
ABSTRACT
PT-112 is a novel platinum-pyrophosphate conjugate under clinical development for cancer therapy. PT-112 mediates cytostatic and cytotoxic effects against a variety of human and mouse cancer cell lines in vitro. The cytotoxic response to PT-112 is associated with the emission of danger signals underpinning the initiation of anticancer immunity, including calreticulin exposure on the surface of dying cells, as well as ATP and HMGB1 secretion. Consistently, mouse cancer cells succumbing to PT-112 in vitro can be used to provide syngeneic, immunocompetent mice with immunological protection against a subsequent challenge with living tumor cells of the same type. Moreover, PT-112 administration synergizes with PD-1 or PD-L1 blockade in the control of mouse cancers in immunologically competent settings, as it simultaneously recruits immune effector cells and depletes immunosuppressive cells in the tumor microenvironment. Finally, PT-112 employed intratumorally in the context of immune checkpoint inhibition initiates a robust immune response that has systemic outreach and limits the growth of untreated, distant lesions. Thus, PT-112 induces the immunogenic demise of cancer cells, and hence stands out as a promising combinatorial partner of immune checkpoint blockers, especially for the treatment of otherwise immunologically cold tumors.
Introduction
Platinum-based chemotherapeutics such as cisplatin (CDDP), carboplatin and oxaliplatin have extensively been used for the clinical management of numerous neoplasms, including (but not limited to) pulmonary, ovarian and colorectal tumors.Citation1-Citation3 However, platinum derivatives are associated with considerable toxicity and a high incidence of acquired resistance,Citation4,Citation5 calling for the identification of improved chemical entities. R,R-1,2 cyclohexanediamine-pyrophosphato-platinum(II) (PT-112, ) has been developed in this setting, with the specific aim of altering the cellular mechanisms of action of the drug to improve its efficacy and at the same limit its toxicity.Citation6-Citation10 From a chemical perspective, PT-112 differs from other platinum derivatives as Pt2+ ions are chelated by diaminocyclohexane and pyrophosphate moieties. Pyrophosphate exists in the plasma in a di-anionic state, providing PT-112 with improved pharmacokinetic and pharmacodynamic properties, including a considerable tendency to accumulate in the lung, liver and bones (in mice).Citation7,Citation11-Citation13 In line with this notion, multiple individual patients with primary or metastatic lesions in these organs, who failed several lines of conventional and/or experimental therapy, have experienced robust and durable responses upon systemic administration of PT-112 in the context of ongoing, dose-escalation, Phase I clinical trials (NCT02266745, NCT03409458).Citation11,Citation12,Citation14 In particular, PT-112 monotherapy enabled durable responses in three patients with solid tumors, including two individuals who progressed on immune checkpoint blocker (ICB)-based immunotherapy.Citation11,Citation12 Moreover, heavily pretreated men with castration resistant prostate cancer exhibited serologic and radiographic responses to PT-112, employed as standalone therapeutic agentCitation11,Citation12 or combined with avelumab,Citation14 an ICB specific for CD274 (best known as PD-L1)Citation15,Citation16 that is poorly active in such patients.Citation17 Together with existing preclinical data,Citation7,Citation13 these observations suggest that PT-112 treatment may elicit, or at least be compatible with, a tumor-targeting immune response that can be potentiated by ICBs.Citation18,Citation19
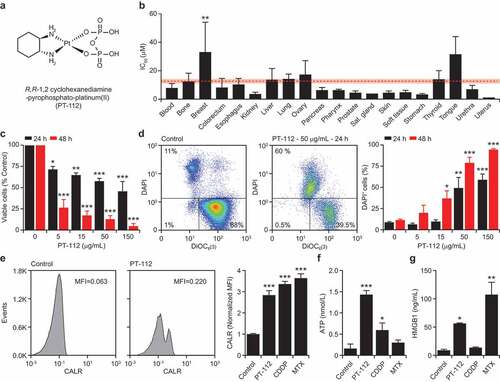
Figure 1. Cell death driven by PT-112 is associated with DAMP emission. (a). Chemical structure of R,R-1,2 cyclohexanediamine-pyrophosphato-platinum(II) (PT-112). (b). IC50 values associated with exposure of 121 human cancer cell lines to PT-112 for 72 hours. Results are means ± SEM, based on cancer cell histology. Mean IC50 ± SEM for all cells is reported in red. **p < .01 (one-way ANOVA), as compared to all other cells confounded. See also . (c). Residual number of mouse colorectal carcinoma CT26 cells upon exposure to the indicated concentration of PT-112 for 24 or 48 hours. Quantitative results (means ± SEM) are reported. n = 2–3 independent experiments; *p < .05, **p < .01, ***p < .001 (one-way ANOVA), as compared to untreated cells at the same time point. (d). Percentage of DAPI+ (dead) mouse mammary carcinoma TSA cells upon exposure to PT-112 in the indicated concentrations for 24 or 48 hours. Representative dotplots (with percentage of events in each quadrant) and quantitative results (means ± SEM) are reported. n = 2–3 independent experiments; *p < .05, **p < .01, ***p < .001 (one-way ANOVA), as compared to untreated cells at the same time point. (e). CALR exposure on PI− TSA cells upon treatment with 50 µg/mL PT-112, 15 µM cisplatin (CDDP), or 2.5 µM mitoxantrone (MTX) for 24 hours. Representative histograms (isotype staining is reported as dashed profile) and quantitative results (mean MFI ± SEM) are reported. n = 2–3 independent experiments; ***p < .001 (one-way ANOVA), as compared to untreated cells. MFI, mean fluorescence intensity. (f,g). ATP (f) and HMGB1 (g) amounts in the supernatant of TSA cells treated as in panel d. Quantitative results (means ± SEM) are reported. n = 2–3 independent experiments; *p < .05, **p < .01, ***p < .001 (one-way ANOVA), as compared to untreated cells
Although conventional platinum derivatives share the ability to cause DNA lesions with some degree of specificity for highly-proliferating cells, including (but not limited to) cancer cells, the immunomodulatory profile of these drugs exhibits considerable variability. In particular, CDDP, carboplatin and oxaliplatin differ in their capacity to elicit bona fide immunogenic cell death (ICD), a specific form of regulated cell death (RCD)Citation20 that is sufficient (in immunocompetent, syngeneic settings) for the initiation of adaptive immunity against dead cell-associated antigens.Citation21 At least in part, this reflects the proficient activation of intracellular stress responses culminating with the emission of adjuvant signals commonly known as damage-associated molecular patterns (DAMPs)Citation22 by oxaliplatin (which is largely considered as a bona fide ICD inducer)Citation23,Citation24 but less so by CDDP and carboplatin (whose immunogenicity remains a matter of debate).Citation25,Citation26
Based on these premises, we set out to investigate the emission of ICD-associated DAMPs including calreticulin (CALR), ATP and high mobility group box 1 (HMBG1) by cancer cells responding to PT-112, as well as the ability of PT-112 to (1) drive bona fide ICD in gold standard vaccination and abscopal models,Citation27,Citation28 and (2) synergize with ICBs in the eradication of established mouse tumors. Here, we report that PT-112 causes a form of cancer cell death that is immunogenic per se. ICD induction by PT-112 potentially explains durable responses to the drug observed in the context of ongoing Phase I/II clinical trials, and suggests that PT-112 can be successfully combined with ICBs for superior therapeutic activity.
Materials and methods
Chemicals and cell culture
Media and supplements for cell culture were obtained from Invitrogen™-Thermo Fisher, unless otherwise noted. All cells were maintained according to ATCC recommendations, and cells between passage 2 and 10 were employed for experimental determinations.
Cell number
Residual number of living cells upon exposure of human cancer cell lines to increasing doses of PT-112 for 72 hours was assessed with the CyQUANT Proliferation Assay (Thermo Fisher), as per the manufacturer’s recommendations.
Cell death
Cell death was assessed by flow cytometry upon co-staining cells with the mitochondrial transmembrane potential-sensitive dye 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3), from Invitrogen™-Thermo Fisher) (40 nM) and either of the vital dyes 4′,6-diamidino-2-phenylindole (DAPI, from Sigma-Aldrich) (25 ng/mL) and propidium iodide (PI, from Sigma-Aldrich) (0.5 µg/mL), as per standard protocols.Citation29 Stained samples were acquired on a MACSQuant® Analyzer 10 (Miltenyi Biotech) and data were analyzed with FlowJo v. 10.6 (FlowJo LLC).
DAMP emission
CALR exposure on the cell surface was measured by flow cytometry upon staining cells with a rabbit antibody specific for CALR (Abcam, #AB2907) at 4°C for 1 hour, followed by incubation with anti-rabbit IgG Alexa Fluor488® conjugates (Invitrogen, #A11070) plus 0.5 µg/mL PI for 30 min. As per gold-standard recommendations,Citation30 PI+ cells were excluded from the analysis. Extracellular ATP and HMGB1 levels were quantified with the luciferase-based Enliten ATP Assay (Promega) and the HMGB1 ELISA Kit (Tecan), respectively, as per manufacturer’s recommendations.
Animal experiments
Mice were maintained in specific pathogen-free conditions, and experiments followed the Guidelines for the Care and Use of Laboratory Animals guidelines. Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Weill Cornell Medical College (n° 2018–0002). Wild-type BALB/c or C57BL/6J mice (4–6 weeks old) were obtained from Taconic Bioscience.
Tumor growth
Female BALB/c or C57BL/6J mice were inoculated s.c. with 0.25 × 106 CT26 or 0.5 × 106 MC38 cells, respectively, in the right flank, and monitored routinely for tumor growth with a common lab caliper. When tumors reached an area of 15–25 mm2 (day 0), mice were randomly allocated to the following treatment groups (n = 7–8 per group): (1) vehicle i.p. biweekly (5 doses for CT26 tumors, 7 doses for MC38 tumors); (2) 90 mg/Kg PT-112 in 50 µL in phosphate buffer i.v. weekly (5 doses); (3) a programmed cell death 1 (PDCD1)-specific antibody (Bio X Cell, clone #RMP1-14), 10 mg/Kg i.p. biweekly (5 doses for CT26 tumors, 7 doses for MC38 tumors); (4) a CD274-specific antibody (Bio X Cell, clone #10F-9G2), 10 mg/Kg i.p. biweekly (5 doses for CT26 tumors, 7 doses for MC38 tumors); (5) PT-112 plus RMP1-14, as per the above; and (6) PT-112 plus 10F-9G2, as per the above. All injections were performed with 200 µL. Tumor growth was monitored routinely, and mice were euthanized when tumor size exceeded ethical limits or with manifestations of systemic disease (hunched posture, anorexia, weight loss). Mice achieving complete eradication of CT26 tumors in the PT-112 plus RMP1-14 treatment group within 35 days after treatment initiation were rechallenged s.c. with 0.25 × 106 CT26 cells to evaluate immunological memory.
Vaccination assays
One x 106 TSA cells treated in vitro with 150 µM CDDP, 2.5 µM mitoxantrone (MTX), or 150 µg/mL PT-112 for 24 hours were washed once and resuspended in 100 μL PBS for subcutaneous inoculation into the lower flank of 7 weeks old female BALB/c mice (vaccination). One week later, mice received 0.1 × 106 untreated TSA cells s.c. into the contralateral flank (challenge). Tumor incidence and growth were monitored routinely with a common lab caliper, and mice were euthanized when tumor size exceeded ethical limits or with manifestations of systemic disease (such as hunched posture, anorexia, and weight loss). Mice rejecting the challenge injection were re-challenged 60 days later with 0.1 × 106 untreated TSA cells in one flank, as a control for vaccination durability.
Abscopal assays
Two neoplastic lesions were established by inoculating 0.1 × 106 TSA cells s.c. into either lower flanks of female 4–9 weeks old BALB/c mice three days apart. Mice were routinely monitored for tumor growth at both sites by means of common lab caliper, and once primary tumors reached a surface area of 15–25 mm2 (day 0), mice were allocated to either of the following treatment groups: (1) 100 µL vehicle i.p. on days 2, 5 and 8; (2) a cytotoxic T lymphocyte associated protein 4 (CTLA4)-specific antibody (Bio X Cell, clone #9H10), 200 µg/mouse in 100 µL i.p., on days 2, 5 and 8; (3) 150 mg/Kg PT-112 in 50 µL phosphate buffer i.t. on day 0; and (4) 150 mg/Kg PT-112 in 50 µL phosphate buffer i.t. on day 0 plus 9H10, 200 µg/mouse in 100 µL PBS i.p., on days 2, 5 and 8. Mice were monitored for tumor growth at both disease sites and signs of systemic toxicity as above.
Immune infiltration
CT26 and MC38 tumors treated as above were harvested on day 12 and dissociated according to standard procedures for the assessment of immune cell infiltration,Citation31 upon staining with cocktails of fluorescent antibodies specific for CD3 (BioLegend, clone #17A2), CD4 (BioLegend, clone #GK1-5), CD8 (BioLegend, clone #53-6.7), CD11b (BioLegend, clone #M1/70), CD11c (BioLegend, clone #N418), CD25 (BioLegend, clone #PC61), CD45 (BioLegend, clone #30-F11), F4/80 (BioLegend, clone #BM8), FOXP3 (eBioscience, clone #FJK-16s), and a live/dead (L/D) stain (BioLegend, #423101). Stained samples were acquired on a FACSCalibur (Becton Dickinson) and data were analyzed with FlowJo v. 10.6 (FlowJo LLC).
Statistical analyses
Statistical significance on cell death, DAMP emission and immune infiltration was assessed by one-way ANOVA. Tumor surface was calculated as S = (π × A × B)/4, where A and B are the longest and shortest lesion diameter, respectively. Statistical significance on growth curves was assessed by two-way ANOVA, while statistical significance on Kaplan-Meier curves was assessed by hazard ratio (Log-rank) and Mantel-Cox tests.
Results
PT-112 exerts cytotoxic effects that are accompanied by the emission of immunostimulatory DAMPs
To characterize the cytostatic and cytotoxic activity of PT-112, we harnessed a commercial DNA-based test to estimate residual cell number upon exposing a large panel of 121 human cancer cell lines of various histological derivation to increasing concentrations of the drug for 72 hours. As expected, we identified a spectrum of sensitivities to PT-112, with IC50 values ranging from 0.287 µM (for human gastric adenocarcinoma AGS cells) to 222.14 µM (for human breast carcinoma MDAMB415 cells) (). Interestingly, mean IC50 values for cell lines of different histological derivation exhibited limited variation as compared to mean IC50 value for all other cell lines confounded (), with the sole exception of breast carcinoma cells, largely due to the extraordinary resistance of MDAMB415 cells. Since human cancer cells are intrinsically incompatible with in vivo immuno-oncology studies,Citation32 we decided to switch to utilize murine systems.
Table 1. Cytostatic and cytotoxic effects of PT-112 against human cancer cell lines (IC50, µM)
The proliferation of mouse colorectal carcinoma CT26 cells was virtually arrested upon exposure to PT-112 in vitro (), mostly in the absence of overt cytotoxicity (data not shown). Conversely, PT-112 efficiently killed mouse mammary carcinoma TSA cells in a dose- and time-dependent manner, as determined by flow cytometry upon co-staining with fluorescent probes for mitochondrial outer membrane permeabilization and plasma membrane rupture (). We therefore selected mouse TSA cells to investigate the ability of PT-112 to cause the emission of DAMPs that have been mechanistically linked to the activation of anticancer immunity by RCD.Citation21 We employed CDDP (which in our hands is unable to cause bona fide ICD)Citation24 as a negative control, and MTX (a potent ICD inducer)Citation33 as a positive control.
In line with previous findings from the Kroemer laboratory,Citation34 TSA cells responding to MTX exposed CALR on the outer leaflet of the plasma, and secreted abundant amounts of HMGB1 (–). Unexpectedly, exposure of TSA cells to CDDP also caused CALR exposure and ATP release, but poor HMBG1 secretion (–). Of note, CALR exposure by cancer cells exposed to CDDP has not been observed with mouse colorectal carcinoma CT26 cells,Citation24 but reportedly occurs in mouse ovarian carcinoma 2F8 cells,Citation35 pointing to some degree of variability across different cell types. Irrespective of this partially unexpected finding, PT-112 was highly efficient at causing the emission of ICD-associated DAMPs from TSA cells (–). Similar results have previously been obtained with human colorectal carcinoma HCT 116 cells,Citation7 suggesting that PT-112 may constitute a novel bona fide inducer of ICD in both mouse and human tumor models.
PT-112 causes bona fide ICD in vivo
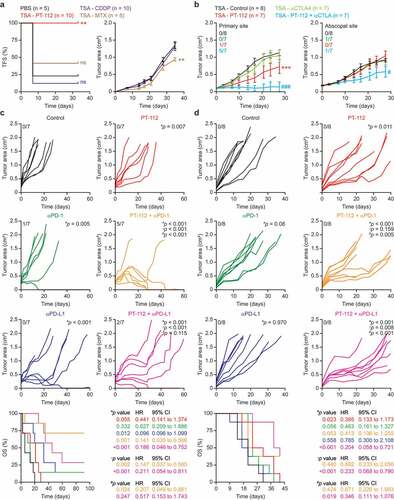
As surface CALR exposure, ATP release and HMGB1 secretion are all required, but not sufficient, for cancer cell death to be perceived as immunogenic,Citation36 we next set to evaluate the immunogenicity of PT-112-driven RCD in gold-standard vaccination assays.Citation30 To this aim, fully immunocompetent, wild-type BALB/c mice were vaccinated by subcutaneous inoculation of PBS (negative control) or TSA cells pre-exposed in vitro to a cytotoxic dose of CDDP, MTX, or PT-112. One week later, all mice were challenged contralaterally with living TSA cells and monitored over time for the ability of the latter to form progressing tumors. Neither PBS nor CDDP-treated TSA cells conferred a significant degree of immunological protection against the challenge injection (). In this specific sets of experiments, MTX-treated cells exhibited partial (but statistically significant) immunogenicity, as they enabled 40% tumor-free survival 35 days after challenge with living TSA cells, as well as with a reduction in the growth rate of tumors evolving despite vaccination (). Conversely, TSA cells succumbing in vitro to PT-112 conferred 100% immunological protection against the subsequent injection of living TSA cells (). Nine out of ten mice rejecting a first challenge with TSA cells were subcutaneously rechallenged 60 days later with TSA cells to check for the durability of protection. Such living TSA cells failed to generate progressing tumors in 5/9 mice (Suppl. ), suggesting that the immunological protection conferred by PT-112-treated cells is durable.
Figure 2. PT-112 induces bona fide ICD and can be combined with ICBs in vivo. (a). Tumor-free survival (TFS) and tumor area in BALB/c mice vaccinated with PBS or TSA cells exposed in vitro to 150 µg/mL PT-112, 150 µM cisplatin (CDDP), or 2.5 µM mitoxantrone (MTX) for 24 hours, and (one week later) challenged contralaterally with living TSA cells. Number of mice is indicated. Tumor areas are reported as means ± SEM. ns: not significant, **p < .01 (Log-rank for TFS, two-way ANOVA for tumor area), as compared to PBS-vaccinated mice. See also Suppl. Figure 1. (b). Growth of primary and abscopal TSA lesions established in immunocompetent, syngeneic BALB/c mice that were optionally allocated to receive 150 mg/Kg PT-112 i.t. in the context of optional, systemic CTLA4 blockage. Number of mice and incidence of disease eradication are indicated. Tumor growth data are reported as means ± SEM. ***p < .001 (two-way ANOVA), as compared to untreated mice; #p < .05, ###p < .001 (two-way ANOVA), as compared to mice treated with CTLA4 blockers. (c,d). Growth of CT26 (c) or MC38 (d) tumors established in immunocompetent, syngeneic BALB/c or C57BL/6J mice, respectively, that were allocated to receive 90 mg/Kg PT-112 weekly i.v. in the context of optional, biweekly systemic (i.p.) PD-1 or PD-L1 blockage (or PD-1 or PD-L1 blockage alone). Number of mice, incidence of disease eradication, overall survival (OS), hazard ratio (HR) and p values (two-way ANOVA for tumor growth, Mantel-Cox for OS) are reported. *compared to untreated mice; †compared to mice treated with PT-112; #compared to mice treated with PD-1 or PD-L1 blockers
These findings demonstrate that the demise of cancer cells driven by PT-112 administration is sufficiently immunogenic to protect tumor-naïve mice from a challenge with living cancer cells of the same type.
PT-112 causes systemic immune outreach in abscopal tumor models
Since vaccination assays are exquisitely sensitive, we decided to investigate the immunogenicity of PT-112 in abscopal settings, adapting a model and procedures that are generally employed for radiation oncology studies.Citation37 To this aim, BALB/c mice were used as hosts for the establishment of two slightly asynchronous TSA lesions (one on each flank), followed by the intratumoral administration of a systemically inactive dose of PT-112 to a single lesion (primary tumor) in the context of systemic CTLA4 blockage (which per se is also inactive in this model). In this setting, the growth of untreated (abscopal) lesions can be influenced only by the activation of robust immunity with systemic outreach. As intended, PT-112 exhibited limited activity upon intratumoral inoculation into TSA lesions, but such an effect was considerably potentiated by whole-body CTLA4 blockage, resulting in disease eradication at the primary site in 5 out of 7 mice (). Moreover, the growth of PT-112-naïve (secondary) tumors was reduced in mice receiving intratumoral PT-112 (at a systematically inactive dose) to the primary site plus a CTLA4-blocking antibody i.p. ().
These data demonstrate that local PT-112 therapy can synergize with CTLA4 blockage at the reversion of intratumoral immunosuppression and the activation of a potent immune response with systemic outreach affecting untreated disease sites.
PT-112 synergizes with ICBs to eradicate established mouse tumors
Reassured by the ability of PT-112 to trigger bona fide ICD, and to synergize with CTLA4 blockers in the initiation of systemic anticancer immunity (abscopal responses) in the TSA model, we next set to assess the synergism between PT-112 and ICBs targeting programmed cell death 1 (PDCD1, best known PD-1) or its main ligand PD-L1, and hence operating at the effector (rather than priming) phase. To this aim, we selected two mouse cancer cell lines that exhibit incomplete sensitivity to PT-112 and are syngeneic to different mouse strains, namely CT26 cells (syngeneic to BALB/c mice), and mouse colorectal carcinoma MC38 cells (syngeneic to C57BL/6J mice). PT-112 monotherapy reduced the growth of CT26 tumors established in immunocompetent BALB/c mice but was unable to extend overall survival to a statistically significant degree (). PD-L1 blockers synergized with PT-112 at extending the survival of CT26-bearing mice, although PD-L1 blockage mediated some anticancer activity (but virtually no effects on survival) per se (). Conversely, PD-1 blockers had limited therapeutic activity against CT26 lesions when employed as standalone agents (). However, therapeutic effects were pronounced when PT-112 was combined with PD-1 blockage, resulting in significant extensions in overall survival as compared to either therapy alone (). Of note, 5/7 mice receiving PT-112 plus PD-1 blockage achieved complete tumor eradication, four of which occurring within 35 days after initiation of treatment. These four mice rejected a novel challenge with a tumorigenic dose of CT26 cells, suggesting the activation of long-term immunological protection. Similar results were obtained in the MC38 model (). In this setting, however, PT-112 monotherapy was associated with a statistically significant increase in survival that was further enhanced by combination with PD-L1 (but not PD-1) blockers (). Of note, we were unable to document overt signs of toxicity (e.g., anorexia, hunched posture, weight loss) in any treatment group (data not shown).
Of note, both CT26 and MC38 tumors receiving PT-112 monotherapy exhibited a considerable decrease in the relative abundance of CD45+ cells, which was largely accounted for by a reduction in CD11b+ myeloid cells and was not altered by PD-1 or PD-L1 blockage (). In the CT26, (but not in the MC38) model, such a loss of CD11b+ was largely confined to immunosuppressive F4/80+ tumor-associated macrophages (TAMs), while the relative proportion of CD11c+F4/80− dendritic cells (DCs) increased (at least to some degree) (). Consistent with these findings, the microenvironment of PT-112-treated CT26 and MC38 tumors was enriched for CD3+ T cells, with a predominance of CD8+ cytotoxic T lymphocytes (CTLs) over CD4+ helper T cells (). Moreover, the CD4+ compartment of CT26 and MC38 tumors exposed to PT-112 exhibited reduced polarization toward an immunosuppressive CD25+FOXP3+ regulatory T (TREG) phenotype, in particular when PT-112 was combined with PD-1 or PD-L1 blockers ().
Figure 3. Immune infiltration of CT26 and MC38 responding to PT-112 plus ICBs. (a,b). Percentage of CD45+ (over total live), CD11b+ (over CD45+), F4/80+ (over CD11b+), and CD11c+ (over CD11b+) cells, and CD11c+ to F4/80+ cell ratio, in CT26 (a) and MC38 (b) tumors treated with PT-112 in the context of optional PD-1 (a) or (b) PD-L1 blockage. *p < .05, **p < .01, ***p < .001 (one-way ANOVA), as compared to untreated tumors; †p < .05, ††p < .01, †††p < .001 (one-way ANOVA), as compared to tumors treated with PT-112 only; #p < .05, ##p < .01, ###p < .001 (one-way ANOVA), as compared to tumors treated with PD-1 or PD-L1 blockers only, as relevant. See also Suppl. Figure 2a. (c,d). Percentage of CD3+ (over CD45+), CD8+ (over CD3+), CD4+ (over CD3+), and CD25+FOXP3+ (over CD4+) cells, and CD8+ to CD25+FOXP3+ cell ratio, in CT26 (c) and MC38 (d) tumors treated with PT-112 in the context of optional PD-1 (c) or (d) PD-L1 blockage. *p < .05, **p < .01, ***p < .001 (one-way ANOVA), as compared to untreated tumors; †p < .05 (one-way ANOVA), as compared to tumors treated with PT-112 only; #p < .05, ##p < .01, ###p < .001 (one-way ANOVA), as compared to tumors treated with PD-1 or PD-L1 blockers only, as relevant. See also Suppl. Figure 2b
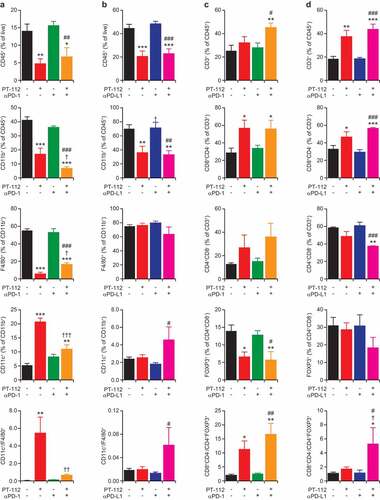
Altogether, these findings indicate that PT-112 favors the establishment of an immunostimulatory tumor microenvironment characterized by increased CD8+ CTL infiltration and reduced TAM- and TREG cell-dependent immunosuppression, and that some of these beneficial alterations can be boosted by PD-1 or PD-L1 blockage along with the activation of therapeutically relevant anticancer immunity.
Discussion
This is the first demonstration that PT-112, a novel platinum-pyrophosphate conjugate under clinical development, causes the emission of immunostimulatory DAMPs by dying cancer cells (), drives bona fide ICD and hence can initiate anticancer immunity per se (), synergizing with ICBs in the context of superior immune infiltration ( and ). These findings are in line with preliminary clinical evidence on the use of PT-112 in patients with solid tumors, either as a standalone therapeutic agent (NCT02266745),Citation11,Citation12 or in combination with the PD-L1 blocker avelumab (NCT03409458).Citation14
While platinum derivatives such as CDDP, carboplatin and oxaliplatin have been extensively employed for the treatment of multiple solid tumors,Citation1-Citation3 they (1) are frequently associated with toxicities and relatively prone to cause acquired resistance,Citation4,Citation5 and (2) have limited activity in bone lesions.Citation38,Citation39 Moreover, the actual value of platinum derivatives in the context of ICB-based immunotherapy remains to be determined. Indeed, ICBs have been successfully combined with standard-of-care platinum-based chemotherapy in patients with a variety of ICB-sensitive tumors, such as non-small cell lung carcinoma.Citation40 However, little benefit has been documented from the addition of ICBs to chemotherapy with current platinum derivatives in ICB-resistant tumors, such as microsatellite stable colorectal tumors.Citation41
Conversely, PT-112 appears to possess a unique combination of factors, including (1) safety in heavily pretreated patients,Citation11,Citation12 (2) an improved pharmacokinetic and pharmacodynamic profile including (but not limited to) a prominent osteotropism,Citation13 (3) monotherapy efficacy in patients with pulmonary tumors, prostate cancer and thymoma,Citation11,Citation12 (4) combinatorial efficacy in the context of PD-L1 blockage in men with castration resistant prostate cancer,Citation14 and (5) activity in immunocompetent mouse models of breast and colorectal cancer linked to the initiation of ICD, as demonstrated in this paper.
Thus, PT-112 stands out as a promising agent for the treatment of solid neoplasms that display limited sensitivity to ICBs and/or originate or tend to metastasize to the bone.Citation42-Citation45 Based on our preclinical findings, it is tempting to speculate that PT-112 may cause a robust wave of ICD associated with an increased abundance of antigenic material from malignant cells as well as with the emission of chemotactic and immunostimulatory signals that altogether (re)activate anticancer immunity, de facto setting the stage for efficacious ICB-based immunotherapy. Additional experiments are required to validate this working model. Irrespective of unknowns related to the molecular mechanism of action of PT-112 and its potential ICD-independent immunomodulatory activity, this novel platinum-pyrophosphate conjugate stands out as a promising agent for the implementation of innovative ICB-based immunochemotherapeutic regimens.
Disclosure of Potential Conflicts of Interest
TDA is a full-time employee of and owns equity in Phosplatin Therapeutics. LG provides remunerated consulting to Astra Zeneca, Boehringer Ingelheim, Inzen and the Luke Heller TECPR2 Foundation, and he is member of Scientific Advisory Committees for Boehringer Ingelheim and OmniSEQ. As per standard operations at Oncoimmunology, LG has been excluded from all steps of editorial evaluation of the present article.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Supplemental Material
Download ()Supplemental Material
Download ()Supplemental Material
Download ()Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Andre T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, Topham C, Zaninelli M, Clingan P, Bridgewater J, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004;350:2343–10. doi:10.1056/NEJMoa032709.
- Arriagada R, Bergman B, Dunant A, Le Chevalier T, Pignon JP, Vansteenkiste J. Cisplatin-based adjuvant chemotherapy in patients with completely resected non-small-cell lung cancer. N Engl J Med. 2004;350:351–360.
- International Collaborative Ovarian Neoplasm G. Paclitaxel plus carboplatin versus standard chemotherapy with either single-agent carboplatin or cyclophosphamide, doxorubicin, and cisplatin in women with ovarian cancer: the ICON3 randomised trial. Lancet. 2002;360:505–515. doi:10.1016/S0140-6736(02)09738-6.
- Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi:10.1038/onc.2011.384.
- Guminski AD, Harnett PR, deFazio A. Scientists and clinicians test their metal-back to the future with platinum compounds. Lancet Oncol. 2002;3:312–318. doi:10.1016/S1470-2045(02)00733-7.
- Bose RN, Maurmann L, Mishur RJ, Yasui L, Gupta S, Grayburn WS, Hofstetter H, Salley T. Non-DNA-binding platinum anticancer agents: cytotoxic activities of platinum-phosphato complexes towards human ovarian cancer cells. Proc Natl Acad Sci U S A. 2008;105:18314–18319. doi:10.1073/pnas.0803094105.
- Ames T, Slusher B, Wozniak K, Takase Y, Shimizu H, Nishibata-Kobayashi K, Kanada-Sonobe RM, Kerns W, Fong KL, Pourquier P, et al. Findings across pre-clinical models in the development of PT-112, a novel investigational platinum-pyrophosphate anti-cancer agent. Eur J Cancer. 2016;69:S153. doi:10.1016/S0959-8049(16)33054-4.
- Yamazaki T, Ames TD, Galluzzi L. Abstract B199: potent induction of immunogenic cell death by PT-112. Cancer Immunol Res. 2019;7:B199–B.
- Corte-Rodriguez M, Espina M, Sierra LM, Blanco E, Ames T, Montes-Bayon M, Sanz-Medel A. Quantitative evaluation of cellular uptake, DNA incorporation and adduct formation in cisplatin sensitive and resistant cell lines: comparison of different Pt-containing drugs. Biochem Pharmacol. 2015;98:69–77. doi:10.1016/j.bcp.2015.08.112.
- Bose RN, Moghaddas S, Belkacemi L, Tripathi S, Adams NR, Majmudar P, McCall K, Dezvareh H, Nislow C. Absence of activation of DNA repair genes and excellent efficacy of phosphaplatins against human ovarian cancers: implications to treat resistant cancers. J Med Chem. 2015;58:8387–8401. doi:10.1021/acs.jmedchem.5b00732.
- Karp DD, Camidge DR, Bryce AH, Jimeno J, Infante JR. A phase I study of PT-112 in advanced solid tumors. J Clin Oncol. 2017;35:2519. doi:10.1200/JCO.2017.35.15_suppl.2519.
- Karp DD, Camidge DR, Infante JR, Ames TD, Jimeno JM, Bryce AH. PT-112: A well-tolerated novel immunogenic cell death (ICD) inducer with activity in advanced solid tumors. Ann Oncol. 2018;29. doi:10.1093/annonc/mdy279.424.
- Ames TD, Sharik ME, Rather GM, Hochart G, Bonnel D, Linehan S, Stauber J, Wing RA, Jimeno JJ, Medina D, et al. Translational research of PT-112, a clinical agent in advanced phase i development: evident bone tropism, synergy in vitro with bortezomib and lenalidomide, and potent efficacy in the Vk*MYC mouse model of multiple myeloma. Blood. 2017;130:1797.
- Bryce AH, Dronca RS, Costello BA, Infante JR, Ames TD, Jimeno JM, Karp DD. PT-112 in advanced metastatic castrate-resistant prostate cancer (mCRPC), as monotherapy or in combination with PD-L1 inhibitor avelumab: findings from two phase I studies. J Clin Oncol. 2020;38.
- Galluzzi L, Chan TA, Kroemer G, Wolchok JD, Lopez-Soto A. The hallmarks of successful anticancer immunotherapy. Sci Transl Med. 2018;10:eaat7807. doi:10.1126/scitranslmed.aat7807.
- Tang J, Yu JX, Hubbard-Lucey VM, Neftelinov ST, Hodge JP, Lin Y. Trial watch: the clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat Rev Drug Discov. 2018;17:854–855. doi:10.1038/nrd.2018.210.
- Taghizadeh H, Marhold M, Tomasich E, Udovica S, Merchant A, Krainer M. Immune checkpoint inhibitors in mCRPC - rationales, challenges and perspectives. Oncoimmunology. 2019;8:e1644109. doi:10.1080/2162402X.2019.1644109.
- Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi:10.1016/j.cell.2015.03.030.
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi:10.1126/science.aaa8172.
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
- Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi:10.1038/nri.2016.107.
- Galluzzi L, Yamazaki T, Kroemer G. Linking cellular stress responses to systemic homeostasis. Nat Rev Mol Cell Biol. 2018;19:731–745. doi:10.1038/s41580-018-0068-0.
- Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–491. doi:10.1038/onc.2009.356.
- Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, Michaud M, Menger L, Gdoura A, Tajeddine N, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. 2011;30:1147–1158. doi:10.1038/onc.2010.500.
- Tatsuno K, Yamazaki T, Hanlon D, Han P, Robinson E, Sobolev O, Yurter A, Rivera-Molina F, Arshad N, Edelson RL, et al. Extracorporeal photochemotherapy induces bona fide immunogenic cell death. Cell Death Dis. 2019;10:578. doi:10.1038/s41419-019-1819-3.
- Park SJ, Ye W, Xiao R, Silvin C, Padget M, Hodge JW, Van Waes C, Schmitt NC. Cisplatin and oxaliplatin induce similar immunogenic changes in preclinical models of head and neck cancer. Oral Oncol. 2019;95:127–135. doi:10.1016/j.oraloncology.2019.06.016.
- Humeau J, Levesque S, Kroemer G, Pol JG. Gold standard assessment of immunogenic cell death in oncological mouse models. Methods Mol Biol. 2019;1884:297–315.
- Yamazaki T, Martinez AB, Rybstein M, Chen J, Sato A, Galluzzi L. Methods to detect immunogenic cell death in vivo. Methods Mol Biol. 2020;2055:433–452.
- Hoffmann J, Vitale I, Buchmann B, Galluzzi L, Schwede W, Senovilla L, Skuballa W, Vivet S, Lichtner RB, Vicencio JM, et al. Improved cellular pharmacokinetics and pharmacodynamics underlie the wide anticancer activity of sagopilone. Cancer Res. 2008;68:5301–5308. doi:10.1158/0008-5472.CAN-08-0237.
- Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3:e955691. doi:10.4161/21624011.2014.955691.
- Fucikova J, Rakova J, Hensler M, Kasikova L, Belicova L, Hladikova K, Truxova I, Skapa P, Laco J, Pecen L, et al. TIM-3 Dictates Functional Orientation of the Immune Infiltrate in Ovarian Cancer. Clin Cancer Res. 2019;25:4820–4831. doi:10.1158/1078-0432.CCR-18-4175.
- Buque A, Galluzzi L. Modeling tumor immunology and immunotherapy in mice. Trends Cancer. 2018;4:599–601. doi:10.1016/j.trecan.2018.07.003.
- Garg AD, More S, Rufo N, Mece O, Sassano ML, Agostinis P, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology. 2017;6:e1386829. doi:10.1080/2162402X.2017.1386829.
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi:10.1038/nm1523.
- Grabosch S, Bulatovic M, Zeng F, Ma T, Zhang L, Ross M, Brozick J, Fang Y, Tseng G, Kim E, et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene. 2019;38:2380–2393. doi:10.1038/s41388-018-0581-9.
- Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, Yamazaki T, Sukkurwala AQ, Michaud M, Mignot G, et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med. 2012;4:143ra99. doi:10.1126/scitranslmed.3003807.
- Rodriguez-Ruiz ME, Buque A, Hensler M, Chen J, Bloy N, Petroni G, Sato A, Yamazaki T, Fucikova J, Galluzzi L, et al. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology. 2019;8:e1655964. doi:10.1080/2162402X.2019.1655964.
- Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12:6243s–9s. doi:10.1158/1078-0432.CCR-06-0931.
- Coleman RE. Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treat Rev. 2001;27:165–176. doi:10.1053/ctrv.2000.0210.
- Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, Gentzler RD, Martins RG, Stevenson JP, Jalal SI, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016;17:1497–1508. doi:10.1016/S1470-2045(16)30498-3.
- Morse MA, Hochster H, Benson A. Perspectives on treatment of metastatic colorectal cancer with immune checkpoint inhibitor therapy. Oncologist. 2019. doi:10.1634/theoncologist.2019-0176.
- Rugo HS, Delord JP, Im SA, Ott PA, Piha-Paul SA, Bedard PL, Sachdev J, Tourneau CL, van Brummelen EMJ, Varga A, et al. Safety and antitumor activity of pembrolizumab in patients with estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer. Clin Cancer Res. 2018;24:2804–2811. doi:10.1158/1078-0432.CCR-17-3452.
- Tucker MD, Zhu J, Marin D, Gupta RT, Gupta S, Berry WR, Ramalingam S, Zhang T, Harrison M, Wu Y, et al. Pembrolizumab in men with heavily treated metastatic castrate-resistant prostate cancer. Cancer Med. 2019;8:4644–4655. doi:10.1002/cam4.v8.10.
- Aroldi F, Zaniboni A. Immunotherapy for pancreatic cancer: present and future. Immunotherapy. 2017;9:607–616. doi:10.2217/imt-2016-0142.
- Marino S, Petrusca DN, Roodman GD. Therapeutic targets in myeloma bone disease. Br J Pharmacol. 2019. doi:10.1111/bph.14889.