-47), and the combination of ASTX660 + TNFα for 24–72 hours and analyzed by flow cytometry. (a-b) Quantification of % cells expressing surface CRT (a) and HSP70 (b) after 24 hours (UMSCC-46; more sensitive) or 48 hours (UMSCC-47; less sensitive). Results from viable, Zombie Yellow-negative cells are shown. (c) Quantification of % cells with low levels of intracellular HMGB1 by flow cytometry on fixed, permeabilized cells after 48 hours (UMSCC-46; more sensitive) or 72 hours (UMSCC-47; less sensitive). (d) Measurement of extracellular HMGB1 in cell culture supernatants by ELISA, expressed as fold-change of the control. Data are mean + SEM, n = 6 from 2 independent experiments. *p < .05, **p < .01 versus control. TNFα, tumor necrosis factor α; ICD, immunogenic cell death; CRT, calreticulin; HSP70, heat shock protein 70. MTX, mitoxantrone; HMGB1, high mobility group box 1
Figure 2. ASTX660 alters expression of DAMPs in murine cell lines and modestly enhances XRT-induced ICD to reject tumor formation in vivo. (a-b) MOC1 and MEER cell lines were treated for 24 hours with mitoxantrone (MTX, 1 μg/ml) or ASTX660 (1 μM) +TNFα (20 ng/ml), then stained for surface calreticulin and HSP70. Results from viable, Zombie Yellow-negative cells are shown. (c). MOC1 and MEER cells were treated for 72 hours with control media or ASTX660+ TNFα, then radiated (100 Gy), fixed, and stained for intracellular HMGB1. Gating strategies are shown in Supplemental Data.(d-g) Mice were inoculated with sham saline (negative control) or 2 × 106 MOC1 or MEER cells killed in vitro by the following: radiation (100 Gy, positive control), MTX (1 μg/mL x 24 hours, positive control), ASTX660 (1 μM x 72 hours) + TNFα (20 ng/mL x 72 hours), ASTX660 (x 72 hours) + TNFα (x 72 hours) + radiation (100 Gy). This was followed by re-challenge with respective live MOC1 (3x106 cells) or MEER (1x106 cells) one week later. (d) Treatment schematic. (e) MOC1 and (f) MEER tumor growth of individual animals. (g) Corresponding Kaplan-Meier curves for % tumor free mice (n = 10–11). For both MOC1 and MEER, all treatments significantly delayed or rejected tumor growth compared to controls (p < .01). XRT, radiation; MTX, mitoxantrone; TNFα, tumor necrosis factor α
We also assessed the release of HMGB1 by flow cytometry of intracellular protein levels and by ELISA of treated cell culture supernatants (,). UMSCC-47 cells were treated for 72 hours compared to 48 hours for UMSCC-46 due to cell line differences in sensitivity and timing of cell death. In both UMSCC-46 and UMSCC-47 cells, treatment with ASTX660 + TNFα induced HMGB1 secretion, as evidenced by decreased intracellular levels ()) and increased extracellular levels ()). TNFα alone and ASTX660 alone also increased extracellular HMGB1 in UMSCC-46 cells ()). To further explore the temporal relationship of our treatments and HMGB1 secretion, we also analyzed intracellular HMGB1 levels at multiple time points for both UMSCC-46 (24, 48, 72 hrs) and UMSCC-47 (48, 72, 96 hrs) cells. Interestingly, we found that intracellular HMGB1 increased prior to its release from the cells (Suppl. Figure S4). Consistent with their susceptibilities to ASTX660 + TNFα, UMSCC-47 exhibited delayed and less robust release of intracellular HMGB1 as compared to UMSCC-46. Taken together, these data suggest that ASTX660 + TNFα is able to modulate immunostimulatory mediators of immunogenic cell death in tumor cells that are sensitive to this treatment. This effect is also likely time and/or dose dependent based on tumor cell susceptibility. Other cell lines that are insensitive to ASTX660 + TNFα in vitro did not demonstrate an increase in DAMPs after treatment (data not shown).
ASTX660 combined with XRT modestly promotes ICD in vivo
With our observation that ASTX660 + TNFα induces expression of immunogenic cell death associated DAMPs in vitro, we next wanted to assess its ability to induce immunogenic cell death in murine models. We used two syngeneic mouse models: mouse oral cancer 1 (MOC1), a carcinogen-induced model, and MEER, a mouse model engineered to express HPV oncoproteins E6 and E7.Citation36,Citation37 For in vitro experiments, we exposed these murine cell lines to ASTX660 ± TNFα or radiation (XRT) or mitoxantrone (positive control) and stained for surface CRT/HSP70 or intracellular HMGB1. We noted that surface CRT/HSP70 were increased in MOC1, but not MEER cells (,). The percent of cells with low HMGB1 at 72 hours was increased by ASTX660 and further increased by adding XRT in MOC1 cells; in contrast, MEER cells were low in HMGB1 after ASTX660 or radiation, with no added benefit from the combined treatment ()). To determine whether ASTX660 could enhance radiation-induced ICD in vivo, we inoculated mice subcutaneously with MOC1 or MEER cells treated in vitro with MTX (positive control), ASTX660 + TNFα, XRT, or ASTX660 + XRT and rechallenged on the opposite flank with live cells 7 days later ()). Compared with the small dose of TNFα added in vitro (20 ng/mL), XRT treatment is known to induce the release of robust amounts of TNFα.Citation38 In MOC1, cells killed by XRT induced a robust immunogenic response with rejection of tumor formation in 50% (5/10) of mice (,). In comparison, cells killed by ASTX660 + XRT induced an even greater immune response with rejection of tumor formation in 72% (8/11) of mice, though the increase in % tumor-free mice did not reach statistical significance (p = .24). In MEER, cells treated with either XRT or ASTX660 + XRT induced similar robust immune responses, with the combination showing no difference from the tumor rejection rate of 80% (8/10) already observed with XRT alone (,). However, there was a slight tumor growth delay in the ASTX660 + XRT vaccination group compared to the XRT-only vaccination group. Treatment with ASTX660 + TNFα was unable to kill MOC1 or MEER to a significant degree in vitro, leading to tumor engraftment at the vaccination site (Suppl. Figure S5). Similar results were also observed after vaccinating with live tumor cells (data not shown), indicating that ASTX660 alone had minimal effects on subsequent tumor-cell rechallenge. These results suggest that ASTX660 may modestly promote ICD in vivo and works best in combination with XRT. It is important to note, however, that this effect may be variable across different tumor cell types.
ASTX660 combined with XRT enhances clonal expansion of antigen specific T cells
Given our finding that ASTX660 may promote ICD under some circumstances, we next wanted to assess how direct treatment with ASTX660 with or without radiation affects antigen-specific immune responses. We treated tumor-bearing mice with ASTX660, XRT, or combination ASTX660 + XRT using the MEER model. Tumors, spleens, and draining lymph nodes were harvested and analyzed by flow cytometry for immune correlates ()). While there were no significant differences in intratumoral CD11b+CD11c+ dendritic cell numbers among treatment groups, expression of co-stimulatory molecule CD80 was significantly increased in the XRT group and approached significance in the ASTX660 + XRT group (p = .058, Suppl. Figure S6). We also investigated CD8+ T lymphocyte populations in tumor, spleen, and draining lymph nodes. Radiation alone caused a decrease in CD8 + T cells within the tumors ()), possibly due to direct toxicity of radiation to the T cells. The CD8 + T cell numbers in the spleen increased significantly in the animals treated with ASTX660 alone and in a subset of animals treated with radiation ± ASTX660 ()). The number of CD8+ T lymphocytes increased to a significant degree in the draining lymph nodes of animals treated with XRT alone, and to a near-significant degree in animals treated with combination therapy ()).
Figure 3. Treatment with ASTX660 and XRT enhances T cell number and function. Using the MEER syngeneic mouse model, engrafted tumors were treated with ASTX660 by oral gavage (16 mg/kg), XRT (single dose of 8 Gy), or the combination of both. Tumors, spleens, and DLNs were harvested and analyzed by flow cytometry for CD8+ lymphocytes. A subset of tumors were digested and magnetically sorted for T cells and co-cultured with inactivated dendritic cells presenting p15E peptide. Subsequent IFN-γ production was quantified using ELISpot assay. (a) Mouse treatment schema. (b-d) CD8+ T-lymphocytes per 10,000 cells in tumors, spleens, and DLNs. (e-f) Quantification of IFN-γ-producing T cells among various treatment groups with representative ELISpot plate images. PMA/ionomycin was the positive control and ovalbumin was the negative control. (g) MHC class I expression on CD45− tumor cells. *p < .05, **p < .01 versus control. DLN, draining lymph node; TIL, tumor infiltrating lymphocyte; IFN-γ, interferon-γ; MHC, major histocompatibility complex, MFI, mean fluorescence intensity
In addition to counting the number of intratumoral T lymphocytes, we assessed the functional capacity of antigen-specific immune responses. Tumor infiltrating lymphocytes (TIL) from subsets of tumors in each treatment condition were cultured with IL-2 and magnetically isolated for ELISpot analysis to quantify the number of TIL producing IFN-γ in response to the endogenous retroviral antigen p15E peptide.Citation39,Citation40 Expression of p15E in MEER cells was quantified by qPCR and found to be much higher than in MOC1 cells, which are known to express p15E (Suppl. Figure S7).Citation39,Citation40 Interestingly, we observed a significant increase in the amount of IFN-γ-generating TIL in the ASTX660 + XRT treatment group, in contrast to significant decreases in IFN-γ-generating TIL from animals treated with ASTX660 or XRT alone (,). Though the reasons for this stark contrast are unclear, we suspect that the combination therapy was much better able to induce the expansion of p15E-specific T cells prior to tumor harvest, whereas the individual treatments may have simply been mildly toxic to both tumor cells and T cells. Lastly, we also observed a significant increase in MHC class I expression on intratumoral nonimmune cells in the ASTX660 + XRT treatment group ()). Taken together, these results suggest that early increases in dendritic cell activation and enhanced MHC class I expression in response to ASTX660 + XRT may subsequently lead to clonal expansion of tumor antigen-specific TIL.
ASTX660 enhances TIL-mediated tumor cell killing in the absence of dendritic cells
In addition to assessing ASTX660 for its ability to induce immunogenic cell death, we next performed experiments to evaluate other immune mechanisms by which ASTX660 exerts its anti-tumor immunity at the tumor cell level. We used the xCELLigence impedance platform in three murine tumor cell lines to record cell density over time, previously shown to reflect tumor cell killing mediated by T cells enriched from tumor infiltrating lymphocytes (TIL)Citation41 (). Cultured T cells from day 7–14 MOC1, MEER, or MOC2 tumors were magnetically sorted and plated at various effector:target (E:T) ratios with or without ASTX660. In all cell lines, ASTX660 alone was not cytotoxic. TIL added in a 1:1 ratio of effector (TIL) cells to target (tumor) cells had a significant effect in MOC1, moderate effects in MEER, and no effect on tumor cell killing in MOC2. In all cell lines, the addition of ASTX660 enhanced TIL killing of tumor cells. In additional impedance experiments in MEER cells, the addition of MHC class I blocking antibody abrogated ASTX660-induced enhancement of tumor cell killing by T cells (Suppl. Figure S8). Taken together, these data suggest that in addition to immunogenic cell death, ASTX660 is able to enhance tumor cell killing through a process involving MHC class I in an environment devoid of dendritic cells.
Figure 4. ASTX660 enhances TIL-mediated killing of HNSCC cell lines. TIL were cultured from MOC1, MEER, and MOC2 tumor fragments, enriched, and magnetically sorted for T cells. Tumor cells were plated and allowed to grow for 24 hours before addition of ASTX660 (500 nM) and effector T cells at a 1:1 effector:target (E:T) ratio. Impedance lines are graphed as averages of 3 replicates normalized to a cell index of 1.0 at 24 hours when ASTX660 and/or T cells were added. TIL, tumor infiltrating lymphocytes
ASTX660 in the presence of TNFα differentially alters APM in a cell-line-dependent fashion
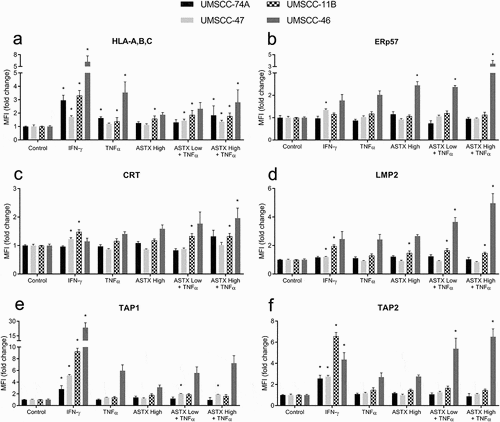
Given our observations that ASTX660 + XRT upregulates tumor cell MHC class I expression in murine models in vivo and that MHC class I blocking antibody partially reverses ASTX660-mediated enhancement of T cell killing of murine tumor cells ex vivo, we next investigated whether the increase in T-cell-dependent killing may be associated with increased APM expression in human HNSCC cell lines. We treated 4 human HNSCC cell lines (3 HPV-, 1 HPV+) with IFN-γ (positive control, 10 ng/mL), TNFα (20 ng/mL), ASTX660 (500 nM, 1 μM) alone, and ASTX660 (250 nM, 500 nM, 1 μM) + TNFα for 48 hours in vitro prior to analysis of intracellular APM components by flow cytometry. APM components included HLA-A,B,C; ERp57; CRT (intracellular); LMP2; TAP1; and TAP2. Across all 4 cell lines, HLA-A,B,C expression was consistently increased with ASTX660 + TNFα treatment ()). The other APM components were variably altered across the different cell lines (-). These results suggest that ASTX660 + TNFα may enhance tumor cell killing partly due to functionally increased antigen processing and subsequent presentation on MHC class I, with HLA-A,B,C commonly upregulated across all cell lines. Other components of the cellular APM may be differentially enhanced or defective across different cancers, possibly due to genetic heterogeneity.
Figure 5. Schematic for intracellular APM components. Antigen processing is an intracellular pathway whereby endogenous or foreign proteins are broken down into peptides by the proteasome, transported into the endoplasmic reticulin, loaded onto MHC class I, and transported to the cell surface for recognition by the adaptive immune system. Several proteases, transporters and chaperones are involved
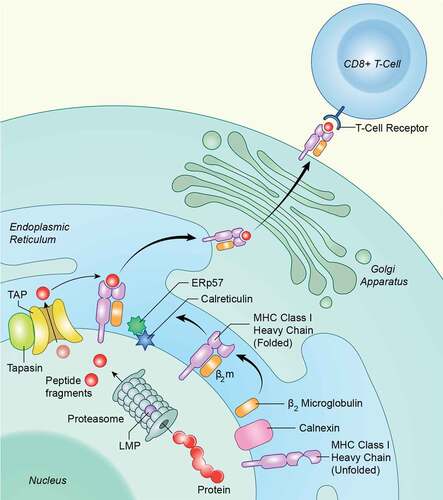
Discussion
Previous studies suggest that IAP antagonists, in addition to their ability to enhance sensitivity to TNF-mediated apoptosis of cancer cells, are able to modulate diverse innate and adaptive anti-tumor immune responses.Citation8,Citation18,Citation20,Citation21,Citation25,Citation26,Citation42 Promising pre-clinical data across multiple cancer models and numerous ongoing clinical trials involving IAP antagonists (NCT02503423, NCT02649673, NCT03111992) highlight the great potential for combining these novel agents with chemotherapy, radiation and/or immunotherapies such as checkpoint inhibitors.Citation20,Citation21,Citation26 Despite significant advances in understanding how IAP antagonists enhance anti-tumor functions of immune cells,Citation20,Citation21,Citation25 the tumor-cell-intrinsic mechanisms involved in inducing these immune responses remain largely unexplored, particularly in HNSCC. Thus, we sought to characterize the effects of ASTX660, with or without radiation, on tumor cell ICD-related DAMPs, APM, and antigen specific immunity.
Based on prior work by our group and others showing that IAP antagonists enhance dendritic cell activation,Citation8,Citation20,Citation26 we investigated whether these agents are capable of inducing ICD, which modulates DAMPs that activate dendritic-cell-mediated immune responses. We found that ASTX660 in the presence of TNFα, known to be induced by XRT with or without IAP antagonists, promoted the expression of classic markers for ICD in a subset of susceptible HNSCC cell lines in vitro. We have previously published data on the types of cell death induced by combination of ASTX660 + TNFα in the human cell lines included in the present study.Citation43 In UMSCC-47, cell death was attenuated primarily by pan-caspase and caspase 8 inhibitors, while in UMSCC-46, the necroptosis inhibitor necrostatin attenuated cell death. These data suggest that either apoptosis or necroptosis may be associated with increased DAMPs. These ICD markers were absent in other cell lines that were resistant to ASTX660 + TNFα, emphasizing the importance of treatment-induced cell death in this process. Because treatment with ASTX660 alone is often unable to induce cell death, the addition of ASTX660 to XRT or other cytotoxic therapies may act to enhance ICD. In other experiments using the gold standard in vivo mouse vaccination paradigm for the detection of ICD,Citation34 we showed that the combination of ASTX660 and XRT exhibited improvement over XRT alone in the MOC1 syngeneic mouse model, providing further rationale for combination ASTX660 + XRT therapy. However, this additive effect was not seen in the MEER model, for reasons that are unclear, suggesting that biomarkers may be needed to determine which tumors may be most responsive to ASTX660 + XRT.
Additional ex vivo experiments combining tumor cells and TIL in an environment devoid of dendritic cells demonstrated that ASTX660 enhances cytotoxic TIL-dependent killing across multiple cell lines, suggesting additional mechanistic pathways for enhancement of anti-tumor immunity that are independent from ICD. Consistent with our results, a prior study showed that knockdown of neuronal apoptosis inhibitory protein (NAIP), another member of the IAP family, sensitized prostate cancer cells to antigen-specific T-cell killing.Citation44 As a possible mechanism for this enhanced T-cell killing, we hypothesized that ASTX660 might enhance antigen presentation on tumor cells, facilitating T lymphocyte recognition and subsequent cytotoxic killing. Further experiments showed that ASTX660 + TNFα reliably increased the expression of HLA-A,B,C across multiple human HNSCC cell lines in vitro, and ASTX660 + XRT increased murine MHC class I expression on tumor cells in vivo. In addition, ELISpot analysis demonstrated a prominent antigen-specific functional immune response to the combination treatment. ASTX660 + XRT, but neither agent alone, significantly enhanced clonal proliferation of antigen-specific TIL generating IFN-γ, clarifying possible immune-stimulating mechanisms of ASTX660 and XRT.
Although initially shown to target anti-apoptotic signaling in cancers, IAP antagonists have now been found to play significant roles in enhancing anti-tumor immunity with diverse effects on B-cell survival, dendritic cell activation, and T-cell costimulation.Citation8,Citation20,Citation21,Citation24–Citation26,Citation45 One recent study demonstrated that IAP antagonist LCL-161 was able to kill pancreatic cancer cells in vivo but not in vitro, an effect that was highly dependent on dendritic cells and T lymphocytes.Citation46 Another study demonstrated that intratumoral delivery of lentiviral vectors encoding cytosolic SMAC mimetic LV-tSMAC into tumor-bearing mice resulted in growth delay/regression, dendritic cell activation and improved tumor-specific CD8+ T cell response.Citation47 The authors found that transduction of LV-tSMAC in vitro resulted in tumor cell apoptosis and exposure of calreticulin, but did not induce release HMGB1 or ATP. Our study builds upon these prior findings by showing that ASTX660 can induce expression of classic ICD markers in sensitive cell lines in vitro, suggesting that this mechanism of anti-tumor immunity is intact for a subset of HNSCC tumors. Another study showed synergistic anti-tumor immunity when combining IAP antagonist Debio 1143 with XRT in lung cancer models,Citation48 a relationship that was also established between ASTX660 and XRT in HNSCC in our prior published work.Citation8 Our current study significantly builds upon these prior findings and clarifies mechanisms of IAP antagonist-induced anti-tumor immunity by showing that ASTX660 + XRT is able to enhance clonal expansion of tumor-specific cytotoxic T lymphocytes in vivo, improve TIL-mediated killing in vitro, and differentially upregulate MHC class I and other APM components.
Our study has several limitations. Our ICD experiments demonstrated the expression of classical ICD markers only in cell lines that were susceptible to ASTX660 in the presence of TNFα in vitro. Other cell lines that did not exhibit measurable cell death in vitro in response to treatment did not display markers for ICD, reinforcing the rationale for combining IAP antagonists with radiotherapy and other cancer therapies. We also did not look at ATP, another classic DAMP associated with ICD, due to technical challenges with rapid ATP degradation in our HNSCC cell lines. Other studies have shown that HNSCC tumor cells overexpress ectonucleoside triphosphate diphosphohydrolase 1 (CD39), which degrades extracellular ATP,Citation49 possibly explaining these findings. In our in vivo immune correlate flow cytometry analysis, tumor-bearing mice were only treated with one dose of ASTX660 (16 mg/kg) and one round of radiation (8 Gy) at 24 hours and 14 hours prior to harvest, respectively. This was done to prevent the complete tumor regression that occurred in a prior study.Citation8 However, later time points may have yielded more robust immune responses. Although we observed statistically significant increases in splenic and DLN CD8+ T cell numbers, as well as in tumor MHC class I expression in response to treatment with ASTX660 ± XRT, due to logistical constraints, large numbers of animals could not be used to detect more subtle changes.
The present study has important clinical implications as an increasing number of immune-based therapies are under investigation in preclinical and clinical settings. Cisplatin, the most common systemic drug for HNSCC, has several adverse effects including severe nausea/vomiting, nephrotoxicity, peripheral neuropathy and hearing loss.Citation50 Thus, more targeted approaches involving one or more immunomodulatory agents along with other standard therapies such as radiotherapy are under active investigation. A recent study demonstrated that the IAP antagonist Debio 1143 enhances the response to anti-PD-L1 in a mouse model of bladder cancer, with results suggesting a possible synergistic interaction.Citation51 This combination (Debio 1143 and the anti-PD-L1 antibody avelumab) is currently under investigation in a phase-IIb trial (NCT03270176) for recurrent/metastatic solid tumors including HNSCC. Another phase I/II trial of cisplatin chemoradiation + Debio 1143 or placebo for previously untreated HNSCC is also currently underway (NCT02022098). Preliminary results from another recent study showed that neoadjuvant Debio 1143 monotherapy increased CD8 + T cell infiltration in HNSCC surgical specimens, validating preclinical work from our group and others suggesting enhanced immune infiltration of HNSCC tumors with IAP antagonists.Citation52 Our prior work showed that the efficacy of ASTX660 and radiation was enhanced with the addition of anti-PD-1,Citation8 suggesting that the combination of ASTX660, radiotherapy, and anti-PD-1/PD-L1 immune checkpoint blockade may be a promising therapeutic strategy for the treatment of HNSCC.
In conclusion, ASTX660 in combination with XRT in vivo, or TNFα in vitro, modestly promotes immunogenic cell death in sensitive cell lines, providing an avenue for enhanced dendritic cell activation and adaptive immunity. This combination treatment also enhances tumor-specific cytotoxic lymphocyte proliferation and killing, and variably upregulates APM components across different cell lines. These results suggest that IAP antagonists have widespread effects across multiple mechanistic pathways that contribute to anti-tumor immunity. Use of IAP antagonists in combination with immunotherapies and radiotherapy merits further investigation in clinical trials.
Materials and methods
Cell lines
Human cell lines UMSCC-46, −47 (HPV-positive), 11B, and 74A were acquired from Dr. T. Carey at the University of Michigan, authenticated, and maintained as previously described.Citation53 Murine cell lines MOC1 and 2 were obtained from Dr. R. Uppaluri at the Washington University School of Medicine, and MEER from Dr. W. Spanos at the University of South Dakota School of Medicine, authenticated, and maintained as previously described.Citation39,Citation51,Citation54 All cell lines were stored in liquid nitrogen, regularly tested for Mycoplasma, and cultured for no longer than 6 months or 20 passages.
Antibodies and reagents
ASTX660 was acquired from Astex Pharmaceuticals through a cooperative research and development agreement (CRADA) with the National Institute on Deafness and Other Communication Disorders (NIDCD). Pharmaceutical grade mitoxantrone (MTX) was obtained from the National Institutes of Health veterinary pharmacy. Recombinant human IFN-γ, and human and mouse TNFα were obtained from BioLegend. Fluorescent-conjugated antibodies for mouse tumor flow cytometry were obtained from eBioscience (CD3) and BioLegend (CD45.2, CD8a, H-2Kb/H-2Db, CD11b, CD11c, CD80, CD86, I-A/I-E). Viability dyes were obtained from BD Biosciences (7AAD) and Biolegend (Zombie Yellow, Zombie NIR). Unconjugated antibodies for flow cytometry in human cell lines were obtained from LSBio (TAP1) and Abcam (TAP2, LMP2) with corresponding secondary antibodies obtained from BioLegend. Fluorescent-conjugated antibodies for flow cytometry in human cell lines were obtained from Abcam (CRT, HSP70, HMGB1, and ERp57) and Biolegend (HLA-A,B,C). The in vivo anti-mouse MHC class I antibody used for ex vivo T cell impedance assays was from BioXCell (clone M1/42.3.9.8). Antibodies and concentrations used for ICD and APM flow panels are detailed in Supplemental Methods.
Flow cytometry
For ICD DAMP analysis, cells were plated at 100,000–200,000 cells per well in 6 well plates and allowed to adhere overnight prior to treatment with MTX (0.25 μg/mL or 1 μg/mL), TNFα (20 ng/mL), ASTX660 (500 nM or 1 μM), and ASTX660 (500 nM or 1 μM) + TNFα (20 ng/mL). Cells were harvested at 24, 48, or 72 hours post-treatment with Trypsin-EDTA, stained for surface markers CRT and HSP70 followed by Zombie Yellow viability dye, then fixed and permeabilized with the eBioscience Intracellular Fixation and Permeabilization Buffer Set prior to staining for intracellular marker HMGB1. Using isotype controls, conservative gating measures were used to assess % cells positive for surface markers CRT and HSP70 and % cells low for intracellular HMGB1. For CRT and HSP70, Zombie Yellow staining was used to gate out non-viable cells.
For APM analysis, cells were plated the same way as above but treated with IFN-γ (10 ng/mL), TNFα (20 ng/mL), ASTX660 (500 nM or 1 μM), and ASTX660 (250 nM, 500 nM, or 1 μM) + TNFα (20 ng/mL). Cells were harvested 48 hours post-treatment with Trypsin-EDTA, stained with Zombie Yellow viability dye, fixed and permeabilized as above, and stained for intracellular APM markers. Mean fluorescence intensity (MFI) values for isotype controls were subtracted from primary stains for each sample and values were graphed as fold-change over the control. Zombie Yellow viability dye (BioLegend) was used to gate out dead cells. Cytometry sample acquisition was completed on a BD LSRFortessa cytometer with subsequent analysis done using FlowJo software. “Fluorescence minus one” controls were tested for all multicolor panels.
Gating strategies and the cell viability assay used in Supplemental Figures S1/S2 are further described in Supplemental Methods.
HMGB1 ELISA
Supernatants from cells cultured with MTX and ASTX660 ± TNFα were collected and stored at −80°C. Samples were later assessed using ELISA kits for HMGB1 (IBL International) according to manufacturer specifications and analyzed with a Biotek uQuant microplate reader.
Impedance assays
For impedance assays, MOC1, MEER, MOC2, and LLC tumors were harvested from mice, cultured with IL-2 (100 U/mL) to expand tumor infiltrating lymphocytes (TIL), and magnetically sorted using the Pan T Cell Isolation Kit II (Miltenyi) to generate effector TIL. 10,000–20,000 MOC1, MEER, MOC2, and LLC target cells were plated in 96-well E-plates and allowed to adhere overnight. Respective TIL were subsequently added with or without ASTX660 at a 1:1 effector-to-target cell (E:T) ratios. Changes in impedance were recorded using the xCELLigence RTCA platform as previously described.Citation8,Citation55,Citation56
In vivo mouse experiments
All animal experiments were approved by the Animal Care and Use Committee at NIDCD. Wildtype, female C57BL/6 mice aged 6–8 weeks were acquired from Taconic and housed in a pathogen-free animal facility. For ICD vaccination experiments, MOC1 and MEER cells were treated in vitro with ASTX660 + TNFα for 72 hours alone or in combination with 100 Gy radiation at the end. Additional cells were treated with radiation alone, or MTX alone for 24 hours as described in the literature.Citation34,Citation57–Citation59 Dead/dying cells were rinsed in PBS and then injected in the left flank (2 x 106 for both MOC1 and MEER). One week later, mice were injected in the right flank with live MOC1 (3 x 106) or MEER (2.5 x 105) cells. Tumor growth and mouse weight were measured 2 to 3 times weekly with tumor volume measured with calipers and calculated as (length2 x width)/2.
For mouse tumor flow experiments, 1 × 106 live MEER cells were injected into the right leg and allowed to grow for 6 days. Mice were randomized, and then treated with one dose of ASTX660 (oral gavage, 16 mg/kg, 24 hours prior to sacrifice), one dose of radiation (8 Gy, 14 hours prior to sacrifice), or the combination of both. Tumors, spleens, and draining lymph nodes were harvested on day 7 and digested into single cell suspensions as previously described,Citation40 then stained and analyzed by flow cytometry, using 7AAD to viability dye to gate out dead cells. Cells from tumor draining lymph nodes were magnetically sorted for T cells using the Pan T Cell Isolation Kit II (Miltenyi Biotec) prior to analysis. In addition, tumor fragments from each treatment group were plated and cultured in IL-2 (100 U/mL) to expand the TIL for ELISPot assays.
For ELISpot assays, after 5–7 days of culture, cells were harvested and sorted for T cells as above. TIL were incubated at 20,000 cells with p15E peptide and 40,000 naïve splenocytes pulsed with 20 Gy radiation in an ELISpot plate pre-coated with anti-IFN-γ antibody. ELISpot was performed per manufacturer specifications and read on ELISpot reader.
Statistical analyses
Data were analyzed using GraphPad Prism 7 software, using p < .05 as the cutoff for statistical significance. In vitro data were analyzed with one- or two- way ANOVA where appropriate. Percent tumor free curves were generated using the Kaplan-Meier method with Mantel-Cox log rank testing for comparisons.
Conflict of Interest Disclosure
Astex Pharmaceuticals provided ASTX660 and research funding to Dr. Schmitt and Dr. Van Waes under a Cooperative Research and Development Agreement (CRADA) with NIDCD, NIH.
Supplemental Material
Download ()Acknowledgments
The authors acknowledge Christopher Silvin and Yvette Robbins for technical assistance, James Mitchell and his lab for assistance with irradiation treatments, James Hodge and Wojciech Mydlarz for critical review of the manuscript, and the NIH Medical Arts team for illustration of . This research was made possible in part through the NIH Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation (DDCF Grant #2014194), the American Association for Dental Research, the Colgate-Palmolive Company, Genentech, Elsevier, and other private donors.
Figure 6. ASTX660 combined with TNFα differentially alters APM across various human HNSCC cell lines. UMSCC-74A (HPV-), −11B(HPV-), −47(HPV+), −46 (HPV-) cells were treated with IFN-γ (10 ng/mL, positive control), TNFα (20 ng/mL), ASTX660 (500 nM or 1 μM), and ASTX660 (250 nM, 500 nM, or 1 μM) + TNFα for 48 hours prior to staining and analysis by flow cytometry. Data are represented as mean + SEM, n = 6–9 from at least 2 independent experiments. *p < .05 versus control. APM, antigen processing machinery; IFN-γ, interferon-γ; TNFα, tumor necrosis factor α; MFI, mean fluorescence intensity
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–13. doi:10.3322/caac.v69.1.
- Cognetti DM, Weber RS, Lai SY. Head and neck cancer: an evolving treatment paradigm. Cancer. 2008;113(7 Suppl):1911–1932. doi:10.1002/cncr.23654.
- Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. doi:10.1016/j.ejphar.2014.07.025.
- Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–1867. doi:10.1056/NEJMoa1602252.
- Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17(7):956–965. doi:10.1016/S1470-2045(16)30066-3.
- Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–582. doi:10.1038/nature14129.
- Eytan DF, Snow GE, Carlson S, Derakhshan A, Saleh A, Schiltz S, Cheng H, Mohan S, Cornelius S, Coupar J, et al. SMAC mimetic birinapant plus radiation eradicates human head and neck cancers with genomic amplifications of cell death genes FADD and BIRC2. Cancer Res. 2016;76(18):5442–5454. doi:10.1158/0008-5472.CAN-15-3317.
- Xiao R, Allen CT, Tran L, Patel P, Park S-J, Chen Z, Van Waes C, Schmitt NC. Antagonist of cIAP1/2 and XIAP enhances anti-tumor immunity when combined with radiation and PD-1 blockade in a syngeneic model of head and neck cancer. OncoImmunology. 2018;7(9):e1471440. doi:10.1080/2162402X.2018.1471440.
- Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer. 2010;10(8):561–574. doi:10.1038/nrc2889.
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363–367. doi:10.1038/nature09852.
- Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi:10.1016/j.cell.2007.10.030.
- Oberst A, Green DR. It cuts both ways: reconciling the dual roles of caspase 8 in cell death and survival. Nat Rev Mol Cell Biol. 2011;12(11):757–763. doi:10.1038/nrm3214.
- Moquin DM, McQuade T, Chan FK. CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One. 2013;8(10):e76841. doi:10.1371/journal.pone.0076841.
- Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 2007;14(3):400–410. doi:10.1038/sj.cdd.4402085.
- Salvesen GS, Duckett CS. Apoptosis: IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol. 2002;31:5045–5060.
- Derakhshan A, Chen Z, Van Waes C. Therapeutic small molecules target inhibitor of apoptosis proteins in cancers with deregulation of extrinsic and intrinsic cell death pathways. Clin Cancer Res. 2017;23(6):1379–1387. doi:10.1158/1078-0432.CCR-16-2172.
- Brands RC, Herbst F, Hartmann S, Seher A, Linz C, Kübler AC, Müller-Richter UDA. Cytotoxic effects of SMAC-mimetic compound LCL161 in head and neck cancer cell lines. Clin Oral Investig. 2016;20(9):2325–2332. doi:10.1007/s00784-016-1741-3.
- Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11(2):109–124. doi:10.1038/nrd3627.
- Matzinger O, Viertl D, Tsoutsou P, Kadi L, Rigotti S, Zanna C, Wiedemann N, Vozenin MC, Vuagniaux G, Bourhis J. The radiosensitizing activity of the SMAC-mimetic, debio 1143, is TNFalpha-mediated in head and neck squamous cell carcinoma. Radiother Oncol. 2015;116(3):495–503. doi:10.1016/j.radonc.2015.05.017.
- Dougan SK, Dougan M. Regulation of innate and adaptive antitumor immunity by IAP antagonists. Immunotherapy. 2018;10(9):787–796. doi:10.2217/imt-2017-0185.
- Beug ST, Beauregard CE, Healy C, Sanda T, St-Jean M, Chabot J, Walker DE, Mohan A, Earl N, Lun X, et al. Smac mimetics synergize with immune checkpoint inhibitors to promote tumour immunity against glioblastoma. Nat Commun. 2017;8. doi:10.1038/ncomms14278.
- Kearney CJ, Lalaoui N, Freeman AJ, Ramsbottom KM, Silke J, Oliaro J. PD-L1 and IAPs co-operate to protect tumors from cytotoxic lymphocyte-derived TNF. Cell Death Differ. 2017;24(10):1705–1716. doi:10.1038/cdd.2017.94.
- Gentle IE, Moelter I, Lechler N, Bambach S, Vucikuja S, Häcker G, Aichele P. Inhibitors of apoptosis proteins (IAPs) are required for effective T-cell expansion/survival during antiviral immunity in mice. Blood. 2014;123(5):659–668. doi:10.1182/blood-2013-01-479543.
- Clancy-Thompson E, Ali L, Bruck PT, Exley MA, Blumberg RS, Dranoff G, Dougan M, Dougan SK. IAP antagonists enhance cytokine production from mouse and human iNKT cells. canimm.0490.2017; 2017.
- Dougan M, Dougan S, Slisz J, Firestone B, Vanneman M, Draganov D, Goyal G, Li W, Neuberg D, Blumberg R, et al. IAP inhibitors enhance co-stimulation to promote tumor immunity. J Exp Med. 2010;207(10):2195–2206. doi:10.1084/jem.20101123.
- Chesi M, Mirza NN, Garbitt VM, Sharik ME, Dueck AC, Asmann YW, Akhmetzyanova I, Kosiorek HE, Calcinotto A, Riggs DL, et al. IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat Med. 2016;22(12):1411–1420. doi:10.1038/nm.4229.
- Ward GA, Lewis EJ, Ahn JS, Johnson CN, Lyons JF, Martins V, Munck JM, Rich SJ, Smyth T, Thompson NT, et al. ASTX660, a novel non-peptidomimetic antagonist of cIAP1/2 and XIAP, potently induces TNFα-dependent apoptosis in cancer cell lines and inhibits tumor growth. Mol Cancer Ther. 2018;17(7):1381–1391. doi:10.1158/1535-7163.MCT-17-0848.
- Kroemer G, Senovilla L, Galluzzi L, Andre F, Zitvogel L. Natural and therapy-induced immunosurveillance in breast cancer. Nat Med. 2015;21(10):1128–1138. doi:10.1038/nm.3944.
- Terenzi A, Pirker C, Keppler BK, Berger W. Anticancer metal drugs and immunogenic cell death. J Inorg Biochem. 2016;165:71–79. doi:10.1016/j.jinorgbio.2016.06.021.
- Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, Galluzzi L, Kepp O, Kroemer G. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. 2015;6:187.
- Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97–111. doi:10.1038/nri.2016.107.
- Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003. doi:10.1084/jem.20101158.
- Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208(10):2005–2016. doi:10.1084/jem.20101159.
- Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3(9):e955691. doi:10.4161/21624011.2014.955691.
- Montico B, Nigro A, Casolaro V, Dal Col J. Immunogenic apoptosis as a novel tool for anticancer vaccine development. Int J Mol Sci. 2018;19(2):594. doi:10.3390/ijms19020594.
- Hoover AC, Spanos WC, Harris GF, Anderson ME, Klingelhutz AJ, Lee JH. The role of human papillomavirus 16 E6 in anchorage-independent and invasive growth of mouse tonsil epithelium. Arch Otolaryngol Head Neck Surg. 2007;133(5):495–502. doi:10.1001/archotol.133.5.495.
- Judd NP, Allen CT, Winkler AE, Uppaluri R. Comparative analysis of tumor-infiltrating lymphocytes in a syngeneic mouse model of oral cancer. Otolaryngol Head Neck Surg. 2012;147(3):493–500. doi:10.1177/0194599812442037.
- Hallahan DE, Spriggs DR, Beckett MA, Kufe DW, Weichselbaum RR. Increased tumor necrosis factor alpha mRNA after cellular exposure to ionizing radiation. Proc Natl Acad Sci U S A. 1989;86:10104–10107. doi:10.1073/pnas.86.24.10104.
- Cash H, Shah S, Moore E, Caruso A, Uppaluri R, Van Waes C, Allen C. mTOR and MEK1/2 inhibition differentially modulate tumor growth and the immune microenvironment in syngeneic models of oral cavity cancer. Oncotarget. 2015;6(34):36400–36417. doi:10.18632/oncotarget.v6i34.
- Moore EC, Cash HA, Caruso AM, Uppaluri R, Hodge JW, Van Waes C, Allen CT. Enhanced tumor control with combination mTOR and PD-L1 inhibition in syngeneic oral cavity cancers. Cancer Immunol Res. 2016;4(7):611–620. doi:10.1158/2326-6066.CIR-15-0252.
- Morisada M, Moore EC, Hodge R, Friedman J, Cash HA, Hodge JW, Mitchell JB, Allen CT. Dose-dependent enhancement of T-lymphocyte priming and CTL lysis following ionizing radiation in an engineered model of oral cancer. Oral Oncol. 2017;71:87–94. doi:10.1016/j.oraloncology.2017.06.005.
- Fischer K, Tognarelli S, Roesler S, Boedicker C, Schubert R, Steinle A, Klingebiel T, Bader P, Fulda S, Ullrich E, et al. The smac mimetic BV6 improves NK cell-mediated killing of rhabdomyosarcoma cells by simultaneously targeting tumor and effector cells. Front Immunol. 2017;8:202. doi:10.3389/fimmu.2017.00202.
- Xiao R, An Y, Ye W, Derakhshan A, Cheng H, Yang X, Allen C, Chen Z, Schmitt NC, Van Waes C, et al. Dual antagonist of cIAP/XIAP ASTX660 Sensitizes HPV(-)and HPV(+) head and neck cancers to TNFalpha, TRAIL, and radiation therapy. Clin Cancer Res. 2019;25:6463–6474. doi:10.1158/1078-0432.CCR-18-3802.
- Ardani A, Gameiro S, Kwilas A, Donahue R, Hodge J. Androgen deprivation therapy sensitizes prostate cancer cells to T-cell killing through androgen receptor dependent modulation of the apoptotic pathway. Oncotarget. 2014;5:9335–9348. doi:10.18632/oncotarget.2429.
- Knights AJ, Fucikova J, Pasam A, Koernig S, Cebon JJCI. Inhibitor of apoptosis protein (IAP) antagonists demonstrate divergent immunomodulatory properties in human immune subsets with implications for combination therapy. Cancer Immunol Immunother. 2013;62(2):321–335. doi:10.1007/s00262-012-1342-1.
- Roehle KDM, Dougan S Augmenting immunity with IAP antagonists in PDAC. In: Annual Meeting and Pre-Conference Programs, Society for Immunotherapy of Cancer; 2018 Nov 7-11, Washington, DC. Abstract P467.
- Emeagi PU, Van Lint S, Goyvaerts C, Maenhout S, Cauwels A, McNeish IA, Bos T, Heirman C, Thielemans K, Aerts JL, et al. Proinflammatory characteristics of SMAC/DIABLO-induced cell death in antitumor therapy. Cancer Res. 2012;72(6):1342–1352. doi:10.1158/0008-5472.CAN-11-2400.
- Tao Z, McCall NS, Wiedemann N, Vuagniaux G, Yuan Z, Lu B. SMAC mimetic debio 1143 and ablative radiation therapy synergize to enhance antitumor immunity against lung cancer. Clin Cancer Res. 2019;25(3):1113–1124. doi:10.1158/1078-0432.CCR-17-3852.
- Michaud M, Sukkurwala AQ, Martins I, Shen S, Zitvogel L, Kroemer G. Subversion of the chemotherapy-induced anticancer immune response by the ecto-ATPase CD39. Oncoimmunology. 2012;1(3):393–395. doi:10.4161/onci.19070.
- Ding D, Liu H, Qi W, Jiang H, Li Y, Wu X, Sun H, Gross K, Salvi R. Ototoxic effects and mechanisms of loop diuretics. J Otol. 2016;11(4):145–156. doi:10.1016/j.joto.2016.10.001.
- Vermeer DW, Coppock JD, Zeng E, Lee KM, Spanos WC, Onken MD, Uppaluri R, Lee JH, Vermeer PD. Metastatic model of HPV+ oropharyngeal squamous cell carcinoma demonstrates heterogeneity in tumor metastasis. Oncotarget. 2016;7(17):24194–24207. doi:10.18632/oncotarget.v7i17.
- Gomez-Roca C, Even C, Le Tourneau C, Baste Rotlan N, Delord JP, Sarini J, Vergez S, Teman S, Hoffman C, Rochaix P, et al. Open-label, exploratory pre-operative window-of-opportunity trial to investigate the pharmacokinetics and pharmacodynamics of the SMAC mimetic debio 1143 in patients with resectable squamous cell carcinoma of the head and neck. In: Proceedings, AACR Annual Meeting; 2019, Apr 14-18, 2018; Atlanta, GA. Philadelphia (PA): AACR; Abstract nr 5001.
- Brenner JC, Graham MP, Kumar B, Saunders LM, Kupfer R, Lyons RH, Bradford CR, Carey TE. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck. 2010;32(4):417–426. doi:10.1002/hed.21198.
- Moore E, Clavijo PE, Davis R, Cash H, Van Waes C, Kim Y, Allen C. Established T Cell-Inflamed Tumors Rejected after Adaptive Resistance Was Reversed by Combination STING Activation and PD-1 Pathway Blockade. Cancer Immunol Res. 2016;4(12):1061–1071. doi:10.1158/2326-6066.CIR-16-0104.
- Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, Das R, Silvin C, Van Waes C, Horn LA, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight. 2019;4(7). doi:10.1172/jci.insight.126853.
- Clavijo PE, Friedman J, Robbins Y, Moore EC, Smith E, Zauderer M, Evans EE, Allen CT. Semaphorin4d inhibition improves response to immune-checkpoint blockade via attenuation of mdsc recruitment and function. Cancer Immunol Res. 2019;7(2):282–291. doi:10.1158/2326-6066.CIR-18-0156.
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61. doi:10.1038/nm1523.
- Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, Yamazaki T, Sukkurwala AQ, Michaud M, Mignot G, et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med. 2012;4(143):143ra199. doi:10.1126/scitranslmed.3003807.
- Sukkurwala AQ, Adjemian S, Senovilla L, Michaud M, Spaggiari S, Vacchelli E, Baracco EE, Galluzzi L, Zitvogel L, Kepp O, et al. Screening of novel immunogenic cell death inducers within the NCI mechanistic diversity set. Oncoimmunology. 2014;3:e28473. doi:10.4161/onci.28473.